
Dysregulation of the Bone Marrow Microenvironment in Pediatric Tumors: The Role of Extracellular Vesicles in Acute Leukemias and Neuroblastoma

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Role of EVs in Immune Modulation and Anti-Tumor Responses
3. Acute Leukemia (ALL and AML)
4. Neuroblastoma
5. The BM Niche in Physiological Condition
6. BM Environment in Acute Leukemia
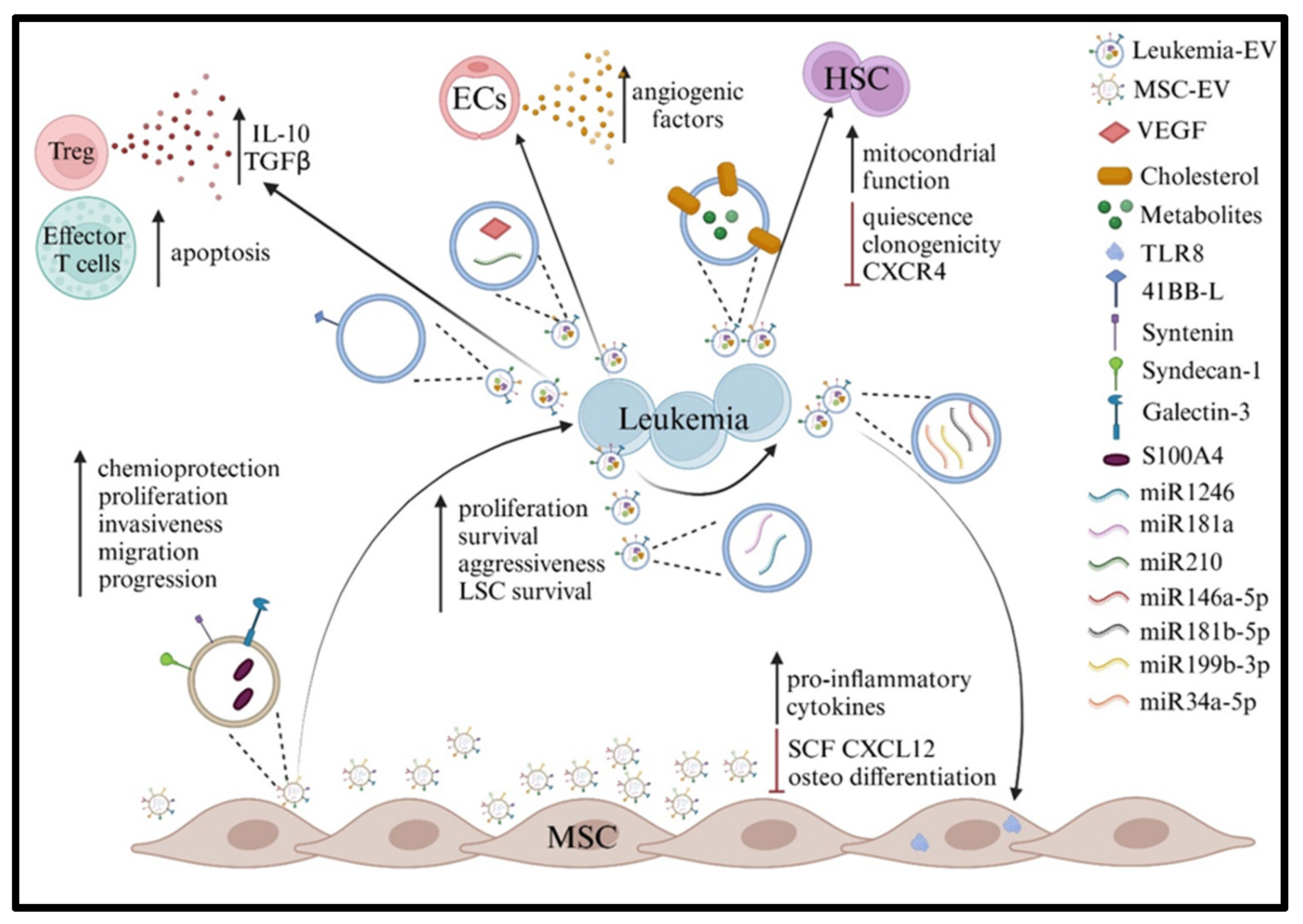
7. Role of EVs in BM Dysregulation in Acute Leukemia
8. BM Environment in Metastatic NB
9. Role of EVs in the Dysregulation of NB BM
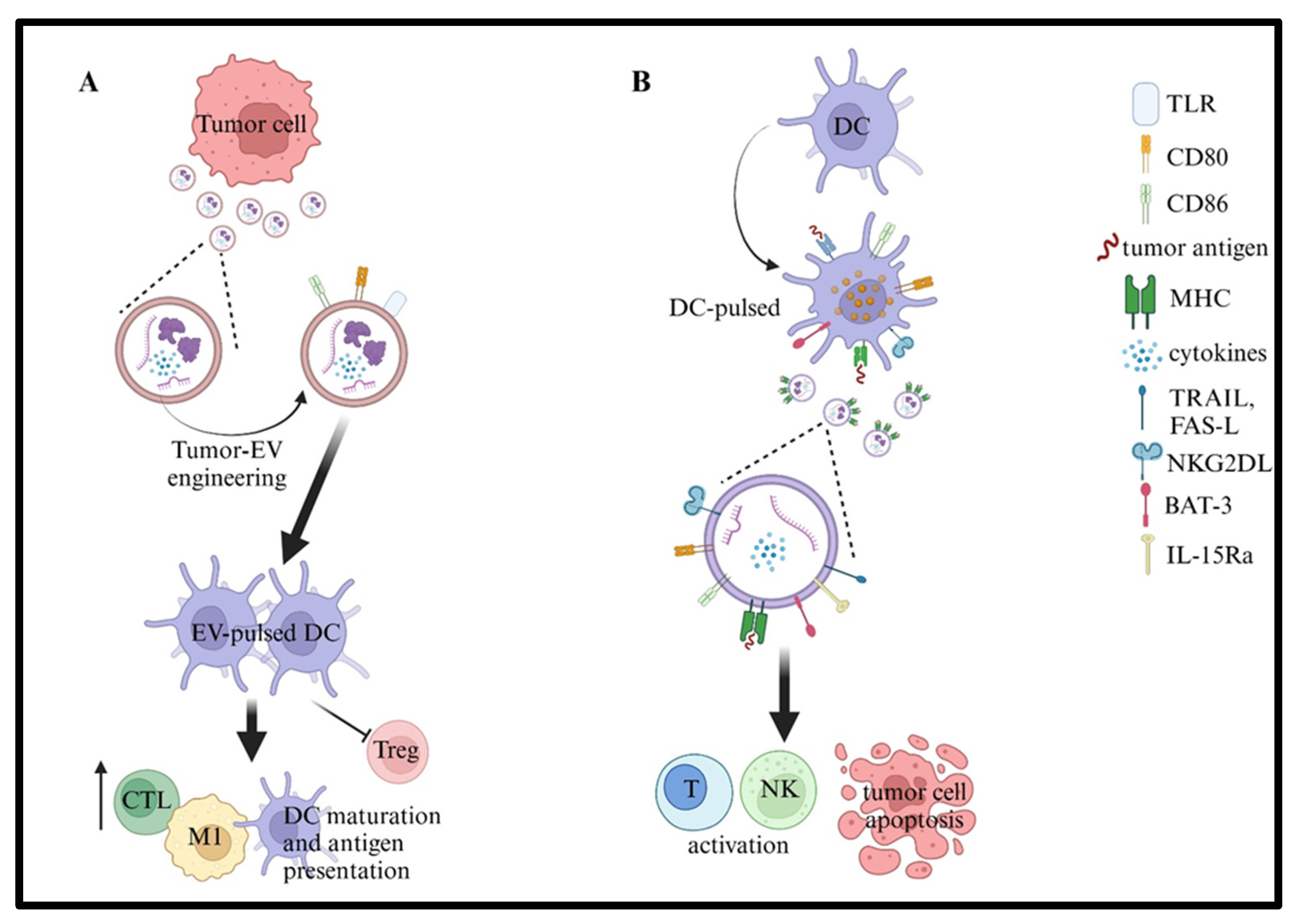
10. Cancer Vaccines Based on EVs
11. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrügger, U.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Zhang, Q.; Franklin, J.L.; Coffey, R.J. Extracellular vesicles and nanoparticles: Emerging complexities. Trends Cell Biol. 2023, 33, 667–681. [Google Scholar] [CrossRef]
- Kalluri, R.; McAndrews, K.M. The role of extracellular vesicles in cancer. Cell 2023, 186, 1610–1626. [Google Scholar] [CrossRef]
- Sheta, M.; Taha, E.A.; Lu, Y.; Eguchi, T. Extracellular Vesicles: New Classification and Tumor Immunosuppression. Biology 2023, 12, 110. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- O’Brien, K.; Breyne, K.; Ughetto, S.; Laurent, L.C.; Breakefield, X.O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 2020, 21, 585–606. [Google Scholar] [CrossRef]
- Dhamdhere, M.R.; Spiegelman, V.S. Extracellular vesicles in neuroblastoma: Role in progression, resistance to therapy and diagnostics. Front. Immunol. 2024, 15, 1385875. [Google Scholar] [CrossRef] [PubMed]
- Fusco, C.; De Rosa, G.; Spatocco, I.; Vitiello, E.; Procaccini, C.; Frigè, C.; Pellegrini, V.; La Grotta, R.; Furlan, R.; Matarese, G.; et al. Extracellular vesicles as human therapeutics: A scoping review of the literature. J. Extracell. Vesicles 2024, 13, e12433. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of extracellular vesicles in immune response and immunity. Immunity 2024, 57, 1752–1768. [Google Scholar] [CrossRef] [PubMed]
- Marar, C.; Starich, B.; Wirtz, D. Extracellular vesicles in immunomodulation and tumor progression. Nat. Immunol. 2021, 22, 560–570. [Google Scholar] [CrossRef]
- Buzas, E.I. The roles of extracellular vesicles in the immune system. Nat. Rev. Immunol. 2023, 23, 236–250. [Google Scholar] [CrossRef]
- Kuang, L.; Wu, L.; Li, Y. Extracellular vesicles in tumor immunity: Mechanisms and novel insights. Mol. Cancer 2025, 24, 45. [Google Scholar] [CrossRef]
- Chen, J.G.; Du, Y.T.; Guan, C.H.; Fan, H.Y.; Liu, Y.A.; Wang, T.; Li, X.; Chen, G. Extracellular Vesicles Derived from Plasmodium-infected Hosts as Stimuli of “Trained” Innate Immunity. Curr. Med. Chem. 2023, 30, 4450–4465. [Google Scholar] [CrossRef]
- Ayre, D.C.; Elstner, M.; Smith, N.C.; Moores, E.S.; Hogan, A.M.; Christian, S.L. Dynamic regulation of CD24 expression and release of CD24-containing microvesicles in immature B cells in response to CD24 engagement. Immunology 2015, 146, 217–233. [Google Scholar] [CrossRef]
- Lundberg, V.; Berglund, M.; Skogberg, G.; Lindgren, S.; Lundqvist, C.; Gudmundsdottir, J.; Thörn, K.; Telemo, E.; Ekwall, O. Thymic exosomes promote the final maturation of thymocytes. Sci. Rep. 2016, 6, 36479. [Google Scholar] [CrossRef]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Balan, S.; Bhardwaj, N. Cross-Presentation of Tumor Antigens Is Ruled by Synaptic Transfer of Vesicles among Dendritic Cell Subsets. Cancer Cell 2020, 37, 751–753. [Google Scholar] [CrossRef] [PubMed]
- Céspedes, P.F.; Jainarayanan, A.; Fernández-Messina, L.; Valvo, S.; Saliba, D.G.; Kurz, E.; Kvalvaag, A.; Chen, L.; Ganskow, C.; Colin-York, H.; et al. T-cell trans-synaptic vesicles are distinct and carry greater effector content than constitutive extracellular vesicles. Nat. Commun. 2022, 13, 3460. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, K.; Llodrá, J.; Roth, E.W.; Tsai, J.; Gordo, S.; Wucherpfennig, K.W.; Kam, L.C.; Stokes, D.L.; Dustin, M.L. Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature 2014, 507, 118–123. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Manning, A.J.; Kuehn, M.J. Contribution of bacterial outer membrane vesicles to innate bacterial defense. BMC Microbiol. 2011, 11, 258. [Google Scholar] [CrossRef]
- Adamczyk, A.M.; Leicaj, M.L.; Fabiano, M.P.; Cabrerizo, G.; Bannoud, N.; Croci, D.O.; Witwer, K.W.; Remes Lenicov, F.; Ostrowski, M.; Pérez, P.S. Extracellular vesicles from human plasma dampen inflammation and promote tissue repair functions in macrophages. J. Extracell. Vesicles 2023, 12, e12331. [Google Scholar] [CrossRef]
- Ramil, C.P.; Xiang, H.; Zhang, P.; Cronin, A.; Cabral, L.; Yin, Z.; Hai, J.; Wang, H.; Ruprecht, B.; Jia, Y.; et al. Extracellular vesicles released by cancer-associated fibroblast-induced myeloid-derived suppressor cells inhibit T-cell function. Oncoimmunology 2024, 13, 2300882. [Google Scholar] [CrossRef]
- Schioppa, T.; Gaudenzi, C.; Zucchi, G.; Piserà, A.; Vahidi, Y.; Tiberio, L.; Sozzani, S.; Del Prete, A.; Bosisio, D.; Salvi, V. Extracellular vesicles at the crossroad between cancer progression and immunotherapy: Focus on dendritic cells. J. Transl. Med. 2024, 22, 691. [Google Scholar] [CrossRef]
- Beltraminelli, T.; Perez, C.R.; De Palma, M. Disentangling the complexity of tumor-derived extracellular vesicles. Cell Rep. 2021, 35, 108960. [Google Scholar] [CrossRef]
- Del Vecchio, F.; Martinez-Rodriguez, V.; Schukking, M.; Cocks, A.; Broseghini, E.; Fabbri, M. Professional killers: The role of extracellular vesicles in the reciprocal interactions between natural killer, CD8+ cytotoxic T-cells and tumour cells. J. Extracell. Vesicles 2021, 10, e12075. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Mitchell, J.P.; Court, J.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef] [PubMed]
- Wieckowski, E.U.; Visus, C.; Szajnik, M.; Szczepanski, M.J.; Storkus, W.J.; Whiteside, T.L. Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J. Immunol. 2009, 183, 3720–3730. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Poliakov, A.; Liu, C.; Liu, Y.; Deng, Z.B.; Wang, J.; Cheng, Z.; Shah, S.V.; Wang, G.J.; Zhang, L.; et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 2009, 124, 2621–2633. [Google Scholar] [CrossRef]
- Huber, V.; Fais, S.; Iero, M.; Lugini, L.; Canese, P.; Squarcina, P.; Zaccheddu, A.; Colone, M.; Arancia, G.; Gentile, M.; et al. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: Role in immune escape. Gastroenterology 2005, 128, 1796–1804. [Google Scholar] [CrossRef]
- Marimpietri, D.; Airoldi, I.; Faini, A.C.; Malavasi, F.; Morandi, F. The Role of Extracellular Vesicles in the Progression of Human Neuroblastoma. Int. J. Mol. Sci. 2021, 22, 3964. [Google Scholar] [CrossRef]
- Marimpietri, D.; Corrias, M.V.; Tripodi, G.; Gramignoli, R.; Airoldi, I.; Morandi, F. Immunomodulatory properties of extracellular vesicles isolated from bone marrow of patients with neuroblastoma: Role of PD-L1 and HLA-G. Front. Immunol. 2024, 15, 1469771. [Google Scholar] [CrossRef]
- Morandi, F.; Marimpietri, D.; Görgens, A.; Gallo, A.; Srinivasan, R.C.; El-Andaloussi, S.; Gramignoli, R. Human Amnion Epithelial Cells Impair T Cell Proliferation: The Role of HLA-G and HLA-E Molecules. Cells 2020, 9, 2123. [Google Scholar] [CrossRef]
- Morandi, F.; Marimpietri, D.; Horenstein, A.L.; Bolzoni, M.; Toscani, D.; Costa, F.; Castella, B.; Faini, A.C.; Massaia, M.; Pistoia, V.; et al. Microvesicles released from multiple myeloma cells are equipped with ectoenzymes belonging to canonical and non-canonical adenosinergic pathways and produce adenosine from ATP and NAD+. Oncoimmunology 2018, 7, e1458809. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef]
- Kato, M.; Manabe, A. Treatment and biology of pediatric acute lymphoblastic leukemia. Pediatr. Int. 2018, 60, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Namayandeh, S.M.; Khazaei, Z.; Lari Najafi, M.; Goodarzi, E.; Moslem, A. GLOBAL Leukemia in Children 0-14 Statistics 2018, Incidence and Mortality and Human Development Index (HDI): GLOBOCAN Sources and Methods. Asian Pac. J. Cancer Prev. APJCP 2020, 21, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Mullighan, C.G. Pediatric acute lymphoblastic leukemia. Haematologica 2020, 105, 2524–2539. [Google Scholar] [CrossRef] [PubMed]
- Rubnitz, J.E.; Kaspers, G.J.L. How I treat pediatric acute myeloid leukemia. Blood 2021, 138, 1009–1018. [Google Scholar] [CrossRef]
- Lejman, M.; Chałupnik, A.; Chilimoniuk, Z.; Dobosz, M. Genetic Biomarkers and Their Clinical Implications in B-Cell Acute Lymphoblastic Leukemia in Children. Int. J. Mol. Sci. 2022, 23, 2755. [Google Scholar] [CrossRef]
- Slayton, W.B.; Schultz, K.R.; Kairalla, J.A.; Devidas, M.; Mi, X.; Pulsipher, M.A.; Chang, B.H.; Mullighan, C.; Iacobucci, I.; Silverman, L.B.; et al. Dasatinib Plus Intensive Chemotherapy in Children, Adolescents, and Young Adults With Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: Results of Children’s Oncology Group Trial AALL0622. J. Clin. Oncol. 2018, 36, 2306–2314. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef]
- Inaba, H.; Pui, C.H. Advances in the Diagnosis and Treatment of Pediatric Acute Lymphoblastic Leukemia. J. Clin. Med. 2021, 10, 1926. [Google Scholar] [CrossRef]
- Bassan, R.; Spinelli, O.; Oldani, E.; Intermesoli, T.; Tosi, M.; Peruta, B.; Rossi, G.; Borlenghi, E.; Pogliani, E.M.; Terruzzi, E.; et al. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL). Blood 2009, 113, 4153–4162. [Google Scholar] [CrossRef]
- Mullighan, C.G. Molecular genetics of B-precursor acute lymphoblastic leukemia. J. Clin. Investig. 2012, 122, 3407–3415. [Google Scholar] [CrossRef]
- Ross, M.E.; Zhou, X.; Song, G.; Shurtleff, S.A.; Girtman, K.; Williams, W.K.; Liu, H.C.; Mahfouz, R.; Raimondi, S.C.; Lenny, N.; et al. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood 2003, 102, 2951–2959. [Google Scholar] [CrossRef] [PubMed]
- Temple, W.C.; Mueller, S.; Hermiston, M.L.; Burkhardt, B. Diagnosis and management of lymphoblastic lymphoma in children, adolescents and young adults. Best Pract. Res. Clin. Haematol. 2023, 36, 101449. [Google Scholar] [CrossRef] [PubMed]
- Dander, E.; Palmi, C.; D’Amico, G.; Cazzaniga, G. The Bone Marrow Niche in B-Cell Acute Lymphoblastic Leukemia: The Role of Microenvironment from Pre-Leukemia to Overt Leukemia. Int. J. Mol. Sci. 2021, 22, 4426. [Google Scholar] [CrossRef] [PubMed]
- Lauten, M.; Möricke, A.; Beier, R.; Zimmermann, M.; Stanulla, M.; Meissner, B.; Odenwald, E.; Attarbaschi, A.; Niemeyer, C.; Niggli, F.; et al. Prediction of outcome by early bone marrow response in childhood acute lymphoblastic leukemia treated in the ALL-BFM 95 trial: Differential effects in precursor B-cell and T-cell leukemia. Haematologica 2012, 97, 1048–1056. [Google Scholar] [CrossRef]
- Reinhardt, D.; Antoniou, E.; Waack, K. Pediatric Acute Myeloid Leukemia-Past, Present, and Future. J. Clin. Med. 2022, 11, 504. [Google Scholar] [CrossRef]
- Tseng, S.; Lee, M.E.; Lin, P.C. A Review of Childhood Acute Myeloid Leukemia: Diagnosis and Novel Treatment. Pharmaceuticals 2023, 16, 1614. [Google Scholar] [CrossRef]
- de Rooij, J.D.; Zwaan, C.M.; van den Heuvel-Eibrink, M. Pediatric AML: From Biology to Clinical Management. J. Clin. Med. 2015, 4, 127–149. [Google Scholar] [CrossRef]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef]
- Tran, T.H.; Hunger, S.P. The genomic landscape of pediatric acute lymphoblastic leukemia and precision medicine opportunities. Semin. Cancer Biol. 2022, 84, 144–152. [Google Scholar] [CrossRef]
- Bakhtiyari, M.; Liaghat, M.; Aziziyan, F.; Shapourian, H.; Yahyazadeh, S.; Alipour, M.; Shahveh, S.; Maleki-Sheikhabadi, F.; Halimi, H.; Forghaniesfidvajani, R.; et al. The role of bone marrow microenvironment (BMM) cells in acute myeloid leukemia (AML) progression: Immune checkpoints, metabolic checkpoints, and signaling pathways. Cell Commun. Signal. CCS 2023, 21, 252. [Google Scholar] [CrossRef]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Sabatini, F.; Podestà, M.; Airoldi, I. Immunotherapeutic Strategies for Neuroblastoma: Present, Past and Future. Vaccines 2021, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Körber, V.; Stainczyk, S.A.; Kurilov, R.; Henrich, K.O.; Hero, B.; Brors, B.; Westermann, F.; Höfer, T. Neuroblastoma arises in early fetal development and its evolutionary duration predicts outcome. Nat. Genet. 2023, 55, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Nong, J.; Su, C.; Li, C.; Wang, C.; Li, W.; Li, Y.; Chen, P.; Li, Y.; Li, Z.; She, X.; et al. Global, regional, and national epidemiology of childhood neuroblastoma (1990-2021): A statistical analysis of incidence, mortality, and DALYs. EClinicalMedicine 2025, 79, 102964. [Google Scholar] [CrossRef]
- Burchill, S.A.; Beiske, K.; Shimada, H.; Ambros, P.F.; Seeger, R.; Tytgat, G.A.; Brock, P.R.; Haber, M.; Park, J.R.; Berthold, F. Recommendations for the standardization of bone marrow disease assessment and reporting in children with neuroblastoma on behalf of the International Neuroblastoma Response Criteria Bone Marrow Working Group. Cancer 2017, 123, 1095–1105. [Google Scholar] [CrossRef]
- Park, J.R.; Bagatell, R.; Cohn, S.L.; Pearson, A.D.; Villablanca, J.G.; Berthold, F.; Burchill, S.; Boubaker, A.; McHugh, K.; Nuchtern, J.G.; et al. Revisions to the International Neuroblastoma Response Criteria: A Consensus Statement From the National Cancer Institute Clinical Trials Planning Meeting. J. Clin. Oncol. 2017, 35, 2580–2587. [Google Scholar] [CrossRef]
- Boulais, P.E.; Frenette, P.S. Making sense of hematopoietic stem cell niches. Blood 2015, 125, 2621–2629. [Google Scholar] [CrossRef]
- Asada, N.; Kunisaki, Y.; Pierce, H.; Wang, Z.; Fernandez, N.F.; Birbrair, A.; Ma’ayan, A.; Frenette, P.S. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat. Cell Biol. 2017, 19, 214–223. [Google Scholar] [CrossRef]
- Ehninger, A.; Trumpp, A. The bone marrow stem cell niche grows up: Mesenchymal stem cells and macrophages move in. J. Exp. Med. 2011, 208, 421–428. [Google Scholar] [CrossRef]
- Man, Y.; Yao, X.; Yang, T.; Wang, Y. Hematopoietic Stem Cell Niche During Homeostasis, Malignancy, and Bone Marrow Transplantation. Front. Cell Dev. Biol. 2021, 9, 621214. [Google Scholar] [CrossRef]
- Le, P.M.; Andreeff, M.; Battula, V.L. Osteogenic niche in the regulation of normal hematopoiesis and leukemogenesis. Haematologica 2018, 103, 1945–1955. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Stiehl, T.; Raffel, S.; Hoang, V.T.; Hoffmann, I.; Poisa-Beiro, L.; Saeed, B.R.; Blume, R.; Manta, L.; Eckstein, V.; et al. Reduced hematopoietic stem cell frequency predicts outcome in acute myeloid leukemia. Haematologica 2017, 102, 1567–1577. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Kobolak, J.; Dinnyes, A.; Memic, A.; Khademhosseini, A.; Mobasheri, A. Mesenchymal stem cells: Identification, phenotypic characterization, biological properties and potential for regenerative medicine through biomaterial micro-engineering of their niche. Methods 2016, 99, 62–68. [Google Scholar] [CrossRef]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef]
- Sun, G.; Gu, Q.; Zheng, J.; Cheng, H.; Cheng, T. Emerging roles of extracellular vesicles in normal and malignant hematopoiesis. J. Clin. Investig. 2022, 132, e160840. [Google Scholar] [CrossRef]
- Lyu, T.; Wang, Y.; Li, D.; Yang, H.; Qin, B.; Zhang, W.; Li, Z.; Cheng, C.; Zhang, B.; Guo, R.; et al. Exosomes from BM-MSCs promote acute myeloid leukemia cell proliferation, invasion and chemoresistance via upregulation of S100A4. Exp. Hematol. Oncol. 2021, 10, 24. [Google Scholar] [CrossRef]
- Crompot, E.; Van Damme, M.; Pieters, K.; Vermeersch, M.; Perez-Morga, D.; Mineur, P.; Maerevoet, M.; Meuleman, N.; Bron, D.; Lagneaux, L.; et al. Extracellular vesicles of bone marrow stromal cells rescue chronic lymphocytic leukemia B cells from apoptosis, enhance their migration and induce gene expression modifications. Haematologica 2017, 102, 1594–1604. [Google Scholar] [CrossRef]
- Tettamanti, S.; Pievani, A.; Biondi, A.; Dotti, G.; Serafini, M. Catch me if you can: How AML and its niche escape immunotherapy. Leukemia 2022, 36, 13–22. [Google Scholar] [CrossRef]
- Kasherwal, V.; Kale, V.; Vaidya, A. Extracellular vesicles secreted by leukemic cells as mediators of dysregulated hematopoiesis: Acute myeloid leukemia as a case in point. Expert Rev. Hematol. 2025, 18, 225–237. [Google Scholar] [CrossRef]
- Balandrán, J.C.; Purizaca, J.; Enciso, J.; Dozal, D.; Sandoval, A.; Jiménez-Hernández, E.; Alemán-Lazarini, L.; Perez-Koldenkova, V.; Quintela-Núñez Del Prado, H.; Rios de Los Ríos, J.; et al. Pro-inflammatory-Related Loss of CXCL12 Niche Promotes Acute Lymphoblastic Leukemic Progression at the Expense of Normal Lymphopoiesis. Front. Immunol. 2016, 7, 666. [Google Scholar] [CrossRef] [PubMed]
- Crippa, S.; Bernardo, M.E. Mesenchymal Stromal Cells: Role in the BM Niche and in the Support of Hematopoietic Stem Cell Transplantation. HemaSphere 2018, 2, e151. [Google Scholar] [CrossRef] [PubMed]
- Corradi, G.; Bassani, B.; Simonetti, G.; Sangaletti, S.; Vadakekolathu, J.; Fontana, M.C.; Pazzaglia, M.; Gulino, A.; Tripodo, C.; Cristiano, G.; et al. Release of IFNγ by Acute Myeloid Leukemia Cells Remodels Bone Marrow Immune Microenvironment by Inducing Regulatory T Cells. Clin. Cancer Res. 2022, 28, 3141–3155. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef]
- Gu, Y.; Xia, J.; Guo, Y.; Tao, L.; Zhang, G.; Xu, J. Leukemia cells remodel bone marrow stromal cells to generate a protumoral microenvironment via the S100A8-NOX2-ROS signaling pathway. Sci. Rep. 2025, 15, 17179. [Google Scholar] [CrossRef]
- Feng, S.; Cen, J.; Huang, Y.; Shen, H.; Yao, L.; Wang, Y.; Chen, Z. Matrix metalloproteinase-2 and -9 secreted by leukemic cells increase the permeability of blood-brain barrier by disrupting tight junction proteins. PLoS ONE 2011, 6, e20599. [Google Scholar] [CrossRef]
- Schneider, P.; Costa, O.; Legrand, E.; Bigot, D.; Lecleire, S.; Grassi, V.; Vannier, J.P.; Vasse, M. In vitro secretion of matrix metalloprotease 9 is a prognostic marker in childhood acute lymphoblastic leukemia. Leuk. Res. 2010, 34, 24–31. [Google Scholar] [CrossRef]
- Vilchis-Ordoñez, A.; Contreras-Quiroz, A.; Vadillo, E.; Dorantes-Acosta, E.; Reyes-López, A.; Quintela-Nuñez del Prado, H.M.; Venegas-Vázquez, J.; Mayani, H.; Ortiz-Navarrete, V.; López-Martínez, B.; et al. Bone Marrow Cells in Acute Lymphoblastic Leukemia Create a Proinflammatory Microenvironment Influencing Normal Hematopoietic Differentiation Fates. BioMed Res. Int. 2015, 2015, 386165. [Google Scholar] [CrossRef]
- Portale, F.; Cricrì, G.; Bresolin, S.; Lupi, M.; Gaspari, S.; Silvestri, D.; Russo, B.; Marino, N.; Ubezio, P.; Pagni, F.; et al. ActivinA: A new leukemia-promoting factor conferring migratory advantage to B-cell precursor-acute lymphoblastic leukemic cells. Haematologica 2019, 104, 533–545. [Google Scholar] [CrossRef]
- Chen, Y.; Hoffmeister, L.M.; Zaun, Y.; Arnold, L.; Schmid, K.W.; Giebel, B.; Klein-Hitpass, L.; Hanenberg, H.; Squire, A.; Reinhardt, H.C.; et al. Acute myeloid leukemia-induced remodeling of the human bone marrow niche predicts clinical outcome. Blood Adv. 2020, 4, 5257–5268. [Google Scholar] [CrossRef]
- Polak, R.; de Rooij, B.; Pieters, R.; den Boer, M.L. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 2015, 126, 2404–2414. [Google Scholar] [CrossRef] [PubMed]
- Veiga, J.P.; Costa, L.F.; Sallan, S.E.; Nadler, L.M.; Cardoso, A.A. Leukemia-stimulated bone marrow endothelium promotes leukemia cell survival. Exp. Hematol. 2006, 34, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Zanetti, C.; Godavarthy, P.S.; Kumar, R.; Minciacchi, V.R.; Pfeiffer, J.; Metzler, M.; Lefort, S.; Maguer-Satta, V.; Nicolini, F.E.; et al. Bone marrow niche-derived extracellular matrix-degrading enzymes influence the progression of B-cell acute lymphoblastic leukemia. Leukemia 2020, 34, 1540–1552. [Google Scholar] [CrossRef] [PubMed]
- Passaro, D.; Di Tullio, A.; Abarrategi, A.; Rouault-Pierre, K.; Foster, K.; Ariza-McNaughton, L.; Montaner, B.; Chakravarty, P.; Bhaw, L.; Diana, G.; et al. Increased Vascular Permeability in the Bone Marrow Microenvironment Contributes to Disease Progression and Drug Response in Acute Myeloid Leukemia. Cancer Cell 2017, 32, 324–341.e6. [Google Scholar] [CrossRef]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef]
- La Spina, E.; Giallongo, S.; Giallongo, C.; Vicario, N.; Duminuco, A.; Parenti, R.; Giuffrida, R.; Longhitano, L.; Li Volti, G.; Cambria, D.; et al. Mesenchymal stromal cells in tumor microenvironment remodeling of BCR-ABL negative myeloproliferative diseases. Front. Oncol. 2023, 13, 1141610. [Google Scholar] [CrossRef]
- Han, G.; Lu, S.L.; Li, A.G.; He, W.; Corless, C.L.; Kulesz-Martin, M.; Wang, X.J. Distinct mechanisms of TGF-beta1-mediated epithelial-to-mesenchymal transition and metastasis during skin carcinogenesis. J. Clin. Investig. 2005, 115, 1714–1723. [Google Scholar] [CrossRef]
- Vicente López, Á.; Vázquez García, M.N.; Melen, G.J.; Entrena Martínez, A.; Cubillo Moreno, I.; García-Castro, J.; Orellana, M.R.; González, A.G. Mesenchymal stromal cells derived from the bone marrow of acute lymphoblastic leukemia patients show altered BMP4 production: Correlations with the course of disease. PLoS ONE 2014, 9, e84496. [Google Scholar] [CrossRef]
- Magalhães-Gama, F.; Malheiros Araújo Silvestrini, M.; Neves, J.C.F.; Araújo, N.D.; Alves-Hanna, F.S.; Kerr, M.W.A.; Carvalho, M.; Tarragô, A.M.; Soares Pontes, G.; Martins-Filho, O.A.; et al. Exploring cell-derived extracellular vesicles in peripheral blood and bone marrow of B-cell acute lymphoblastic leukemia pediatric patients: Proof-of-concept study. Front. Immunol. 2024, 15, 1421036. [Google Scholar] [CrossRef]
- Pando, A.; Reagan, J.L.; Quesenberry, P.; Fast, L.D. Extracellular vesicles in leukemia. Leuk. Res. 2018, 64, 52–60. [Google Scholar] [CrossRef]
- Georgievski, A.; Michel, A.; Thomas, C.; Mlamla, Z.; Pais de Barros, J.P.; Lemaire-Ewing, S.; Garrido, C.; Quéré, R. Acute lymphoblastic leukemia-derived extracellular vesicles affect quiescence of hematopoietic stem and progenitor cells. Cell Death Dis. 2022, 13, 337. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chen, Q.; Shang, Y.; Chen, L.; Myers, J.; Awadallah, A.; Sun, J.; Yu, S.; Umphred-Wilson, K.; Che, D.; et al. Endothelial PERK-ATF4-JAG1 axis activated by T-ALL remodels bone marrow vascular niche. Theranostics 2022, 12, 2894–2907. [Google Scholar] [CrossRef] [PubMed]
- Gholipour, E.; Kahroba, H.; Soltani, N.; Samadi, P.; Sarvarian, P.; Vakili-Samiani, S.; Hosein Pour Feizi, A.A.; Soltani-Zangbar, M.S.; Baghersalimi, A.; Darbandi, B.; et al. Paediatric pre-B acute lymphoblastic leukaemia-derived exosomes regulate immune function in human T cells. J. Cell. Mol. Med. 2022, 26, 4566–4576. [Google Scholar] [CrossRef] [PubMed]
- Swatler, J.; Turos-Korgul, L.; Brewinska-Olchowik, M.; De Biasi, S.; Dudka, W.; Le, B.V.; Kominek, A.; Cyranowski, S.; Pilanc, P.; Mohammadi, E.; et al. 4-1BBL-containing leukemic extracellular vesicles promote immunosuppressive effector regulatory T cells. Blood Adv. 2022, 6, 1879–1894. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Guo, Z.; Zhou, Y.; Ni, J.; Zhu, J.; Fan, X.; Chen, X.; Liu, Y.; Li, Z.; Zhou, H. microRNA-1246-containing extracellular vesicles from acute myeloid leukemia cells promote the survival of leukemia stem cells via the LRIG1-meditated STAT3 pathway. Aging 2021, 13, 13644–13662. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Vaiselbuh, S.R. Silencing of Exosomal miR-181a Reverses Pediatric Acute Lymphocytic Leukemia Cell Proliferation. Pharmaceuticals 2020, 13, 241. [Google Scholar] [CrossRef]
- Licari, E.; Cricrì, G.; Mauri, M.; Raimondo, F.; Dioni, L.; Favero, C.; Giussani, A.; Starace, R.; Nucera, S.; Biondi, A.; et al. ActivinA modulates B-acute lymphoblastic leukaemia cell communication and survival by inducing extracellular vesicles production. Sci. Rep. 2024, 14, 16083. [Google Scholar] [CrossRef]
- Zhai, X.; You, F.; Xiang, S.; Jiang, L.; Chen, D.; Li, Y.; Fan, S.; Han, Z.; Zhang, T.; An, G.; et al. MUC1-Tn-targeting chimeric antigen receptor-modified Vγ9Vδ2 T cells with enhanced antigen-specific anti-tumor activity. Am. J. Cancer Res. 2021, 11, 79–91. [Google Scholar]
- Rios de Los Rios, J.; Enciso, J.; Vilchis-Ordoñez, A.; Vázquez-Ramírez, R.; Ramirez-Ramirez, D.; Balandrán, J.C.; Rodríguez-Martínez, A.; Ruiz-Tachiquín, M.; Pompa-Mera, E.; Mendoza, L.; et al. Acute lymphoblastic leukemia-secreted miRNAs induce a proinflammatory microenvironment and promote the activation of hematopoietic progenitors. J. Leukoc. Biol. 2022, 112, 31–45. [Google Scholar] [CrossRef]
- Yuan, T.; Shi, C.; Xu, W.; Yang, H.L.; Xia, B.; Tian, C. Extracellular vesicles derived from T-cell acute lymphoblastic leukemia inhibit osteogenic differentiation of bone marrow mesenchymal stem cells via miR-34a-5p. Endocr. J. 2021, 68, 1197–1208. [Google Scholar] [CrossRef]
- Chen, T.; Zhang, G.; Kong, L.; Xu, S.; Wang, Y.; Dong, M. Leukemia-derived exosomes induced IL-8 production in bone marrow stromal cells to protect the leukemia cells against chemotherapy. Life Sci. 2019, 221, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Huan, J.; Hornick, N.I.; Goloviznina, N.A.; Kamimae-Lanning, A.N.; David, L.L.; Wilmarth, P.A.; Mori, T.; Chevillet, J.R.; Narla, A.; Roberts, C.T., Jr.; et al. Coordinate regulation of residual bone marrow function by paracrine trafficking of AML exosomes. Leukemia 2015, 29, 2285–2295. [Google Scholar] [CrossRef] [PubMed]
- Karantanou, C.; Minciacchi, V.R.; Kumar, R.; Zanetti, C.; Bravo, J.; Pereira, R.S.; Tascher, G.; Tertel, T.; Covarrubias-Pinto, A.; Bankov, K.; et al. Impact of mesenchymal stromal cell-derived vesicular cargo on B-cell acute lymphoblastic leukemia progression. Blood Adv. 2023, 7, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Fei, F.; Joo, E.J.; Tarighat, S.S.; Schiffer, I.; Paz, H.; Fabbri, M.; Abdel-Azim, H.; Groffen, J.; Heisterkamp, N. B-cell precursor acute lymphoblastic leukemia and stromal cells communicate through Galectin-3. Oncotarget 2015, 6, 11378–11394. [Google Scholar] [CrossRef]
- Fetahu, I.S.; Esser-Skala, W.; Dnyansagar, R.; Sindelar, S.; Rifatbegovic, F.; Bileck, A.; Skos, L.; Bozsaky, E.; Lazic, D.; Shaw, L.; et al. Single-cell transcriptomics and epigenomics unravel the role of monocytes in neuroblastoma bone marrow metastasis. Nat. Commun. 2023, 14, 3620. [Google Scholar] [CrossRef]
- Hochheuser, C.; Windt, L.J.; Kunze, N.Y.; de Vos, D.L.; Tytgat, G.A.M.; Voermans, C.; Timmerman, I. Mesenchymal Stromal Cells in Neuroblastoma: Exploring Crosstalk and Therapeutic Implications. Stem Cells Dev. 2021, 30, 59–78. [Google Scholar] [CrossRef]
- Colletti, M.; Tomao, L.; Galardi, A.; Paolini, A.; Di Paolo, V.; De Stefanis, C.; Mascio, P.; Nazio, F.; Petrini, S.; Castellano, A.; et al. Neuroblastoma-secreted exosomes carrying miR-375 promote osteogenic differentiation of bone-marrow mesenchymal stromal cells. J. Extracell. Vesicles 2020, 9, 1774144. [Google Scholar] [CrossRef]
- Hochheuser, C.; van Zogchel, L.M.J.; Kleijer, M.; Kuijk, C.; Tol, S.; van der Schoot, C.E.; Voermans, C.; Tytgat, G.A.M.; Timmerman, I. The Metastatic Bone Marrow Niche in Neuroblastoma: Altered Phenotype and Function of Mesenchymal Stromal Cells. Cancers 2020, 12, 3231. [Google Scholar] [CrossRef]
- Marimpietri, D.; Petretto, A.; Raffaghello, L.; Pezzolo, A.; Gagliani, C.; Tacchetti, C.; Mauri, P.; Melioli, G.; Pistoia, V. Proteome profiling of neuroblastoma-derived exosomes reveal the expression of proteins potentially involved in tumor progression. PLoS ONE 2013, 8, e75054. [Google Scholar] [CrossRef]
- Cheng, J.; Ji, D.; Ma, J.; Zhang, Q.; Zhang, W.; Yang, L. Proteomic analysis of serum small extracellular vesicles identifies diagnostic biomarkers for neuroblastoma. Front. Oncol. 2024, 14, 1367159. [Google Scholar] [CrossRef]
- Morandi, F.; Airoldi, I.; Marimpietri, D.; Bracci, C.; Faini, A.C.; Gramignoli, R. CD38, a Receptor with Multifunctional Activities: From Modulatory Functions on Regulatory Cell Subsets and Extracellular Vesicles, to a Target for Therapeutic Strategies. Cells 2019, 8, 1527. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Zúñiga, C.D.; Garza-Veloz, I.; Martínez-Rendón, J.; Ureño-Segura, M.; Delgado-Enciso, I.; Martinez-Fierro, M.L. Circulating Biomarkers Associated with the Diagnosis and Prognosis of B-Cell Progenitor Acute Lymphoblastic Leukemia. Cancers 2023, 15, 4186. [Google Scholar] [CrossRef] [PubMed]
- Hornick, N.I.; Huan, J.; Doron, B.; Goloviznina, N.A.; Lapidus, J.; Chang, B.H.; Kurre, P. Serum Exosome MicroRNA as a Minimally-Invasive Early Biomarker of AML. Sci. Rep. 2015, 5, 11295. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.A.; Baba, S.K.; Sadida, H.Q.; Marzooqi, S.A.; Jerobin, J.; Altemani, F.H.; Algehainy, N.; Alanazi, M.A.; Abou-Samra, A.B.; Kumar, R.; et al. Extracellular vesicles as tools and targets in therapy for diseases. Signal Transduct. Target. Ther. 2024, 9, 27. [Google Scholar] [CrossRef]
- Payandeh, Z.; Tangruksa, B.; Synnergren, J.; Heydarkhan-Hagvall, S.; Nordin, J.Z.; Andaloussi, S.E.; Borén, J.; Wiseman, J.; Bohlooly, Y.M.; Lindfors, L.; et al. Extracellular vesicles transport RNA between cells: Unraveling their dual role in diagnostics and therapeutics. Mol. Asp. Med. 2024, 99, 101302. [Google Scholar] [CrossRef]
- Gu, X.; Erb, U.; Büchler, M.W.; Zöller, M. Improved vaccine efficacy of tumor exosome compared to tumor lysate loaded dendritic cells in mice. Int. J. Cancer 2015, 136, E74–E84. [Google Scholar] [CrossRef]
- Khani, A.T.; Sharifzad, F.; Mardpour, S.; Hassan, Z.M.; Ebrahimi, M. Tumor extracellular vesicles loaded with exogenous Let-7i and miR-142 can modulate both immune response and tumor microenvironment to initiate a powerful anti-tumor response. Cancer Lett. 2021, 501, 200–209. [Google Scholar] [CrossRef]
- Huang, F.; Wan, J.; Hu, W.; Hao, S. Enhancement of Anti-Leukemia Immunity by Leukemia-Derived Exosomes Via Downregulation of TGF-β1 Expression. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 44, 240–254. [Google Scholar] [CrossRef]
- Rossowska, J.; Anger, N.; Wegierek, K.; Szczygieł, A.; Mierzejewska, J.; Milczarek, M.; Szermer-Olearnik, B.; Pajtasz-Piasecka, E. Antitumor Potential of Extracellular Vesicles Released by Genetically Modified Murine Colon Carcinoma Cells With Overexpression of Interleukin-12 and shRNA for TGF-β1. Front. Immunol. 2019, 10, 211. [Google Scholar] [CrossRef]
- Huang, F.; Li, Z.; Zhang, W.; Li, J.; Hao, S. Enhancing the anti-leukemia immunity of acute lymphocytic leukemia-derived exosome-based vaccine by downregulation of PD-L1 expression. Cancer Immunol. Immunother. CII 2022, 71, 2197–2212. [Google Scholar] [CrossRef]
- Zhang, D.; Jiang, Y.; Wang, M.; Zhao, J.; Wan, J.; Li, Z.; Huang, D.; Yu, J.; Li, J.; Liu, J.; et al. A novel costimulatory molecule gene-modified leukemia cell-derived exosome enhances the anti-leukemia efficacy of DC vaccine in mouse models. Vaccine 2024, 42, 126097. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Chen, J.; Xu, F.; Chen, H.; Li, Y.; Li, W. Dendritic Cell-Derived Exosomes in Cancer Immunotherapy. Pharmaceutics 2023, 15, 2070. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, X.; Xiang, X.; Pang, X.; Chen, S.; Zhang, Y.; Ren, E.; Zhang, L.; Liu, X.; Lv, P.; et al. A nanovaccine for antigen self-presentation and immunosuppression reversal as a personalized cancer immunotherapy strategy. Nat. Nanotechnol. 2022, 17, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer—Implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef]
- Dong, Q.; Liu, X.; Cheng, K.; Sheng, J.; Kong, J.; Liu, T. Pre-metastatic Niche Formation in Different Organs Induced by Tumor Extracellular Vesicles. Front. Cell Dev. Biol. 2021, 9, 733627. [Google Scholar] [CrossRef]
- Hoshino, A.; Kim, H.S.; Bojmar, L.; Gyan, K.E.; Cioffi, M.; Hernandez, J.; Zambirinis, C.P.; Rodrigues, G.; Molina, H.; Heissel, S.; et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell 2020, 182, 1044–1061.e18. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Amico, G.; Starace, R.; Della Lastra, M.; Marimpietri, D.; Dander, E.; Morandi, F.; Airoldi, I. Dysregulation of the Bone Marrow Microenvironment in Pediatric Tumors: The Role of Extracellular Vesicles in Acute Leukemias and Neuroblastoma. Int. J. Mol. Sci. 2025, 26, 5380. https://doi.org/10.3390/ijms26115380
D’Amico G, Starace R, Della Lastra M, Marimpietri D, Dander E, Morandi F, Airoldi I. Dysregulation of the Bone Marrow Microenvironment in Pediatric Tumors: The Role of Extracellular Vesicles in Acute Leukemias and Neuroblastoma. International Journal of Molecular Sciences. 2025; 26(11):5380. https://doi.org/10.3390/ijms26115380
Chicago/Turabian StyleD’Amico, Giovanna, Rita Starace, Martina Della Lastra, Danilo Marimpietri, Erica Dander, Fabio Morandi, and Irma Airoldi. 2025. "Dysregulation of the Bone Marrow Microenvironment in Pediatric Tumors: The Role of Extracellular Vesicles in Acute Leukemias and Neuroblastoma" International Journal of Molecular Sciences 26, no. 11: 5380. https://doi.org/10.3390/ijms26115380
APA StyleD’Amico, G., Starace, R., Della Lastra, M., Marimpietri, D., Dander, E., Morandi, F., & Airoldi, I. (2025). Dysregulation of the Bone Marrow Microenvironment in Pediatric Tumors: The Role of Extracellular Vesicles in Acute Leukemias and Neuroblastoma. International Journal of Molecular Sciences, 26(11), 5380. https://doi.org/10.3390/ijms26115380