
Advances in Management of Mitochondrial Myopathies

 , , , , ,
, , , , ,  ,
,  and
and
Abstract
1. Introduction
1.1. Pathophysiology
1.2. Genetics
1.3. Epidemiology
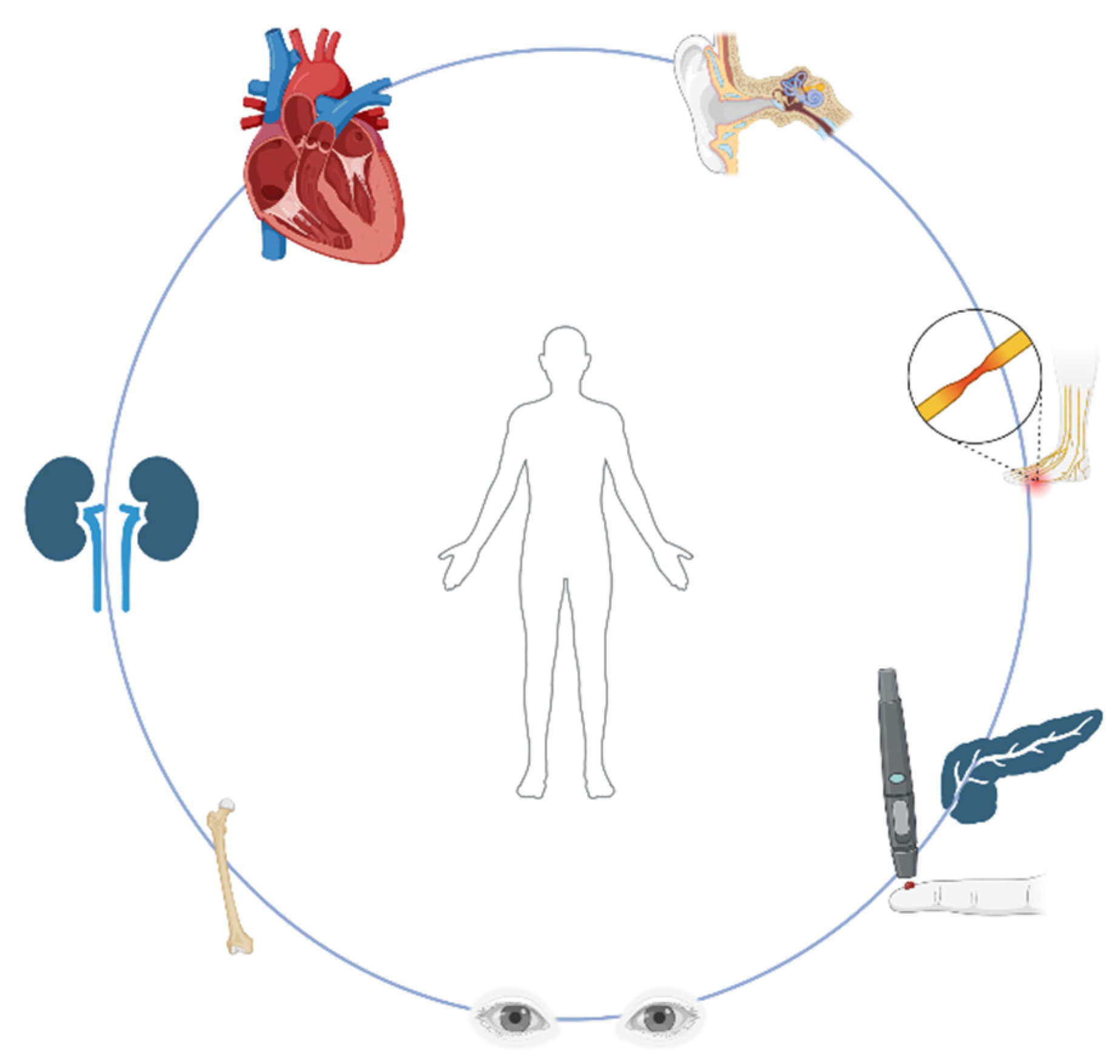
1.4. Clinical Manifestations
Clinical Syndromes
2. Treatment and Management
2.1. Dietary Interventions and Non-Specific Treatments
2.2. Potentially Disease-Modifying Treatments and Therapies Under Investigation
2.3. Emerging Therapeutic Approaches and Ongoing Clinical Trials
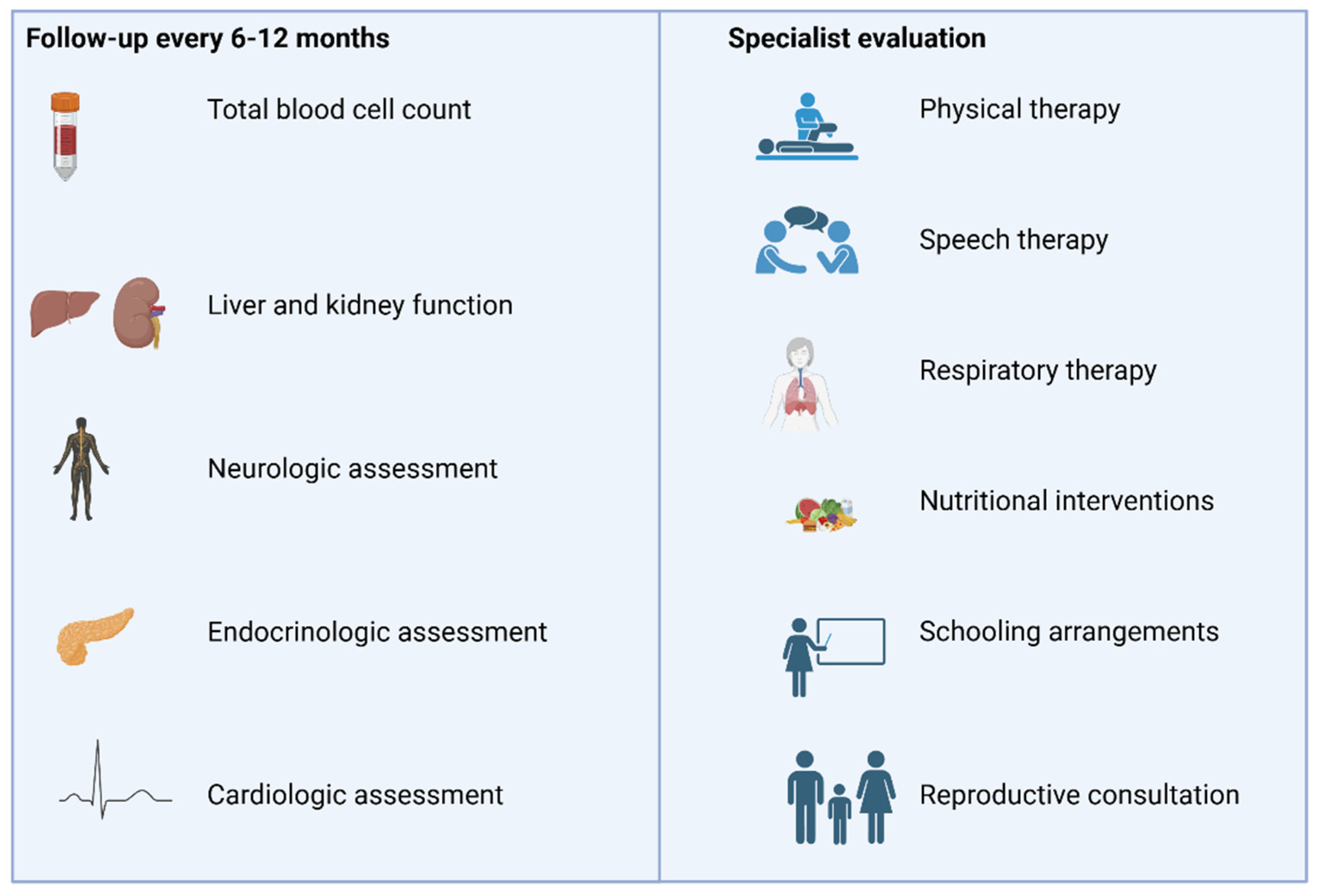
3. Management
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [PubMed]
- Parakatselaki, M.E.; Ladoukakis, E.D. mtDNA Heteroplasmy: Origin, Detection, Significance, and Evolutionary Consequences. Life 2021, 11, 633. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.P.; Letellier, T. Mitochondrial threshold effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhu, M.; Du, J.; Ma, H.; Jin, G.; Dai, J. Genetic variants in nuclear DNA along with environmental factors modify mitochondrial DNA copy number: A population-based exome-wide association study. BMC Genom. 2018, 19, 752. [Google Scholar] [CrossRef]
- National Institute of Neurological Disorders and Stroke (NINDS). NINDS Mitochondrial Myopathy Information Page; National Institute of Health: Bethesda, MD, USA, 2018. Available online: http://www.ninds.nih.gov/ (accessed on 16 June 2022).
- Carroll, J.; Fearnley, I.M.; Skehel, J.M.; Shannon, R.J.; Hirst, J.; Walker, J.E. Bovine complex I is a complex of 45 different subunits. J. Biol. Chem. 2006, 281, 32724–32727. [Google Scholar] [CrossRef]
- Jain-Ghai, S.; Cameron, J.M.; Al Maawali, A.; Blaser, S.; MacKay, N.; Robinson, B.; Raiman, J. Complex II deficiency--a case report and review of the literature. Am. J. Med. Genet. Part A 2013, 161a, 285–294. [Google Scholar] [CrossRef]
- Crofts, A.R. The cytochrome bc1 complex: Function in the context of structure. Annu. Rev. Physiol. 2004, 66, 689–733. [Google Scholar] [CrossRef]
- Fernández-Vizarra, E.; Tiranti, V.; Zeviani, M. Assembly of the oxidative phosphorylation system in humans: What we have learned by studying its defects. Biochim. Et Biophys. Acta 2009, 1793, 200–211. [Google Scholar] [CrossRef]
- Hejzlarová, K.; Mráček, T.; Vrbacký, M.; Kaplanová, V.; Karbanová, V.; Nůsková, H.; Pecina, P.; Houštěk, J. Nuclear genetic defects of mitochondrial ATP synthase. Physiol. Res. 2014, 63, S57–S71. [Google Scholar] [CrossRef]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial genetic medicine. Nat. Genet. 2018, 50, 1642–1649. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.H. Neuromuscular and systemic presentations in adults: Diagnoses beyond MERRF and MELAS. J. Am. Soc. Exp. Neurother. 2013, 10, 227–242. [Google Scholar] [CrossRef]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef]
- McCormick, E.M.; Muraresku, C.C.; Falk, M.J. Mitochondrial Genomics: A complex field now coming of age. Curr. Genet. Med. Rep. 2018, 6, 52–61. [Google Scholar] [CrossRef]
- Muscular Dystrophy Association (MDA). Research. Available online: https://www.mda.org/disease/mitochondrial-myopathies/research (accessed on 29 May 2025).
- Ahmed, S.T.; Craven, L.; Russell, O.M.; Turnbull, D.M.; Vincent, A.E. Diagnosis and Treatment of Mitochondrial Myopathies. J. Am. Soc. Exp. Neurother. 2018, 15, 943–953. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A.; Carelli, V.; Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 2013, 9, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Muscular Dystrophy Association (MDA). Causes/Inheritance. Available online: https://www.mda.org/disease/mitochondrial-myopathies/causes-inheritance (accessed on 29 May 2025).
- Mahmud, S.; Biswas, S.; Afrose, S.; Mita, M.A.; Hasan, M.R.; Shimu, M.S.S.; Paul, G.K.; Chung, S.; Saleh, M.A.; Alshehri, S.; et al. Use of Next-Generation Sequencing for Identifying Mitochondrial Disorders. Curr. Issues Mol. Biol. 2022, 44, 1127–1148. [Google Scholar] [CrossRef]
- Schaefer, A.M.; Taylor, R.W.; Turnbull, D.M.; Chinnery, P.F. The epidemiology of mitochondrial disorders--past, present and future. Biochim. Et Biophys. Acta 2004, 1659, 115–120. [Google Scholar] [CrossRef]
- Elliott, H.R.; Samuels, D.C.; Eden, J.A.; Relton, C.L.; Chinnery, P.F. Pathogenic mitochondrial DNA mutations are common in the general population. Am. J. Hum. Genet. 2008, 83, 254–260. [Google Scholar] [CrossRef]
- Powledge, T. Three Parent Baby Debate: FDA Ponders Mitochondrial Manipulation and, Perhaps, Germline Modification Too. 2014. Available online: https://geneticliteracyproject.org/ (accessed on 16 June 2022).
- Korkiamäki, P.; Kervinen, M.; Karjalainen, K.; Majamaa, K.; Uusimaa, J.; Remes, A.M. Prevalence of the primary LHON mutations in Northern Finland associated with bilateral optic atrophy and tobacco-alcohol amblyopia. Acta Ophthalmol. 2013, 91, 630–634. [Google Scholar] [CrossRef]
- Cohen, B.H. Mitochondrial and Metabolic Myopathies. Continuum 2019, 25, 1732–1766. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.D.; McFarland, R. Mitochondrial myopathies in adults and children: Management and therapy development. Curr. Opin. Neurol. 2014, 27, 576–582. [Google Scholar] [CrossRef]
- Safdar, A.; Saleem, A.; Tarnopolsky, M.A. The potential of endurance exercise-derived exosomes to treat metabolic diseases. Nat. Rev. Endocrinol. 2016, 12, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Muscular Dystrophy Association (MDA). Signs and Symptoms. Available online: https://www.mda.org/disease/mitochondrial-myopathies/signs-and-symptoms (accessed on 29 May 2025).
- Moraes, C.T.; Ricci, E.; Bonilla, E.; DiMauro, S.; Schon, E.A. The mitochondrial tRNA(Leu(UUR)) mutation in mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS): Genetic, biochemical, and morphological correlations in skeletal muscle. Am. J. Hum. Genet. 1992, 50, 934–949. [Google Scholar] [PubMed]
- Campbell, G.; Krishnan, K.J.; Deschauer, M.; Taylor, R.W.; Turnbull, D.M. Dissecting the mechanisms underlying the accumulation of mitochondrial DNA deletions in human skeletal muscle. Hum. Mol. Genet. 2014, 23, 4612–4620. [Google Scholar] [CrossRef]
- Wong, L.J. Mitochondrial syndromes with leukoencephalopathies. Semin. Neurol. 2012, 32, 55–61. [Google Scholar] [CrossRef]
- Kenny, D.; Wetherbee, J. Kearns-Sayre syndrome in the elderly: Mitochondrial myopathy with advanced heart block. Am. Heart J. 1990, 120, 440–443. [Google Scholar] [CrossRef]
- Mashima, Y.; Kigasawa, K.; Hasegawa, H.; Tani, M.; Oguchi, Y. High incidence of pre-excitation syndrome in Japanese families with Leber’s hereditary optic neuropathy. Clin. Genet. 1996, 50, 535–537. [Google Scholar] [CrossRef]
- Cohen, B.H.; Naviaux, R.K. The clinical diagnosis of POLG disease and other mitochondrial DNA depletion disorders. Methods 2010, 51, 364–373. [Google Scholar] [CrossRef]
- Goldstein, A. Chapter 20-MPV17-Related Hepatocerebral Mitochondrial DNA (mtDNA) Depletion Syndrome. In Mitochondrial Case Studies; Saneto, R.P., Parikh, S., Cohen, B.H., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 179–185. [Google Scholar]
- Morris, A.A. Mitochondrial respiratory chain disorders and the liver. Liver 1999, 19, 357–368. [Google Scholar] [CrossRef]
- Emma, F.; Bertini, E.; Salviati, L.; Montini, G. Renal involvement in mitochondrial cytopathies. Pediatr. Nephrol. 2012, 27, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Bindoff, L. Mitochondrial gastroenterology. In Mitochondrial Medicine; Informa Healthcare: Abingdon, UK, 2006; pp. 143–159. [Google Scholar]
- Karaa, A.; Goldstein, A. The spectrum of clinical presentation, diagnosis, and management of mitochondrial forms of diabetes. Pediatr. Diabetes 2015, 16, 1–9. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Emrick, L.T.; Hsu, J.W.; Chanprasert, S.; Jahoor, F.; Scaglia, F.; Craigen, W.J. Glucose metabolism derangements in adults with the MELAS m.3243A>G mutation. Mitochondrion 2014, 18, 63–69. [Google Scholar] [CrossRef]
- Mazzaccara, C.; Iafusco, D.; Liguori, R.; Ferrigno, M.; Galderisi, A.; Vitale, D.; Simonelli, F.; Landolfo, P.; Prisco, F.; Masullo, M.; et al. Mitochondrial diabetes in children: Seek and you will find it. PLoS ONE 2012, 7, e34956. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.K. Presentation and Diagnosis of Mitochondrial Disorders in Children. Pediatr. Neurol. 2008, 38, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.; Falk, M.J. Mitochondrial DNA Deletion Syndromes; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Eds.; GeneReviews®: Seattle, WA, USA, 1993–2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1203/ (accessed on 16 June 2022).
- Johns, D.R. Seminars in medicine of the Beth Israel Hospital, Boston. Mitochondrial DNA and disease. N. Engl. J. Med. 1995, 333, 638–644. [Google Scholar] [CrossRef]
- DiMauro, S.; Emmanuele, V. Chapter 27- Mitochondrial disorders due to mutations in the mitochondrial genome. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 6th ed.; Rosenberg, R.N., Pascual, J.M., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 401–413. [Google Scholar]
- Liang, C.; Ahmad, K.; Sue, C.M. The broadening spectrum of mitochondrial disease: Shifts in the diagnostic paradigm. Biochim. Biophys. Acta 2014, 1840, 1360–1367. [Google Scholar] [CrossRef]
- Giordano, C.; Sebastiani, M.; De Giorgio, R.; Travaglini, C.; Tancredi, A.; Valentino, M.L.; Bellan, M.; Cossarizza, A.; Hirano, M.; d’Amati, G.; et al. Gastrointestinal dysmotility in mitochondrial neurogastrointestinal encephalomyopathy is caused by mitochondrial DNA depletion. Am. J. Pathol. 2008, 173, 1120–1128. [Google Scholar] [CrossRef]
- Hirano, M.; Carelli, V.; De Giorgio, R.; Pironi, L.; Accarino, A.; Cenacchi, G.; D’Alessandro, R.; Filosto, M.; Martí, R.; Nonino, F.; et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): Position paper on diagnosis, prognosis, and treatment by the MNGIE International Network. J. Inherit. Metab. Dis. 2021, 44, 376–387. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef]
- Lin, Y.; Yang, B.; Huang, Y.; Zhang, Y.; Jiang, Y.; Ma, L.; Shen, Y.-Q. Mitochondrial DNA-targeted therapy: A novel approach to combat cancer. Cell Insight 2023, 2, 100113. [Google Scholar] [CrossRef] [PubMed]
- Nishigaki, Y.; Ueno, H.; Coku, J.; Koga, Y.; Fujii, T.; Sahashi, K.; Nakano, K.; Yoneda, M.; Nonaka, M.; Tang, L.; et al. Extensive screening system using suspension array technology to detect mitochondrial DNA point mutations. Mitochondrion 2010, 10, 300–308. [Google Scholar] [CrossRef]
- Viscomi, C.; Zeviani, M. MtDNA-maintenance defects: Syndromes and genes. J. Inherit. Metab. Dis. 2017, 40, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Heighton, J.N.; Brady, L.I.; Sadikovic, B.; Bulman, D.E.; Tarnopolsky, M.A. Genotypes of chronic progressive external ophthalmoplegia in a large adult-onset cohort. Mitochondrion 2019, 49, 227–231. [Google Scholar] [CrossRef]
- Pfeffer, G.; Chinnery, P.F. Diagnosis and treatment of mitochondrial myopathies. Ann. Med. 2013, 45, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Peristeri, E.; Aloizou, A.-M.; Keramida, P.; Tsouris, Z.; Siokas, V.; Mentis, A.F.A.; Dardiotis, E. Cognitive Deficits in Myopathies. Int. J. Mol. Sci. 2020, 21, 3795. [Google Scholar] [CrossRef]
- Hirano, M.; Ricci, E.; Koenigsberger, M.R.; Defendini, R.; Pavlakis, S.G.; DeVivo, D.C.; DiMauro, S.; Rowland, L.P. Melas: An original case and clinical criteria for diagnosis. Neuromuscul. Disord. 1992, 2, 125–135. [Google Scholar] [CrossRef]
- Yatsuga, S.; Povalko, N.; Nishioka, J.; Katayama, K.; Kakimoto, N.; Matsuishi, T.; Kakuma, T.; Koga, Y. MELAS: A nationwide prospective cohort study of 96 patients in Japan. Biochim. Et Biophys. Acta Gen. Subj. 2012, 1820, 619–624. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Almannai, M.; Scaglia, F. MELAS. In GeneReviews® [Internet]; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 2018. Available online: https://www.ncbi.nlm.nih.gov/sites/books/NBK1233/ (accessed on 16 June 2022).
- Alves, C.; Zandifar, A.; Peterson, J.T.; Tara, S.Z.; Ganetzky, R.; Viaene, A.N.; Andronikou, S.; Falk, M.J.; Vossough, A.; Goldstein, A.C. MELAS: Phenotype Classification into Classic-versus-Atypical Presentations. AJNR Am. J. Neuroradiol. 2023, 44, 602–610. [Google Scholar] [CrossRef]
- Sharma, A.K.; Jain, N.; Kharwar, R.B.; Narain, V.S. Classical triad of Kearns-Sayre syndrome. BMJ Case Rep. 2016, 2016, bcr2016216500. [Google Scholar] [CrossRef]
- Baracca, A.; Solaini, G.; Sgarbi, G.; Lenaz, G.; Baruzzi, A.; Schapira, A.H.; Martinuzzi, A.; Carelli, V. Severe impairment of complex I-driven adenosine triphosphate synthesis in leber hereditary optic neuropathy cybrids. Arch. Neurol. 2005, 62, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Sun, F.; Wallace, D.C. Clustering of Caucasian Leber hereditary optic neuropathy patients containing the 11778 or 14484 mutations on an mtDNA lineage. Am. J. Hum. Genet. 1997, 60, 381–387. [Google Scholar] [PubMed]
- DiMauro, S.; Hirano, M. MERRF. In GeneReviews1 [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1520/ (accessed on 16 June 2022).
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Minetti, C.; Moggio, M.; Mongini, T.; Servidei, S.; et al. Phenotypic heterogeneity of the 8344A>G mtDNA “MERRF” mutation. Neurology 2013, 80, 2049–2054. [Google Scholar] [CrossRef]
- Wang, J.; El-Hattab, A.W.; Wong, L.J.C. TK2-related mitochondrial DNA maintenance defect, myopathic form. In GeneReviews1 [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: https://www.ncbi.nlm.nih.gov/books/NBK114628/ (accessed on 16 June 2022).
- Garone, C.; Taylor, R.W.; Nascimento, A.; Poulton, J.; Fratter, C.; Domínguez-González, C.; Evans, J.C.; Loos, M.; Isohanni, P.; Suomalainen, A.; et al. Retrospective natural history of thymidine kinase 2 deficiency. J. Med. Genet. 2018, 55, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Doimo, M.; Desbats, M.A.; Cerqua, C.; Cassina, M.; Trevisson, E.; Salviati, L. Genetics of coenzyme q10 deficiency. Mol. Syndromol. 2014, 5, 156–162. [Google Scholar] [CrossRef]
- Mantle, D.; Millichap, L.; Castro-Marrero, J.; Hargreaves, I.P. Primary Coenzyme Q10 Deficiency: An Update. Antioxidants 2023, 12, 1652. [Google Scholar] [CrossRef]
- Salviati, L.; Trevisson, E.; Agosto, C.; Doimo, M.; Navas, P. Primary Coenzyme Q(10) Deficiency Overview. In GeneReviews(®); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993; GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.: Seattle (WA). [Google Scholar]
- Forsström, S.; Jackson, C.B.; Carroll, C.J.; Kuronen, M.; Pirinen, E.; Pradhan, S.; Marmyleva, A.; Auranen, M.; Kleine, I.M.; Khan, N.A.; et al. Fibroblast Growth Factor 21 Drives Dynamics of Local and Systemic Stress Responses in Mitochondrial Myopathy with mtDNA Deletions. Cell Metab. 2019, 30, 1040–1054.e1047. [Google Scholar] [CrossRef]
- Pfeffer, G.; Majamaa, K.; Turnbull, D.M.; Thorburn, D.; Chinnery, P.F. Treatment for mitochondrial disorders. Cochrane Database Syst. Rev. 2012, 2012, Cd004426. [Google Scholar] [CrossRef]
- Parikh, S.; Saneto, R.; Falk, M.J.; Anselm, I.; Cohen, B.H.; Haas, R.; Medicine Society, T.M. A modern approach to the treatment of mitochondrial disease. Curr. Treat. Options Neurol. 2009, 11, 414–430. [Google Scholar] [CrossRef]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Y.; Lu, L.; Yang, H. Therapeutic Effects of Idebenone on Leber Hereditary Optic Neuropathy. Curr. Eye Res. 2020, 45, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Carelli, V.; Newman, N.J.; Silva, M.J.; Linden, A.; Van Stavern, G.; Szaflik, J.P.; Banik, R.; Lubiński, W.; Pemp, B.; et al. Therapeutic benefit of idebenone in patients with Leber hereditary optic neuropathy: The LEROS nonrandomized controlled trial. Cell Rep. Med. 2024, 5, 101437. [Google Scholar] [CrossRef]
- Rudolph, G.; Dimitriadis, K.; Büchner, B.; Heck, S.; Al-Tamami, J.; Seidensticker, F.; Rummey, C.; Leinonen, M.; Meier, T.; Klopstock, T. Effects of idebenone on color vision in patients with leber hereditary optic neuropathy. J. Neuro Ophthalmol. 2013, 33, 30–36. [Google Scholar] [CrossRef]
- van de Weijer, T.; Phielix, E.; Bilet, L.; Williams, E.G.; Ropelle, E.R.; Bierwagen, A.; Livingstone, R.; Nowotny, P.; Sparks, L.M.; Paglialunga, S.; et al. Evidence for a direct effect of the NAD+ precursor acipimox on muscle mitochondrial function in humans. Diabetes 2015, 64, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Steele, H.; Gomez-Duran, A.; Pyle, A.; Hopton, S.; Newman, J.; Stefanetti, R.J.; Charman, S.J.; Parikh, J.D.; He, L.; Viscomi, C.; et al. Metabolic effects of bezafibrate in mitochondrial disease. EMBO Mol. Med. 2020, 12, e11589. [Google Scholar] [CrossRef] [PubMed]
- Madsen, K.L.; Buch, A.E.; Cohen, B.H.; Falk, M.J.; Goldsberry, A.; Goldstein, A.; Karaa, A.; Koenig, M.K.; Muraresku, C.C.; Meyer, C.; et al. Safety and efficacy of omaveloxolone in patients with mitochondrial myopathy: MOTOR trial. Neurology 2020, 94, e687–e698. [Google Scholar] [CrossRef]
- Ohsawa, Y.; Hagiwara, H.; Nishimatsu, S.I.; Hirakawa, A.; Kamimura, N.; Ohtsubo, H.; Fukai, Y.; Murakami, T.; Koga, Y.; Goto, Y.I.; et al. Taurine supplementation for prevention of stroke-like episodes in MELAS: A multicentre, open-label, 52-week phase III trial. J. Neurol. Neurosurg. Psychiatry 2019, 90, 529–536. [Google Scholar] [CrossRef]
- Martinelli, D.; Catteruccia, M.; Piemonte, F.; Pastore, A.; Tozzi, G.; Dionisi-Vici, C.; Pontrelli, G.; Corsetti, T.; Livadiotti, S.; Kheifets, V.; et al. EPI-743 reverses the progression of the pediatric mitochondrial disease--genetically defined Leigh Syndrome. Mol. Genet. Metab. 2012, 107, 383–388. [Google Scholar] [CrossRef]
- Sadun, A.A.; Chicani, C.F.; Ross-Cisneros, F.N.; Barboni, P.; Thoolen, M.; Shrader, W.D.; Kubis, K.; Carelli, V.; Miller, G. Effect of EPI-743 on the clinical course of the mitochondrial disease Leber hereditary optic neuropathy. Arch. Neurol. 2012, 69, 331–338. [Google Scholar] [CrossRef]
- Kerr, D.S. Review of clinical trials for mitochondrial disorders: 1997–2012. J. Am. Soc. Exp. Neurother. 2013, 10, 307–319. [Google Scholar] [CrossRef]
- Rodriguez, M.C.; MacDonald, J.R.; Mahoney, D.J.; Parise, G.; Beal, M.F.; Tarnopolsky, M.A. Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve 2007, 35, 235–242. [Google Scholar] [CrossRef]
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef]
- Muscular Dystrophy Association (MDA). Medical Management. Available online: https://www.mda.org/disease/mitochondrial-myopathies/medical-management (accessed on 29 May 2025).
- Khan, N.A.; Auranen, M.; Paetau, I.; Pirinen, E.; Euro, L.; Forsström, S.; Pasila, L.; Velagapudi, V.; Carroll, C.J.; Auwerx, J.; et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 2014, 6, 721–731. [Google Scholar] [CrossRef]
- Koga, Y.; Povalko, N.; Inoue, E.; Nakamura, H.; Ishii, A.; Suzuki, Y.; Yoneda, M.; Kanda, F.; Kubota, M.; Okada, H.; et al. Therapeutic regimen of L-arginine for MELAS: 9-year, prospective, multicenter, clinical research. J. Neurol. 2018, 265, 2861–2874. [Google Scholar] [CrossRef]
- Stefanetti, R.J.; Ng, Y.S.; Errington, L.; Blain, A.P.; McFarland, R.; Gorman, G.S. l-Arginine in Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like Episodes: A Systematic Review. Neurology 2022, 98, e2318–e2328. [Google Scholar] [CrossRef]
- Taivassalo, T.; Shoubridge, E.A.; Chen, J.; Kennaway, N.G.; DiMauro, S.; Arnold, D.L.; Haller, R.G. Aerobic conditioning in patients with mitochondrial myopathies: Physiological, biochemical, and genetic effects. Ann. Neurol. 2001, 50, 133–141. [Google Scholar] [CrossRef]
- Jeppesen, T.D.; Schwartz, M.; Olsen, D.B.; Wibrand, F.; Krag, T.; Dunø, M.; Hauerslev, S.; Vissing, J. Aerobic training is safe and improves exercise capacity in patients with mitochondrial myopathy. Brain A J. Neurol. 2006, 129, 3402–3412. [Google Scholar] [CrossRef]
- Taivassalo, T.; Gardner, J.L.; Taylor, R.W.; Schaefer, A.M.; Newman, J.; Barron, M.J.; Haller, R.G.; Turnbull, D.M. Endurance training and detraining in mitochondrial myopathies due to single large-scale mtDNA deletions. Brain A J. Neurol. 2006, 129, 3391–3401. [Google Scholar] [CrossRef]
- Taivassalo, T.; Jensen, T.D.; Kennaway, N.; DiMauro, S.; Vissing, J.; Haller, R.G. The spectrum of exercise tolerance in mitochondrial myopathies: A study of 40 patients. Brain A J. Neurol. 2003, 126, 413–423. [Google Scholar] [CrossRef]
- Cejudo, P.; Bautista, J.; Montemayor, T.; Villagómez, R.; Jiménez, L.; Ortega, F.; Campos, Y.; Sánchez, H.; Arenas, J. Exercise training in mitochondrial myopathy: A randomized controlled trial. Muscle Nerve 2005, 32, 342–350. [Google Scholar] [CrossRef]
- Murphy, J.L.; Blakely, E.L.; Schaefer, A.M.; He, L.; Wyrick, P.; Haller, R.G.; Taylor, R.W.; Turnbull, D.M.; Taivassalo, T. Resistance training in patients with single, large-scale deletions of mitochondrial DNA. Brain A J. Neurol. 2008, 131, 2832–2840. [Google Scholar] [CrossRef]
- Bates, M.G.D.; Newman, J.H.; Jakovljevic, D.G.; Hollingsworth, K.G.; Alston, C.L.; Zalewski, P.; Klawe, J.J.; Blamire, A.M.; MacGowan, G.A.; Keavney, B.D.; et al. Defining cardiac adaptations and safety of endurance training in patients with m.3243A>G-related mitochondrial disease. Int. J. Cardiol. 2013, 168, 3599–3608. [Google Scholar] [CrossRef]
- Zeviani, M. Train, train, train! No pain, just gain. Brain A J. Neurol. 2008, 131, 2809–2811. [Google Scholar] [CrossRef]
- Ahola-Erkkilä, S.; Carroll, C.J.; Peltola-Mjösund, K.; Tulkki, V.; Mattila, I.; Seppänen-Laakso, T.; Oresic, M.; Tyynismaa, H.; Suomalainen, A. Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum. Mol. Genet. 2010, 19, 1974–1984. [Google Scholar] [CrossRef]
- Ahola, S.; Auranen, M.; Isohanni, P.; Niemisalo, S.; Urho, N.; Buzkova, J.; Velagapudi, V.; Lundbom, N.; Hakkarainen, A.; Muurinen, T.; et al. Modified Atkins diet induces subacute selective ragged-red-fiber lysis in mitochondrial myopathy patients. EMBO Mol. Med. 2016, 8, 1234–1247. [Google Scholar] [CrossRef]
- Zweers, H.; van Wegberg, A.M.J.; Janssen, M.C.H.; Wortmann, S.B. Ketogenic diet for mitochondrial disease: A systematic review on efficacy and safety. Orphanet J. Rare Dis. 2021, 16, 295. [Google Scholar] [CrossRef]
- Klopstock, T.; Yu-Wai-Man, P.; Dimitriadis, K.; Rouleau, J.; Heck, S.; Bailie, M.; Atawan, A.; Chattopadhyay, S.; Schubert, M.; Garip, A.; et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain A J. Neurol. 2011, 134, 2677–2686. [Google Scholar] [CrossRef]
- Gueven, N. Idebenone for Leber’s hereditary optic neuropathy. Drugs Today 2016, 52, 173–181. [Google Scholar] [CrossRef]
- Pemp, B.; Kircher, K.; Reitner, A. Visual function in chronic Leber’s hereditary optic neuropathy during idebenone treatment initiated 5 to 50 years after onset. Graefe’s Arch. Clin. Exp. Ophthalmol. 2019, 257, 2751–2757. [Google Scholar] [CrossRef]
- Ishikawa, H.; Masuda, Y.; Ishikawa, H.; Shikisima, K.; Goseki, T.; Kezuka, T.; Terao, M.; Miyazaki, A.; Matsumoto, K.; Nishikawa, H.; et al. Characteristics of Japanese patients with Leber’s hereditary optic neuropathy and idebenone trial: A prospective, interventional, non-comparative study. Jpn. J. Ophthalmol. 2021, 65, 133–142. [Google Scholar] [CrossRef]
- Esmaeil, A.; Ali, A.; Behbehani, R. Leber’s hereditary optic neuropathy: Update on current diagnosis and treatment. Front. Ophthalmol. 2022, 2, 1077395. [Google Scholar] [CrossRef]
- Zhang, Y.; Tian, Z.; Yuan, J.; Liu, C.; Liu, H.L.; Ma, S.Q.; Li, B. The Progress of Gene Therapy for Leber’s Optic Hereditary Neuropathy. Curr. Gene. Ther. 2017, 17, 320–326. [Google Scholar] [CrossRef]
- Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Moster, M.L.; Biousse, V.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Sadun, A.A.; Barboni, P.; et al. Efficacy and Safety of Intravitreal Gene Therapy for Leber Hereditary Optic Neuropathy Treated within 6 Months of Disease Onset. Ophthalmology 2021, 128, 649–660. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Newman, N.J.; Carelli, V.; Moster, M.L.; Biousse, V.; Sadun, A.A.; Klopstock, T.; Vignal-Clermont, C.; Sergott, R.C.; Rudolph, G.; et al. Bilateral visual improvement with unilateral gene therapy injection for Leber hereditary optic neuropathy. Sci. Transl. Med. 2020, 12, eaaz7423. [Google Scholar] [CrossRef]
- Newman, N.; Yu-Wai-Man, P.; Carelli, V.; Subramanian, P.; Moster, M.; Wang, A.-G.; Donahue, S.; Leroy, B.; Biousse, V.; Vignal-Clermont, C.; et al. The Phase III REFLECT Trial: Efficacy and Safety of Bilateral Gene Therapy for Leber Hereditary Optic Neuropathy (LHON) (P17-12.002). Neurology 2022, 98, 928. [Google Scholar] [CrossRef]
- Bax, B.E. Mitochondrial neurogastrointestinal encephalomyopathy: Approaches to diagnosis and treatment. J. Transl. Genet. Genom. 2020, 4, 1–16. [Google Scholar] [CrossRef]
- Bax, B.E.; Bain, M.D.; Scarpelli, M.; Filosto, M.; Tonin, P.; Moran, N. Clinical and biochemical improvements in a patient with MNGIE following enzyme replacement. Neurology 2013, 81, 1269–1271. [Google Scholar] [CrossRef]
- Levene, M.; Bain, M.D.; Moran, N.F.; Nirmalananthan, N.; Poulton, J.; Scarpelli, M.; Filosto, M.; Mandel, H.; MacKinnon, A.D.; Fairbanks, L.; et al. Safety and Efficacy of Erythrocyte Encapsulated Thymidine Phosphorylase in Mitochondrial Neurogastrointestinal Encephalomyopathy. J. Clin. Med. 2019, 8, 457. [Google Scholar] [CrossRef]
- Garone, C.; Garcia-Diaz, B.; Emmanuele, V.; Lopez, L.C.; Tadesse, S.; Akman, H.O.; Tanji, K.; Quinzii, C.M.; Hirano, M. Deoxypyrimidine monophosphate bypass therapy for thymidine kinase 2 deficiency. EMBO Mol. Med. 2014, 6, 1016–1027. [Google Scholar] [CrossRef]
- Akman, H.O.; Dorado, B.; López, L.C.; García-Cazorla, A.; Vilà, M.R.; Tanabe, L.M.; Dauer, W.T.; Bonilla, E.; Tanji, K.; Hirano, M. Thymidine kinase 2 (H126N) knockin mice show the essential role of balanced deoxynucleotide pools for mitochondrial DNA maintenance. Hum. Mol. Genet. 2008, 17, 2433–2440. [Google Scholar] [CrossRef]
- Domínguez-González, C.; Madruga-Garrido, M.; Mavillard, F.; Garone, C.; Aguirre-Rodríguez, F.J.; Donati, M.A.; Kleinsteuber, K.; Martí, I.; Martín-Hernández, E.; Morealejo-Aycinena, J.P.; et al. Deoxynucleoside Therapy for Thymidine Kinase 2-Deficient Myopathy. Ann. Neurol. 2019, 86, 293–303. [Google Scholar] [CrossRef]
- Lopez-Gomez, C.; Sanchez-Quintero, M.J.; Lee, E.J.; Kleiner, G.; Tadesse, S.; Xie, J.; Akman, H.O.; Gao, G.; Hirano, M. Synergistic Deoxynucleoside and Gene Therapies for Thymidine Kinase 2 Deficiency. Ann. Neurol. 2021, 90, 640–652. [Google Scholar] [CrossRef]
- Karaa, A.; Haas, R.; Goldstein, A.; Vockley, J.; Weaver, W.D.; Cohen, B.H. Randomized dose-escalation trial of elamipretide in adults with primary mitochondrial myopathy. Neurology 2018, 90, e1212–e1221. [Google Scholar] [CrossRef]
- Karaa, A.; Bertini, E.; Carelli, V.; Cohen, B.H.; Enns, G.M.; Falk, M.J.; Goldstein, A.; Gorman, G.S.; Haas, R.; Hirano, M.; et al. Efficacy and Safety of Elamipretide in Individuals with Primary Mitochondrial Myopathy: The MMPOWER-3 Randomized Clinical Trial. Neurology 2023, 101, e238–e252. [Google Scholar] [CrossRef]
- Fisher, F.M.; Maratos-Flier, E. Understanding the Physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.S. GDF15 as a central mediator for integrated stress response and a promising therapeutic molecule for metabolic disorders and NASH. Biochim. Et Biophys. Acta. Gen. Subj. 2021, 1865, 129834. [Google Scholar] [CrossRef]
- Foo, A.S.C.; Soong, T.W.; Yeo, T.T.; Lim, K.L. Mitochondrial Dysfunction and Parkinson’s Disease-Near-Infrared Photobiomodulation as a Potential Therapeutic Strategy. Front. Aging Neurosci. 2020, 12, 89. [Google Scholar] [CrossRef]
- Salehpour, F.; Mahmoudi, J.; Kamari, F.; Sadigh-Eteghad, S.; Rasta, S.H.; Hamblin, M.R. Brain Photobiomodulation Therapy: A Narrative Review. Mol. Neurobiol. 2018, 55, 6601–6636. [Google Scholar] [CrossRef]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef]
- Torres, A.K.; Fleischhart, V.; Inestrosa, N.C. Mitochondrial unfolded protein response (UPR(mt)): What we know thus far. Front. Cell Dev. Biol. 2024, 12, 1405393. [Google Scholar] [CrossRef]
- Yi, H.-S.; Chang, J.Y.; Shong, M. The mitochondrial unfolded protein response and mitohormesis: A perspective on metabolic diseases. J. Mol. Endocrinol. 2018, 61, R91–R105. [Google Scholar] [CrossRef]
- Cilleros-Holgado, P.; Gómez-Fernández, D.; Piñero-Pérez, R.; Reche-López, D.; Álvarez-Córdoba, M.; Munuera-Cabeza, M.; Talaverón-Rey, M.; Povea-Cabello, S.; Suárez-Carrillo, A.; Romero-González, A.; et al. mtUPR Modulation as a Therapeutic Target for Primary and Secondary Mitochondrial Diseases. Int. J. Mol. Sci. 2023, 24, 1482. [Google Scholar] [CrossRef]
- Perry, E.A.; Bennett, C.F.; Luo, C.; Balsa, E.; Jedrychowski, M.; O’Malley, K.E.; Latorre-Muro, P.; Ladley, R.P.; Reda, K.; Wright, P.M.; et al. Tetracyclines promote survival and fitness in mitochondrial disease models. Nat. Metab. 2021, 3, 33–42. [Google Scholar] [CrossRef]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Reche-López, D.; Cilleros-Holgado, P.; et al. UPR(mt) activation improves pathological alterations in cellular models of mitochondrial diseases. Orphanet J. Rare Dis. 2022, 17, 204. [Google Scholar] [CrossRef]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Romero-González, A.; Gómez-Fernandez, D.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; et al. Pterostilbene in Combination with Mitochondrial Cofactors Improve Mitochondrial Function in Cellular Models of Mitochondrial Diseases. Front. Pharmacol 2022, 13, 862085. [Google Scholar] [CrossRef]
- Parikh, S.; Goldstein, A.; Karaa, A.; Koenig, M.K.; Anselm, I.; Brunel-Guitton, C.; Christodoulou, J.; Cohen, B.H.; Dimmock, D.; Enns, G.M.; et al. Patient care standards for primary mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Anesth. Analg. 2017, 19, 1380–1397. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Rahman, S. Emerging aspects of treatment in mitochondrial disorders. J. Inherit. Metab. Dis. 2015, 38, 641–653. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial DNA depletion syndromes: Review and updates of genetic basis, manifestations, and therapeutic options. J. Am. Soc. Exp. Neurother. 2013, 10, 186–198. [Google Scholar] [CrossRef]
- Millhouse-Flourie, T. MITO 101 Therapies for Mitochondrial Disease Symptoms; United Mitochondrial Disease Foundation: Pittsburgh, PA, USA, 2016; Available online: https://www.umdf.org/wp-content/uploads/2017/11/mito101_Therapies_Millhouse-Flourie.pdf (accessed on 29 May 2025).
- Millhouse-Flourie, T.J. Physical, occupational, respiratory, speech, equine and pet therapies for mitochondrial disease. Mitochondrion 2004, 4, 549–558. [Google Scholar] [CrossRef]
- Camp, K.M.; Krotoski, D.; Parisi, M.A.; Gwinn, K.A.; Cohen, B.H.; Cox, C.S.; Enns, G.M.; Falk, M.J.; Goldstein, A.C.; Gopal-Srivastava, R.; et al. Nutritional interventions in primary mitochondrial disorders: Developing an evidence base. Mol. Genet. Metab. 2016, 119, 187–206. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Collins, A.; Cohen, B.H.; DeBrosse, S.D.; et al. Practice patterns of mitochondrial disease physicians in North America. Part 2: Treatment, care and management. Mitochondrion 2013, 13, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Herbert, M.; Murdoch, A.; Murphy, J.; Lawford Davies, J.; Turnbull, D.M. Research into Policy: A Brief History of Mitochondrial Donation. Stem Cells 2016, 34, 265–267. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance | Doses | Clinical Outcomes | Reference |
|---|---|---|---|
| Ubiquinol | In PMM with Q10 deficiency adults: p.o. 5–600 mg/daily pediatric: p.o. 2–8 mg/kg/daily divided in 2 doses | Amelioration of symptoms in syndromes characterized by Q10 deficiency, antioxidant and pro-oxidant role | [55,58] |
| Alpha lipoic acid | 50–200 mg/daily | Antioxidant role | [55] |
| Rifoflavin (B2) | p.o. 50–400 mg/daily | Amelioration of symptoms, slowing of disease progression | [60] |
| Nicotinamide riboside (B3) | p.o. 400 mg/kg/daily | Delay of early- and late-stage disease progression | [69] |
| Carnitine | adults: 3 g/daily in 3 doses pediatric: 100 mg/kg/daily in 3 doses | Restoration of free carnitine levels and removal of accumulating toxic acyl compounds | [70] |
| Creatinine | adults: 10 g p.o. daily divided 2–3 times pediatric: 0.1g/kg p.o. daily | Increase in high-intensity, isometric, anaerobic and aerobic power | [60] |
| L-arginine | Acute (1–3 days): iv 500 mg/kg/daily Maintenance: p.o. or iv 150–300 mg/kg/daily in 3 doses | Reduction in the severity and frequency of metabolic strokes in patients with MELAS and other forms of mitochondrial disease | [55,71] |
| Elamipretide | iv 0.25 mg/kg/h over 2 h for 5 consecutive days | Increase in exercise performance, improvement in the distance walked on the 6MWT | [72] |
| Deoxynucleoside monophosphates, deoxynucleosides | 300–400 mg/kg/daily | Halt in early-onset disease progression, amelioration of muscle weakness, reversion of early onset tetraplegia, improvements in mechanical ventilation | [73] |
| Idebenone | 900 mg/day divided into three doses | Improvement in visual acuity, protection from loss of color vision | [74,75,76] |
| Acipimox | 250 mg three times daily for 2 weeks | Enhanced mitochondrial respiration, increased expression of mitochondrial genes | [77] |
| Bezafibrate | 600–1200 mg daily for 12 weeks | Improved cardiac function, reduced proportion of complex IV-deficient muscle fibers | [78] |
| Omaveloxolone | Up to 160 mg for 12 weeks | Reduced heart rate and lowered plasma lactate levels during submaximal exercise | [79] |
| Taurine | 9 g or 12 g per day for 52 weeks | Reduced frequency of stroke-like episodes in MELAS patients | [80] |
| EPI-743 | 100 mg three times daily | Improved neuromuscular function in children with Leigh syndrome | [81] |
| 100–400 mg three times daily | Reversed vision loss in most patients with LHON | [82] |
| Intervention | Phase Study | Participants | ClinicalTrials.gov ID |
|---|---|---|---|
| Nicotinamide riboside (B3) | II | Adults with mitochondrial myopathy | NCT05590468 |
| Deoxycytidine and deoxythymidine | II | Pediatric patients with mtDNA depletion syndromes | NCT04802707 |
| Deoxycytidine and deoxythymidine | II | Adults with TK2 disease | NCT06754098 |
| Autologous mesoangioblasts | II | Adults with m.3243A>G mutation | NCT05962333 |
| MNV-201 | I | Pediatric patients with Pearson syndrome | NCT06017869 |
| KL1333 | II | Adult patients with primary mitochondrial disease | NCT05650229 |
| Sonlicromanol | III | Adults with a genetically confirmed mtDNA tRNALeu(UUR) m.3243A>G Variant | NCT06451757 |
| Zagociguat | II | Adults with MELAS syndrome | NCT06402123 |
| TTI-0102 | II | Patients > 16 years old with MELAS syndrome | NCT06644534 |
| Ketogenic Diet | ΝA | Patients with MELAS syndrome | NCT06013397 |
| Glycerol tributyrate | I | Adults with MELAS or LHON-Plus | NCT06792500 |
| PYC-001 | Ι | Adults with confirmed OPA1 mutation | NCT06461286 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bangeas, A.; Poulidou, V.; Liampas, I.; Marogianni, C.; Aloizou, A.-M.; Tsouris, Z.; Sgantzos, M.; Arnaoutoglou, M.; Bogdanos, D.P.; Dardiotis, E.; et al. Advances in Management of Mitochondrial Myopathies. Int. J. Mol. Sci. 2025, 26, 5411. https://doi.org/10.3390/ijms26115411
Bangeas A, Poulidou V, Liampas I, Marogianni C, Aloizou A-M, Tsouris Z, Sgantzos M, Arnaoutoglou M, Bogdanos DP, Dardiotis E, et al. Advances in Management of Mitochondrial Myopathies. International Journal of Molecular Sciences. 2025; 26(11):5411. https://doi.org/10.3390/ijms26115411
Chicago/Turabian StyleBangeas, Athanasios, Vasiliki Poulidou, Ioannis Liampas, Chrysa Marogianni, Athina-Maria Aloizou, Zisis Tsouris, Markos Sgantzos, Marianthi Arnaoutoglou, Dimitrios P. Bogdanos, Efthimios Dardiotis, and et al. 2025. "Advances in Management of Mitochondrial Myopathies" International Journal of Molecular Sciences 26, no. 11: 5411. https://doi.org/10.3390/ijms26115411
APA StyleBangeas, A., Poulidou, V., Liampas, I., Marogianni, C., Aloizou, A.-M., Tsouris, Z., Sgantzos, M., Arnaoutoglou, M., Bogdanos, D. P., Dardiotis, E., & Siokas, V. (2025). Advances in Management of Mitochondrial Myopathies. International Journal of Molecular Sciences, 26(11), 5411. https://doi.org/10.3390/ijms26115411