1. Introduction
Breast cancer poses a significant global health challenge, with its complexity stemming from diverse molecular subtypes that exhibit distinct responses to various treatment modalities [
1]. Among the five different subtypes, two prominent challenges have emerged in the management of breast cancer: resistance to endocrine therapy in estrogen receptor ER(+) tumors [
2] and heterogeneous response to anti-HER2 therapy in HER2(+) tumors [
3]. In ER(+) breast cancer, resistance to endocrine therapy often develops, limiting the effectiveness of treatment [
4]. Additionally, patients with HER2(+) breast cancer may exhibit varying responses to anti-HER2 targeted therapies, with some developing resistance over time [
5]. These challenges highlight the urgent need for novel predictive biomarkers and therapeutic targets to guide treatment decisions and improve outcomes in patients with breast cancer.
Loss of cell–cell adhesion is a hallmark of cancer progression and metastasis, allowing tumor cells to disseminate from the primary site [
6]. Tight junction proteins, notably the claudin (CLDN) family, are critical for maintaining epithelial cell adhesion, polarity, and barrier function [
7]. Aberrant expression of claudins can compromise junctional integrity and has been implicated in tumor invasion and metastasis in various cancers, including breast cancer [
8]. Indeed, several claudin family members have been identified as prognostic indicators or mediators of breast cancer progression. For example, reduced CLDN1 expression correlates with higher recurrence rates and poorer survival in breast cancer [
9,
10]. Conversely, CLDN2 overexpression has been shown to promote breast cancer metastasis to the liver through interactions between tumor cells and hepatocytes [
11,
12]. Alterations in other claudins further underscore their diverse roles: elevated CLDN4 expression is associated with lymph node metastasis and enhanced cancer cell stemness, while loss of CLDN6 can increase breast cancer cell motility and resistance to apoptosis, facilitating invasion [
13,
14,
15,
16].Another study suggested that CLDN6 is transcriptionally upregulated by HIF-1α under hypoxic conditions [
17]. Loss of CLDN6 may lead to increased HIF-1α-driven breast cancer metastasis in a SUMOylation-dependent manner [
18]. Martin et al. [
19] demonstrated that overexpression of CLDN20 in breast cancer cells decreases TER and thus increases their motility and reduced trans-epithelial resistance. Collectively, these findings suggest that the dysregulation of tight junction molecules is an important contributor to breast cancer aggressiveness and that specific claudins may serve as valuable prognostic biomarkers.
Among the Claudin family, CLDN8 remains relatively unexplored in breast cancer. CLDN8 is known to participate in tight junction formation and epithelial cohesion; however, its function in tumor biology is not well defined [
8]. Early evidence indicates that CLDN8 expression is frequently downregulated in breast tumors [
20]. Clinically, low CLDN8 levels have been associated with a higher incidence of lymph node metastasis and poorer outcomes in patients with breast cancer [
20]. Notably, CLDN8 expression appears to positively correlate with androgen receptor (AR) status in breast tumors, and patients with concomitantly low CLDN8 and low AR expression have particularly unfavorable prognoses [
20]. These observations suggest that CLDN8 may act as a context-dependent tumor suppressor or marker of a less aggressive, more differentiated tumor phenotype. However, the role of CLDN8 in influencing the response to therapy remains unclear. Given the persistent challenges of endocrine resistance in ER(+) disease and treatment failure in some HER2(+) cases, understanding whether CLDN8 impacts sensitivity to these therapies could be key to improving treatment strategies. In this study, we set out to investigate the significance of CLDN8 in breast cancer outcomes and treatment response. We hypothesized that CLDN8 expression may stratify patient prognosis and predict their responsiveness to different therapies. To test this hypothesis, we analyzed clinical breast cancer specimens to correlate CLDN8 expression with disease-free survival and treatment efficacy across various patient subgroups. We further performed in vitro experiments using breast cancer cell lines with modulated CLDN8 expression (knockdown and control) to assess the influence of CLDN8 on cellular behavior and drug sensitivity. A panel of nine therapeutic agents was selected to reflect the major treatment modalities in breast cancer care: chemotherapeutic drugs (Docetaxel, Paclitaxel, Cisplatin, and Methotrexate), HER2-targeted tyrosine kinase inhibitors (Lapatinib and Neratinib), and endocrine therapies (Tamoxifen, Fulvestrant, and Anastrozole). By examining CLDN8’s impact on sensitivity to these treatments, we aimed to bridge our laboratory findings with clinical relevance. Although our mechanistic studies were conducted in vitro, the chosen drugs mirror standard clinical therapies, allowing us to infer how alterations in CLDN8 might affect patient responses in real-world settings. Through this comprehensive approach, we address whether CLDN8 can serve as a predictive biomarker for therapy outcomes and explore the potential of targeting CLDN8 in future breast cancer treatment strategies. Our work thereby seeks to clarify the clinical value of CLDN8 in breast cancer and lay a foundation for more personalized therapeutic interventions.
3. Discussion
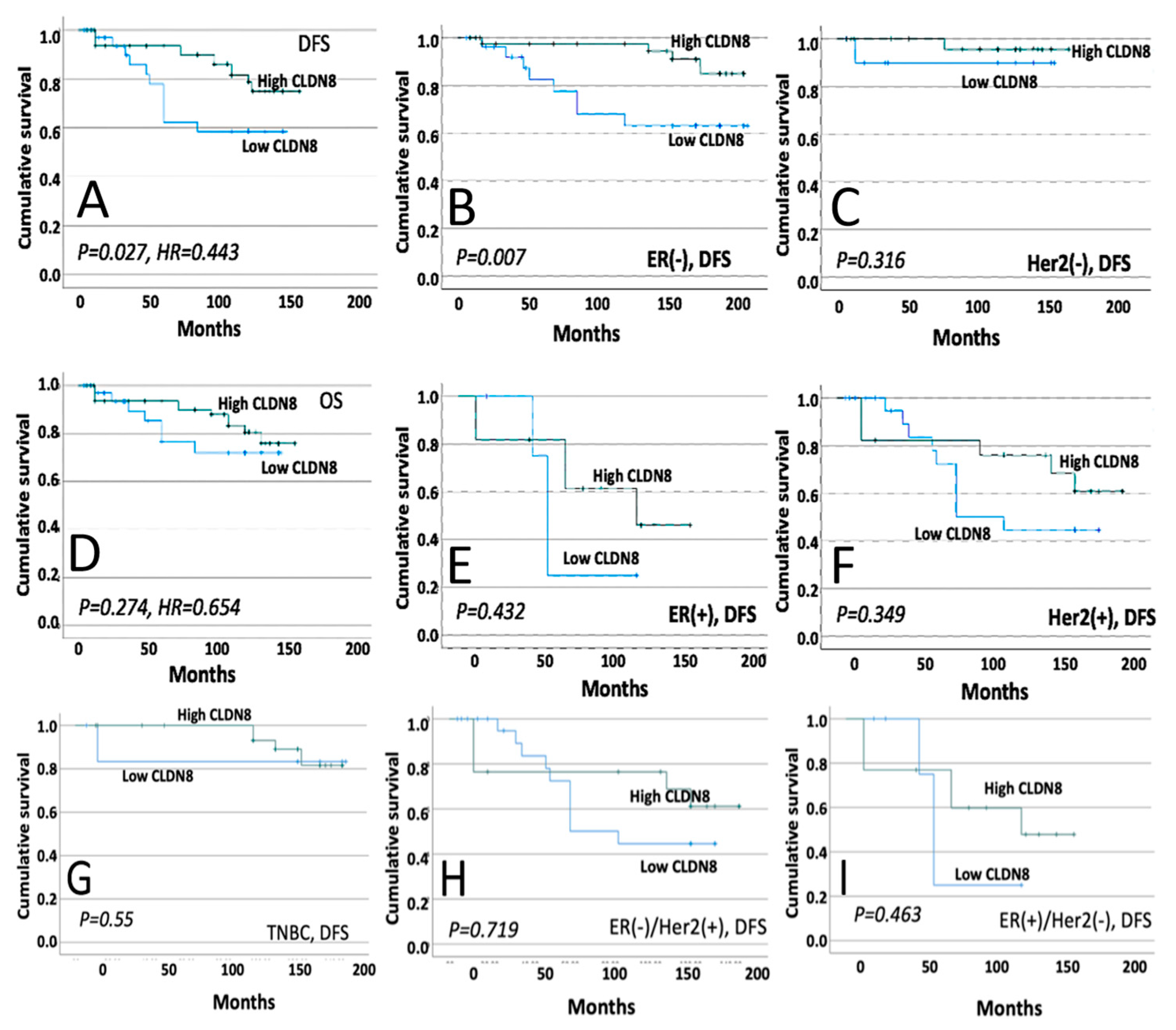
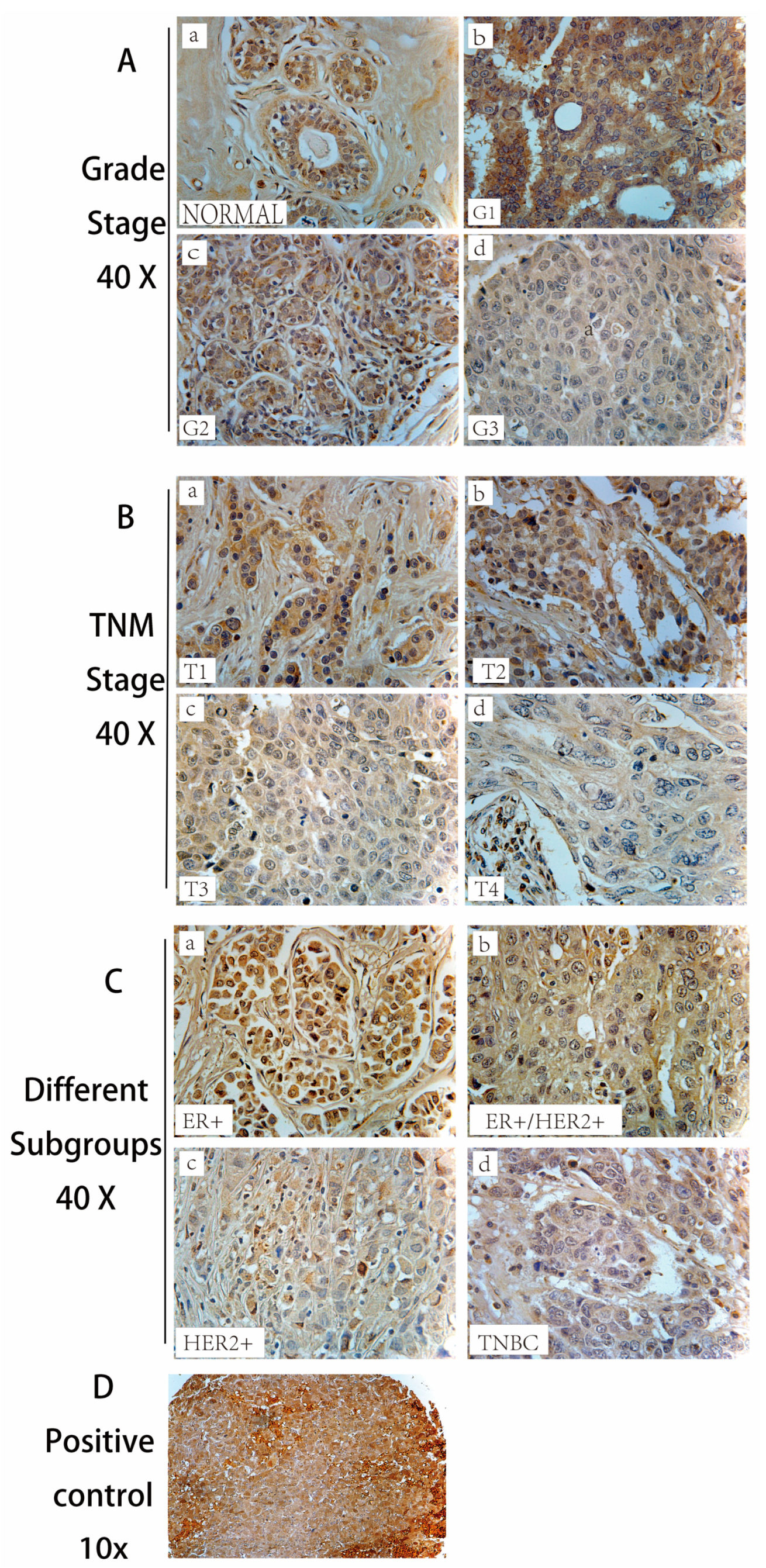
This study systematically explored the role of CLDN8 in breast cancer progression, prognosis, and treatment responses. Immunohistochemistry (IHC) analysis revealed a significant reduction in CLDN8 expression as the tumor grade and TNM stage increased. In normal breast tissue, CLDN8 is primarily localized to the cell membrane, whereas in high-grade tumors, its expression is diffuse and cytoplasmic. This shift in localization suggests a loss of tight junction integrity, which may contribute to increased tumor aggressiveness and metastatic potential. Furthermore, CLDN8 expression varied among different molecular subtypes, with ER(+) and HER2(+) tumors exhibiting moderate expression levels, whereas TNBC displayed significantly lower or undetectable expression levels. These findings highlight the complex role of CLDN8 in maintaining epithelial structure and regulating tumor progression. Clinical data analysis demonstrated that high CLDN8 expression was associated with improved disease-free survival (DFS), particularly in ER- patients, whereas no significant association was observed with overall survival (OS). The lack of correlation with OS suggests that while CLDN8 may influence early tumor progression, other factors may contribute to long-term survival outcomes.
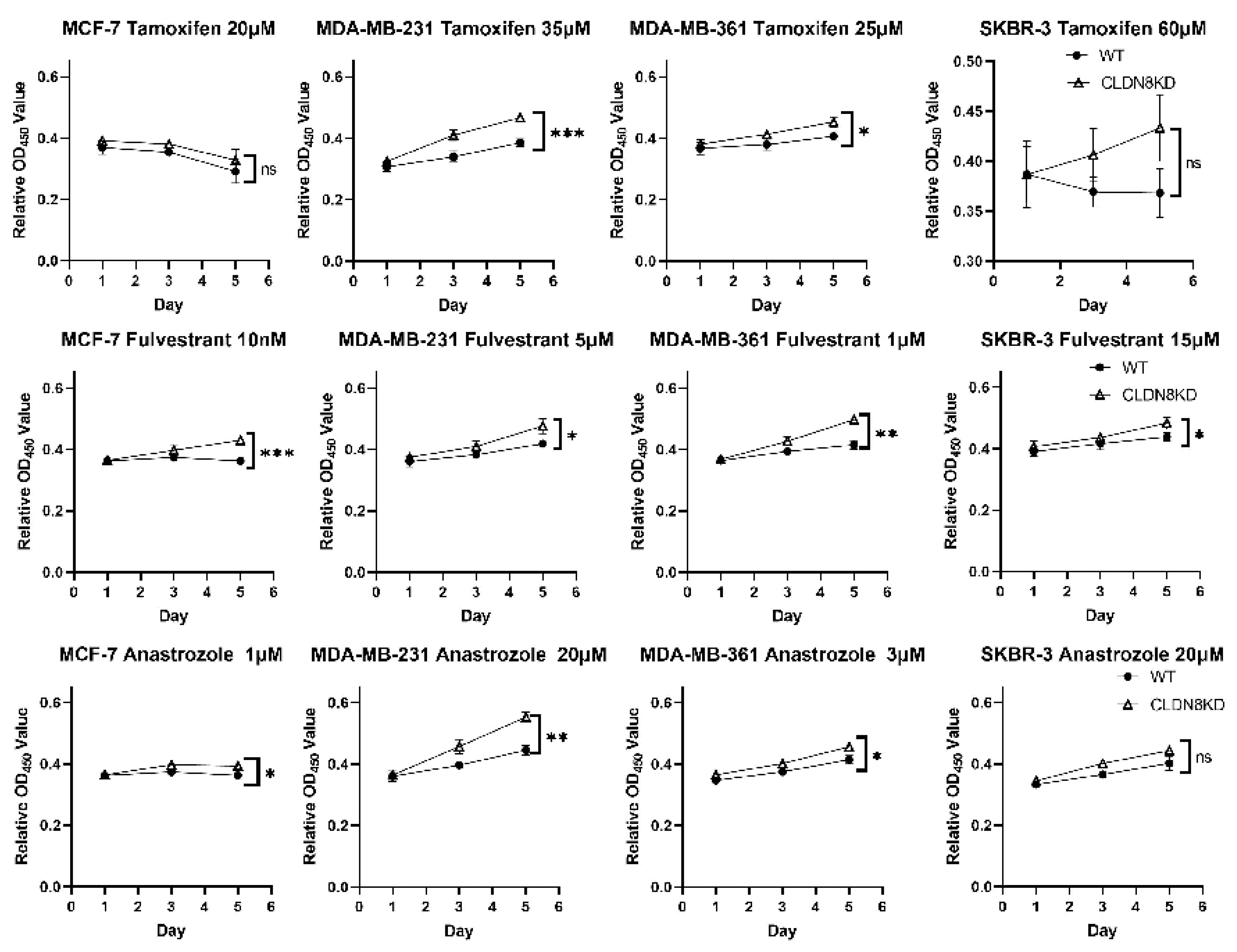
High CLDN8 expression appears to enhance hormone-driven tumor differentiation while buffering cells against cytotoxic stress, explaining its context-dependent effects. In one study, CLDN8 was enriched in luminal breast cancers (ER+/PR+), correlating with low grade, low Ki-67, and better prognosis [
21]. This tight junction protein helps maintain epithelial polarity and cell–cell adhesion, which supports an endocrine-responsive phenotype. For example, CLDN8 knockdown in ER(+) cell models led to reduced efficacy of endocrine treatments (Tamoxifen, Fulvestrant, Anastrozole), suggesting that CLDN8 preserves estrogen receptor signaling and epithelial gene expression needed for anti-estrogen therapies to work. Consistently, patients with high CLDN8 expression have improved survival with endocrine therapy. One reason is that well-differentiated, CLDN8-rich tumors remain dependent on hormone pathways and lack mesenchymal traits, making them more susceptible to growth arrest by endocrine therapy. CLDN8’s co-expression with the androgen receptor (AR) in luminal tumors further underscores its association with a hormonally regulated, less aggressive state [
20]. Notably, AR-driven CLDN8 can activate pro-growth MAPK/AKT signaling in certain contexts, but in ER(+) breast cancer, this may paradoxically reinforce luminal characteristics that endocrine therapy can target effectively [
22].
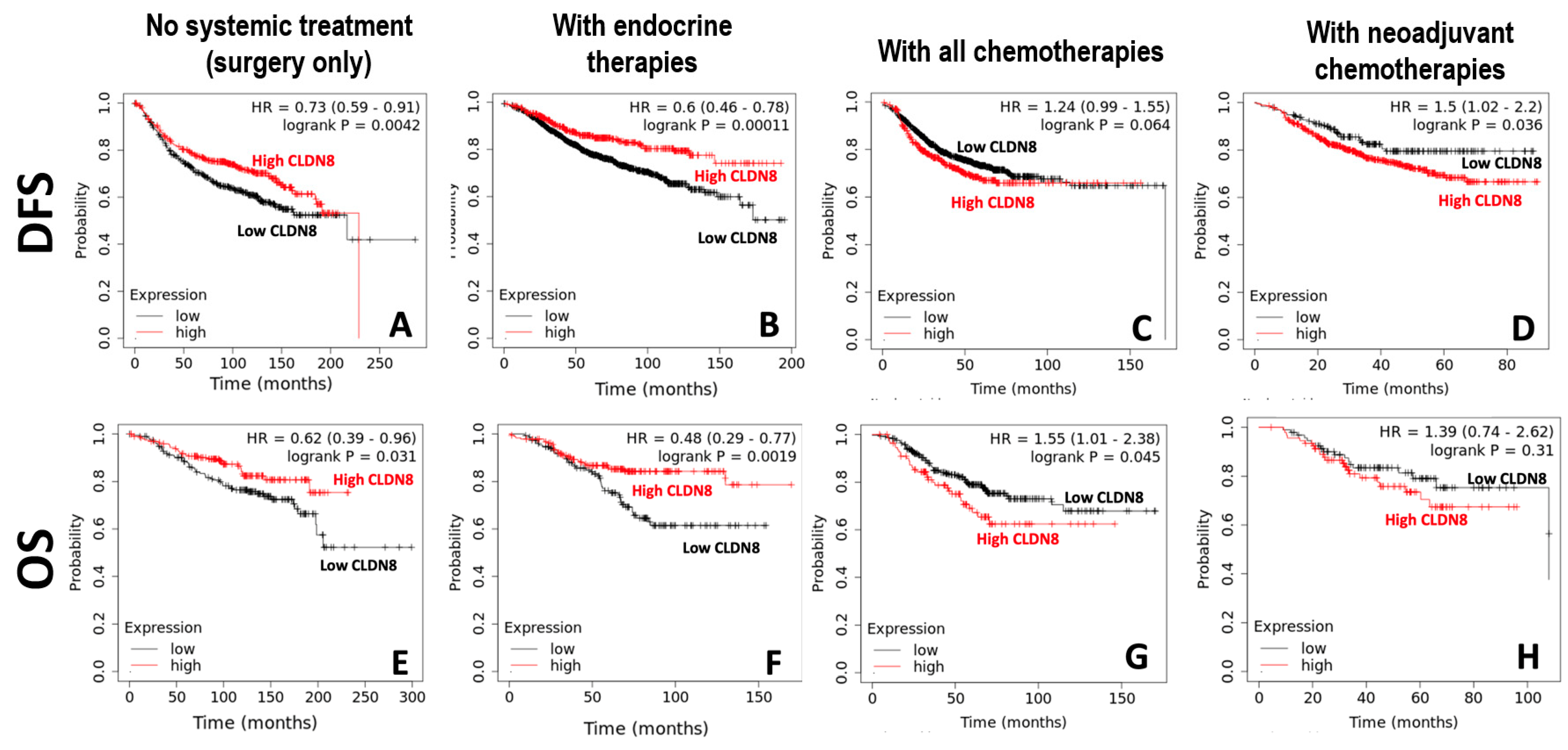
This observation aligns with our in vitro findings, where CLDN8 knockdown led to increased resistance to endocrine therapies, including Tamoxifen, Fulvestrant, and Anastrozole, particularly in ER(+) models. These findings suggest that CLDN8 may play a role in maintaining ER-mediated signaling, thereby influencing the efficacy of endocrine therapy.
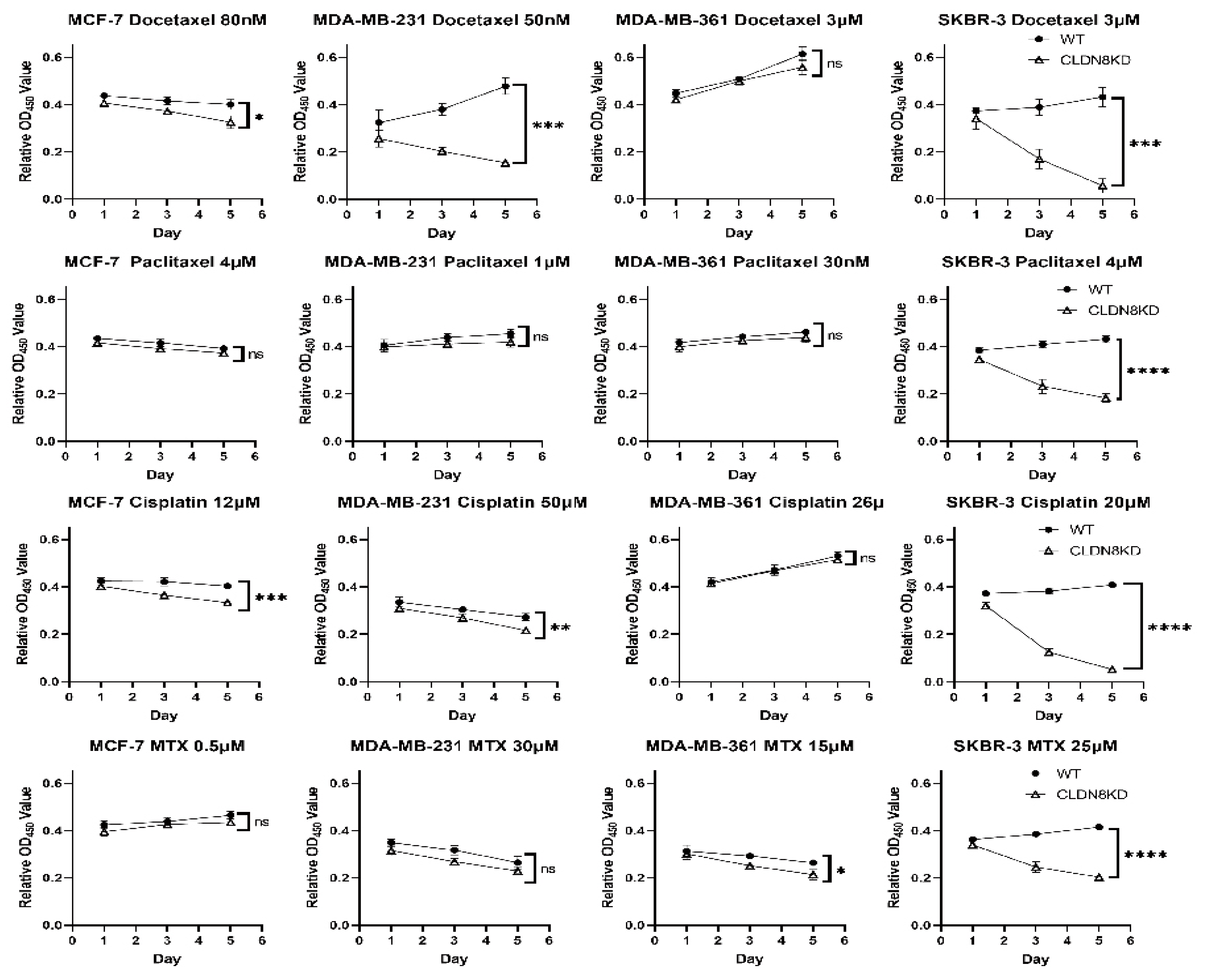
In contrast to its beneficial role in endocrine therapy response, high CLDN8 expression was associated with reduced sensitivity to chemotherapy. Patients with elevated CLDN8 levels demonstrated poorer responses to chemotherapeutic agents, a trend further validated through in vitro experiments. CLDN8 knockdown significantly decreased the IC50 values for Docetaxel, Paclitaxel, Cisplatin, and Methotrexate, indicating that CLDN8 may contribute to chemotherapy resistance by preserving epithelial characteristics and reducing apoptotic susceptibility.
CLDN8-rich tumors tend to be more epithelial and less proliferative, which is unfavorable for chemotherapy response [
23]. Strong tight junctions and cell polarity can impede drug penetration and foster anti-apoptotic signals via cell–cell contact. Indeed, we observed that knocking down CLDN8 significantly increased sensitivity to chemotoxic agents (e.g., Docetaxel and Cisplatin), lowering the IC50 and enhancing cell death, whereas CLDN8-intact cells were relatively chemoresistant. This aligns with reports that high claudin expression can confer drug resistance; for instance, claudin-7 and claudin-1/2 upregulation promotes Cisplatin resistance in pancreatic and lung cancer cells [
22]. Intact junctions may prevent chemotherapy-induced apoptosis by sustaining survival pathways and preventing complete epithelial-to-mesenchymal transition (EMT). In other cancers, CLDN8 suppresses EMT and invasiveness via the AKT pathway, meaning its loss leads to a more mesenchymal, chemosensitive state [
22]. Conversely, CLDN8 retention maintains cells in an adherent, less apoptotic configuration.
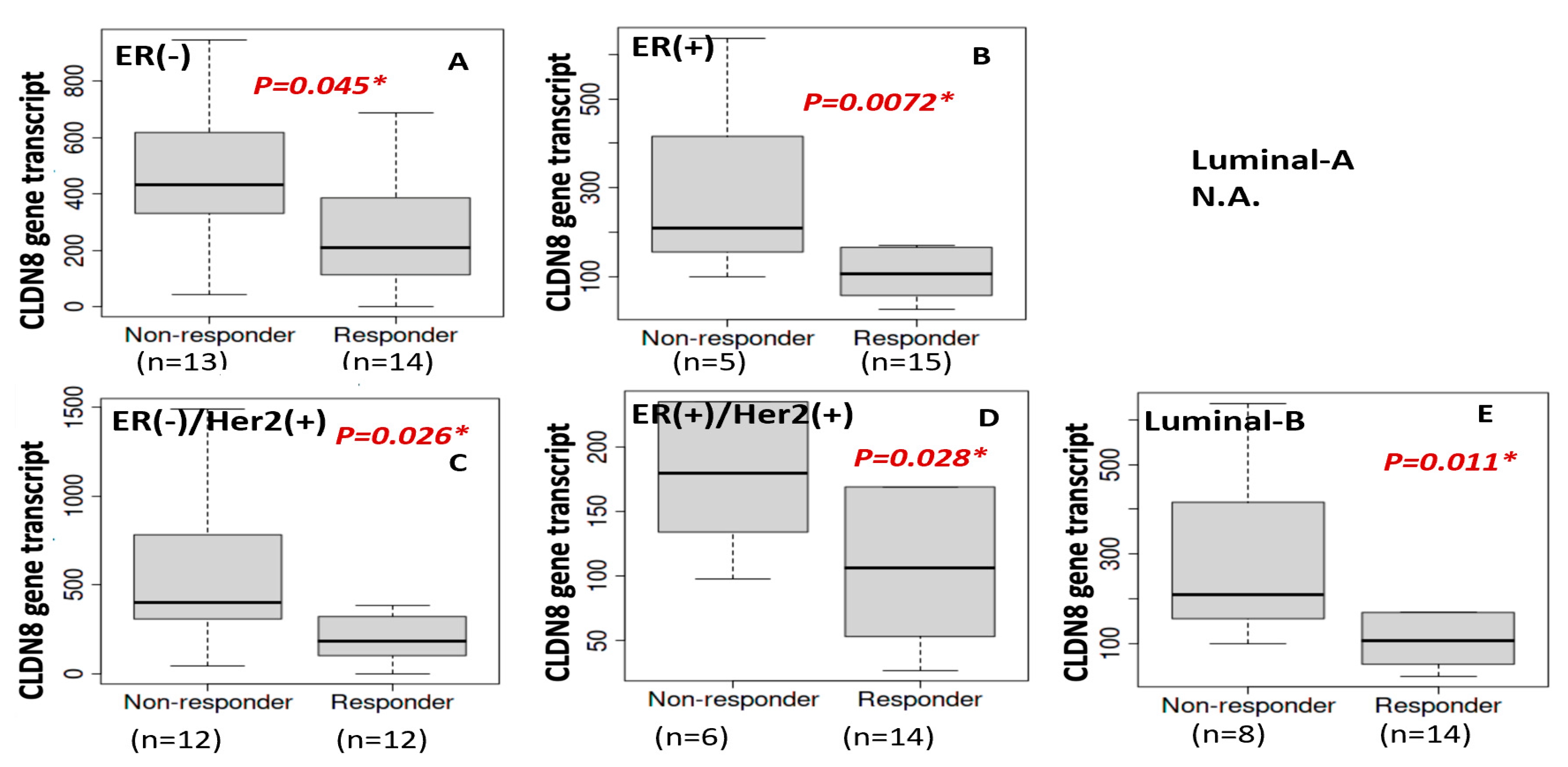
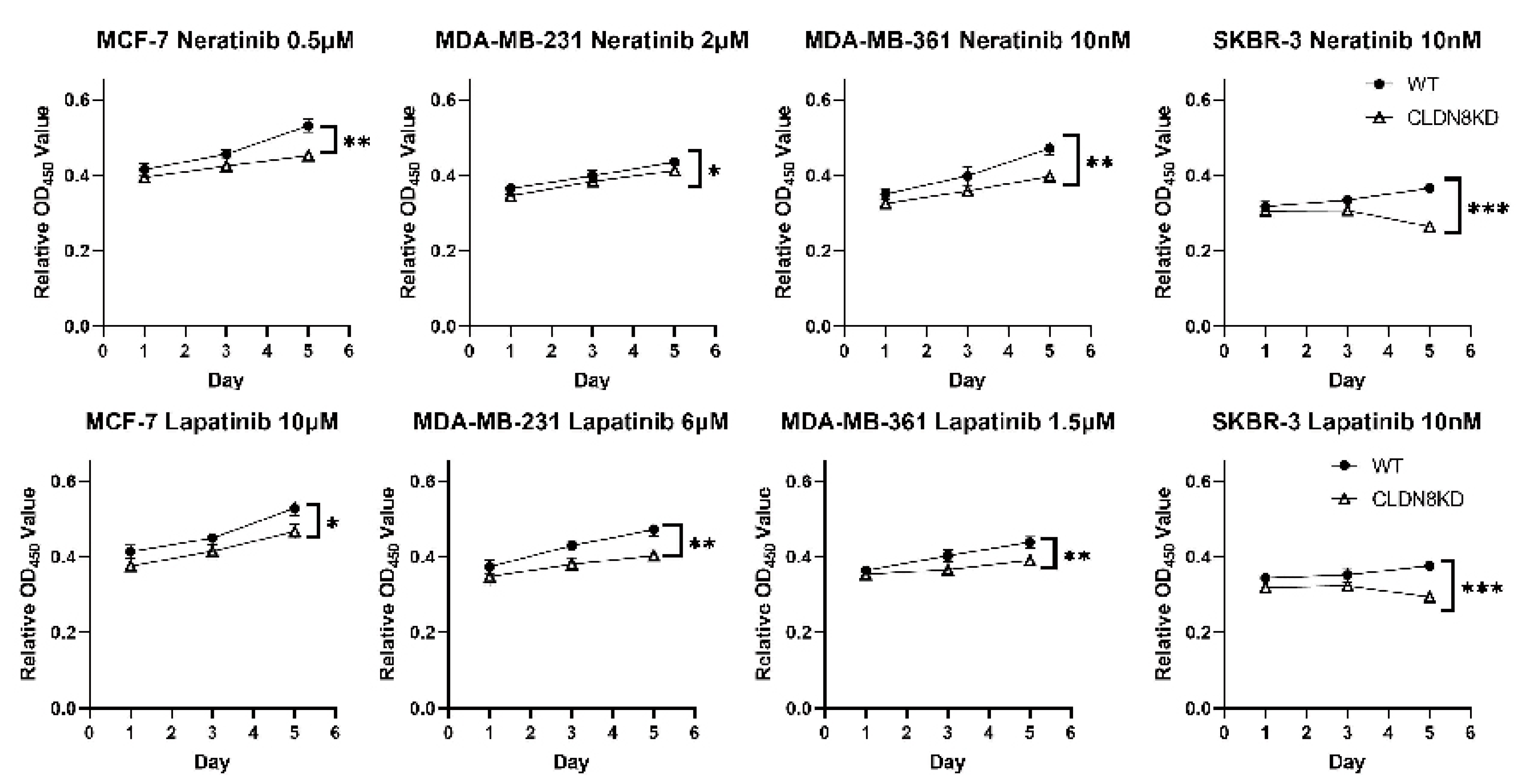
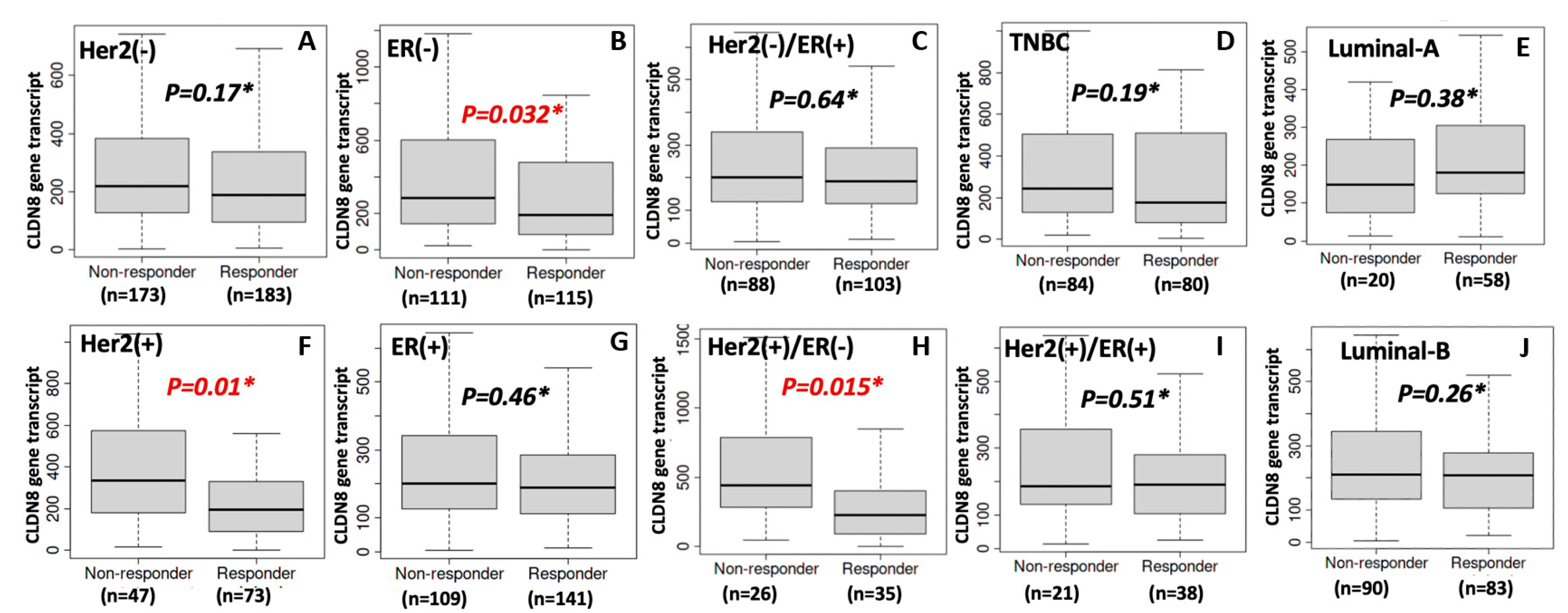
Another critical finding was the inverse relationship between CLDN8 expression and anti-HER2 therapy response. Patients with lower CLDN8 levels exhibited better responses to HER2-targeting agents, and this observation was supported by in vitro experiments, where CLDN8 knockdown led to significantly lower IC50 values for Neratinib and Lapatinib. These results suggest that CLDN8 may modulate HER2 signaling pathways and contribute to resistance mechanisms in HER2(+) breast cancer.
Similarly, a tight junction–rich microenvironment hinders effective HER2-targeted therapy. Anti-HER2 monoclonal antibodies (e.g., trastuzumab) partly rely on immune-mediated mechanisms and tumor accessibility; CLDN8-high tumors, being more “solid” and less infiltrated, may respond suboptimally. Moreover, CLDN8’s association with AR and epithelial differentiation implies that CLDN8-high HER2(+) cells might activate alternate survival pathways or be less “addicted” to HER2 signaling. This could reduce their dependence on HER2 and blunt the efficacy of HER2 blockade. Supporting this, researchers have found that breaking the integrity of tight junctions can improve treatment outcomes. For example, an anti-CLDN4 antibody increased Paclitaxel uptake and apoptosis in breast cancer models, enhancing chemosensitivity and reducing metastasis [
24].
Taken together, these findings suggest that CLDN8 could serve as a biomarker for predicting therapeutic response and guiding personalized treatment strategies. Its biological role shifts with the therapeutic context: it promotes a polarized, hormone-sensitive phenotype (beneficial for endocrine therapy), but that same epithelial fortitude inhibits drug penetration and apoptosis under chemotherapy or HER2-targeted attack, leading to treatment resistance. This dualistic behavior of CLDN8 exemplifies how a tight junction protein can function as a double-edged sword in cancer therapy, reinforcing the need to consider the tumor context when predicting treatment response.
Despite the promising insights gained from this study, several limitations should be acknowledged. The reliance on retrospective clinical data necessitates prospective validation in larger cohorts. Additionally, while in vitro experiments confirmed the functional impact of CLDN8 on drug sensitivity, further mechanistic studies are required to identify the specific pathways through which CLDN8 modulates these effects in vivo. Investigating potential combination therapies targeting CLDN8 alongside conventional treatments could provide novel strategies for overcoming drug resistance in patients with breast cancer.
4. Materials and Methods
4.1. Cell Lines
The cell lines used in this study were obtained from the American Type Culture Collection (ATCC) (LGC standard, Teddington, UK), and comprised four human breast cancer cell lines: MDA-MB-231, MDA-MB-361, MCF-7, and SKBR-3. MDA-MB-231 and MCF-7 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM), while MDA-MB-361 and SKBR-3 cells were cultured in RPMI-1640 Medium. The culture medium was supplemented with 10% fetal calf serum (FCS) (Sigma-Aldrich, Dorset, UK) and 1× antimicrobial solution (Sigma-Aldrich, Dorset, UK). Cells were maintained in a controlled environment with a pH level of 7.3, 95% humidity, 5% CO2, and a temperature of 37 °C within an incubator.
4.2. Drugs and Antibodies
Four chemotherapy agents—Paclitaxel, Docetaxel, Cisplatin, and Methotrexate (MTX)—along with two anti-HER2 inhibitors (Neratinib and Lapatinib) and three endocrine therapies (Tamoxifen, Fulvestrant, and Anastrozole) were obtained from Sigma-Aldrich (Dorset, UK). Drug stock solutions were prepared in DMSO, with a final solvent concentration ≤ 0.1% in all treatments, which is below the cytotoxic threshold for breast cancer cells. Vehicle controls (0.1% DMSO) were included in all experiments to confirm that the solvent exposure did not affect cell viability. For protein blotting, the following antibodies were used: mouse anti-human GAPDH (SC-32233) from Santa Cruz Biotechnologies Inc. (Santa Cruz, CA, USA), and rabbit anti-human CLDN8 (710222) from Thermo Fisher (Oxford, UK).
4.3. Tissue Microarray (TMA) and Immunohistochemistry (IHC)
Breast cancer tissue microarray slides (BR1503f) (
https://tissuearray.com) were used in this project. Immunohistochemistry (IHC) was performed as described previously. The TMA slide was dewaxed in xylene and rehydrated in a graded series of ethanol/distilled water solutions. Heat-induced antigen retrieval was performed using citrate buffer (pH 6.0) for 20 min in a microwave. After cooling, the slides were blocked with PBS containing 5% horse serum for 2 h at RT.
The sections were then incubated overnight at 4 °C with a primary antibody against CLDN8 (1:200; Abcam, Cambridge, UK, ab 211439). After washing thoroughly with PBS, the staining protocol was performed using the Vectastain Universal Elite ABC Kit (cat no. PK-6200; Vectastain Universal Elite ABC Kit, Vector Laboratories, Inc., Newark, NJ, USA) This was followed by the manufacturer’s protocol. Briefly, using the reagent from the kit, the sections were incubated for 30 min with biotinylated secondary antibody, washed with PBS, and then incubated at room temperature for 30 min with ABC tertiary reagent. The staining was then developed using 3,3′-Diaminobenzidine (DAB) substrate for 10 min. Following a brief wash in tap water, the slide was counterstained with Gill’s haematoxylin, then washed in tap water, dehydrated in a graded series of ethanol, cleared in xylene, and finally mounted with DPX (Dibutylphthalate Polystyrene Xylene). A malignant adrenal pheochromocytoma sample, denoted as “Adr” on the tissue microarray slide, was included as a positive control for the IHC experiment. This tissue type has been previously validated to express tight junction proteins, including CLDN8, and was used to confirm the accuracy of antibody binding and consistency of the chromogenic detection process. The presence of distinct positive staining in the control adrenal tissue indicated proper assay performance and ensured the validity of subsequent sample interpretations.
Staining evaluation was performed under a 40× objective based on the percentage of CLDN8-positive cells and staining intensity in two randomly selected fields. The proportion of positive tumor cells was categorized as follows: no positive cells (0), <25% positive cells (1), 25–50% (2), 50–75% (3), and >75% (4). The staining intensity was graded as follows: unstained (0), light-brown (1), brown (2), and dark-brown (3). The staining index (SI) was calculated using the formula SI = staining intensity × proportion of positively stained cells. CLDN8 expression was evaluated using the SI-scored method, with cut-off points of ≤3 and >3. As previously reported, the staining score for CLDN8 was determined by considering the extent of tumor coverage and the proportion of positive staining.
4.4. Tissue Cohort
As previously documented [
25], we used a freshly frozen cohort of breast cancer tissues comprising both tumor and adjacent normal mammary tissues. Transcript abundance of CLDN8 was quantified by real-time PCR (qPCR), with subsequent correlation analyses performed between expression profiles and clinical parameters, including lymph node metastasis status, distant metastasis, histological grade, and 6-year survival outcomes. Written informed consent was obtained from all patients before participation, and the study received ethical approval from the Bro Taf Health Authority (ethics approval No. 01/4303 and 01/4046). Following surgical procedures, patients were monitored in a follow-up study with a median follow-up duration of 120 months post-surgery.
4.5. Patient Response to Chemotherapy and Evaluation
In this study, we utilized an extensive public database containing records of patients with breast cancer along with their corresponding therapeutic interventions (
ROC Plotter—Online ROC analysis (accessed on 1 December 2023). The database used receiver operating characteristic (ROC) curve analysis to classify patients based on their responsiveness to specific therapies. The area under the curve (AUC) values and statistical measures of treatment sensitivity were recorded. Additionally, the gene expression levels of the selected targets were analyzed, with statistical significance assessed using the Mann-Whitney
U test.
4.6. Breast Cancer Cell Model
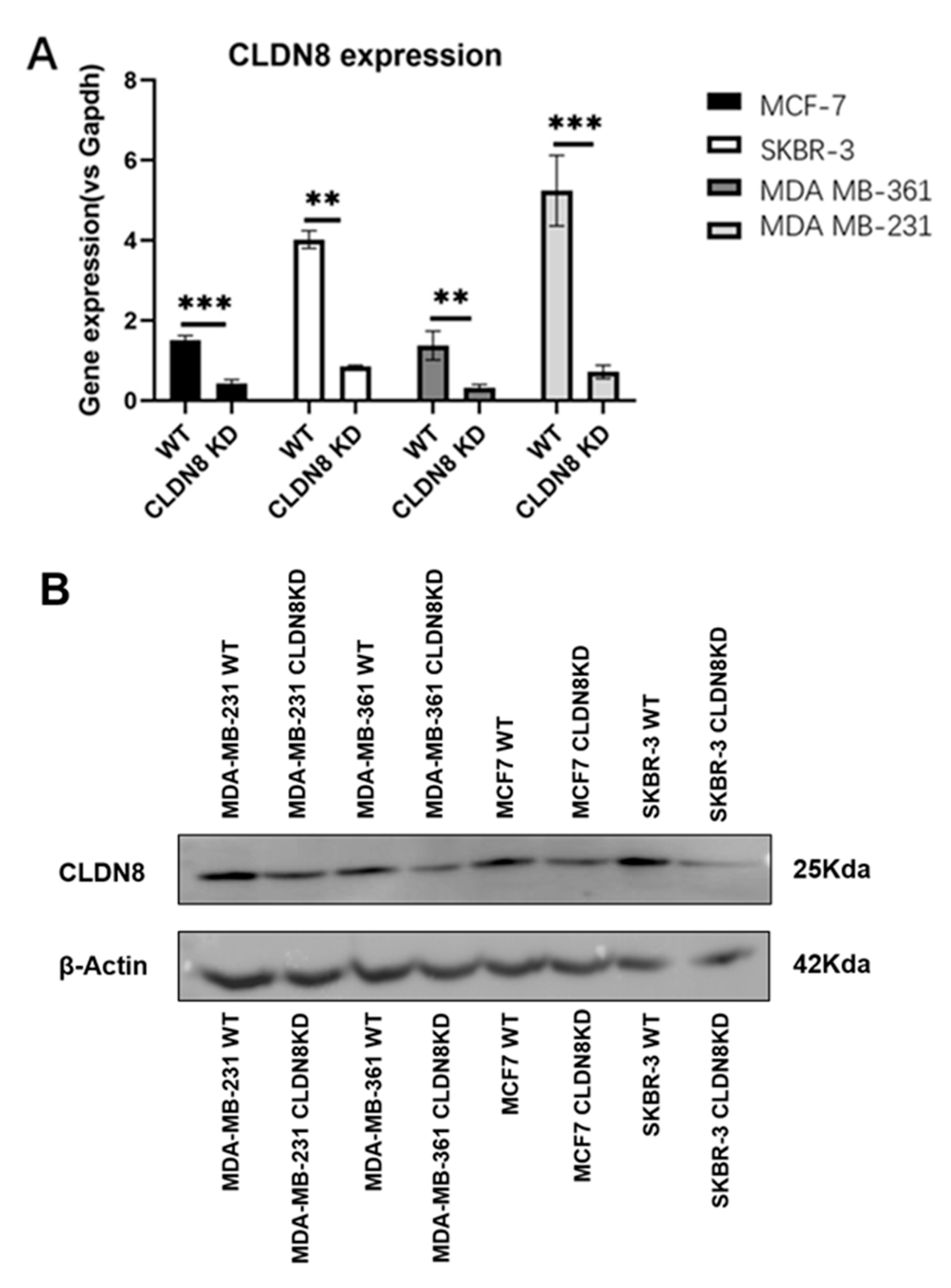
Four breast cancer cell lines, specifically MDA-MB-231, SKBR-3, MCF-7, and MDA-MB-361, were selected for their representation of diverse subtypes within breast cancer pathology. These cell lines were used to create sublines characterized by CLDN8 knockdown, adhering closely to the procedural guidelines provided by the manufacturer. The small interfering RNA (siRNA) designed to target human CLDN8 (SC-44865) was acquired from Santa Cruz Biotechnologies Inc. (Santa Cruz, CA, USA).
4.7. RNA Extraction and Reverse Transcription
Total RNA was extracted from the tissue samples and breast cancer cell lines using the TRI Reagent Kit (Merck KGaA, Darmstadt, Germany) according to the manufacturer’s protocol. After isolation, RNA concentrations were adjusted to 500 ng/μL, and reverse transcription was performed using the GoScript™ Reverse Transcription System Kit (Promega Corporation, Madison, WI, USA) in a SimpliAmp thermocycler (Fisher Scientific UK, Leicestershire, UK). The resulting cDNA was stored at −20 °C until further analysis.
4.8. Real-Time Quantitative PCR (qPCR)
CLDN8 transcript expression was quantified in tissue cohorts using the Amplifluor Uniprimer™ Universal qPCR system (Intergen Inc., Oxford, UK). Forward and reverse primers were designed with a Z sequence (5′-ACTGAACCTGACCGTACA-3′) to enable the incorporation of the FAM-tagged Uniprimer™ probe for fluorescent detection. The primer sequences were as follows: CLDN8 forward primer, ACTGAACCTGACCGTACAAGCTACTGCTCTTTTCGTTG, and Z-tagged reverse primer, ACTGAACCTGACCGTACAAGCTACTGCTCTTTTCGTTG. The internal standard GAPDH forward primer sequences used were 5′-CTGAGTACGTCGTGGAGTCc-3′ and the GAPDH ZR primer sequence was 5′-ACTGA ACCTGACCGTACAGAGATGATGACCCTTTTG-3′. Each qPCR reaction included forward and reverse primers, cDNA from tissue samples, Uniprimer™, and 2× Precision FAST qPCR master mix (Primer Design, Eastleigh, UK). Real-time PCR was performed using a StepOnePlus™ Real-Time PCR System (Thermo Fisher Scientific, Leicestershire, UK) under the following cycling conditions: an initial denaturation step at 95 °C for 10 min, followed by 100 cycles of 95 °C for 10 s, 55 °C for 35 s, and 72 °C for 10 s. Transcript levels were quantified relative to an internal reference gene with known transcript copy numbers. A series of standard samples, ranging from 108 to 101 copies, were included on the same qPCR plates as the test samples, under identical conditions. A standard curve generated from these standards was used to determine the relative transcript copy numbers in unknown samples.
4.9. Western Blotting (WB)
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting were performed as follows. Proteins were extracted from the cultured cells using RIPA buffer and quantified using a Bio-Rad protein quantification kit (Bio-Rad Laboratories, Hertfordshire, UK). The extracted protein samples were mixed with 2× Laemmli sample buffer, heated at 100 °C for 5 min, and loaded onto a 10% SDS-PAGE gel for separation. Following electrophoresis, proteins were transferred onto a pre-activated PVDF membrane using a semi-dry transfer system, with methanol treatment applied prior. The membrane was blocked with 10% milk to prevent non-specific binding, followed by incubation with primary antibodies targeting CLDN8 (1:500; Abcam, ab 211439) and β-actin (1:5000; SANTA CRUZ, sc-47778). After primary antibody incubation, the membrane was treated with horseradish peroxidase (HRP)-conjugated secondary antibody. Protein detection was performed using the EZ-ECL chemiluminescent reagent (Geneflow Ltd., Litchfield, UK).
4.10. In Vitro Drug Sensitivity Test
Cells were seeded into 96-well plates and treated with a series of drug dilutions at a 1:10 ratio. The selected drug concentrations were based on the established IC50 values and previous experimental data. After a 72-h incubation, the cells were fixed with 4% formalin, stained with 0.5% crystal violet, and washed before being solubilized in 10% acetic acid. Absorbance was measured at 595 nm using a spectrophotometer to determine the cell density. Drug toxicity was calculated using the following formula: Percentage drug toxicity = [(Absorbance of untreated control − Absorbance of drug-treated sample)/Absorbance of untreated control] × 100. Scatter plots were generated to visualize the relationship between drug concentration and toxicity, and the IC50 values were determined using the best-fit curve method.
4.11. Thiazolyl Blue Tetrazolium Bromide (MTT) Based Cellular Growth Assay
MTT-based assays were performed to assess the impact of CLDN8 on cell proliferation. Briefly, 2 × 104 cells from each cell model were plated in triplicate onto three separate 96-well plates and incubated at 37 °C with 5% CO2. On days 1, 3, and 5, 22 μL of 5 mg/mL MTT solution (Sigma-Aldrich Co., Poole, Dorset, UK) was added to each well, followed by a 4-h incubation at 37 °C with 5% CO2. After incubation, the medium was removed, and 100 μL of dimethyl sulfoxide (DMSO) (Sigma-Aldrich Co., Poole, Dorset, UK) was added to each well to dissolve the formazan crystals. The plates were then incubated for an additional 10 min at 37 °C with 5% CO2, and absorbance was measured at 540 nm using an LT4500 plate reader (Wolf Laboratories, York, UK).
4.12. Statistical Analysis
Statistical analyses were conducted using the SPSS software (Version 27.0; IBM Corp., New York, NY, USA). Group comparisons were evaluated using the Kruskal−Wallis test and analysis of variance (ANOVA), where applicable. The Mann−Whitney U test was used for pairwise comparisons, as detailed in the text. Survival analysis was performed using the Kaplan−Meier method with log-rank testing. Cox regression modeling was applied for both univariate and multivariate analyses. Classification assessments were performed using Receiver Operating Characteristic (ROC) analysis. A p-value of <0.05 was considered statistically significant.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}