
Transcriptome and Endogenous Hormone Analysis Reveals the Molecular Mechanism of Callus Hyperhydricity in Flax (Linum usitatissimum L.)

Abstract
1. Introduction
2. Results
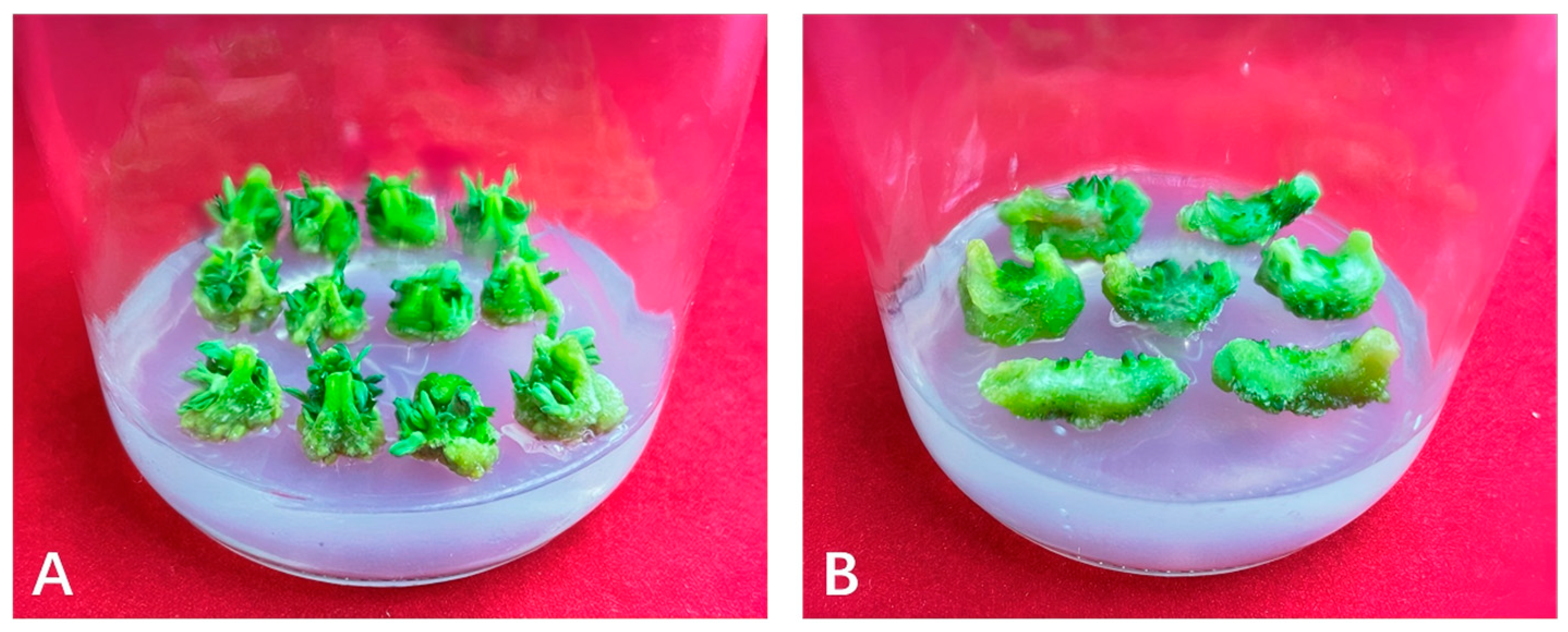
2.1. Morphologic Observation
2.2. Overview of Transcriptome Sequencing Results
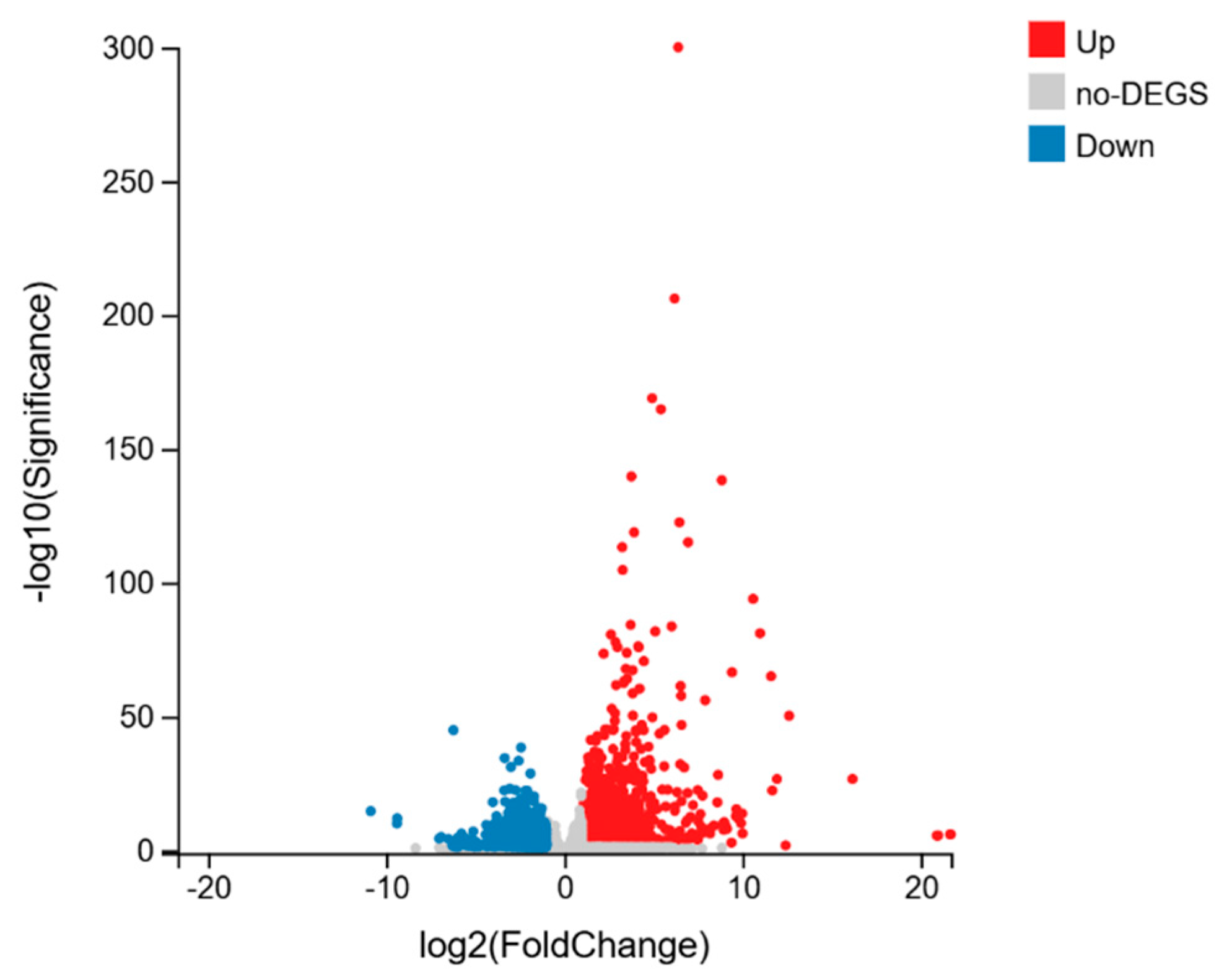
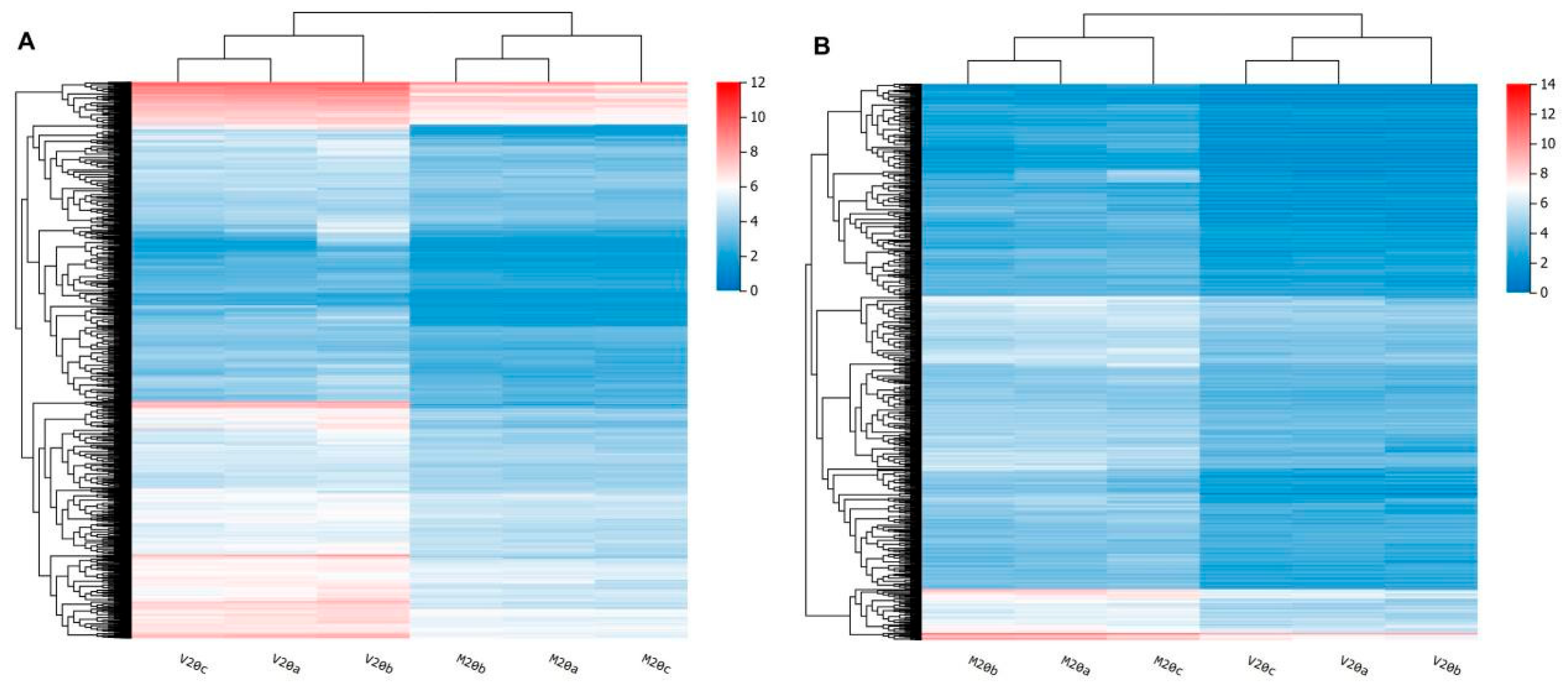
2.3. Identification of Differentially Expressed Genes (DEGs)
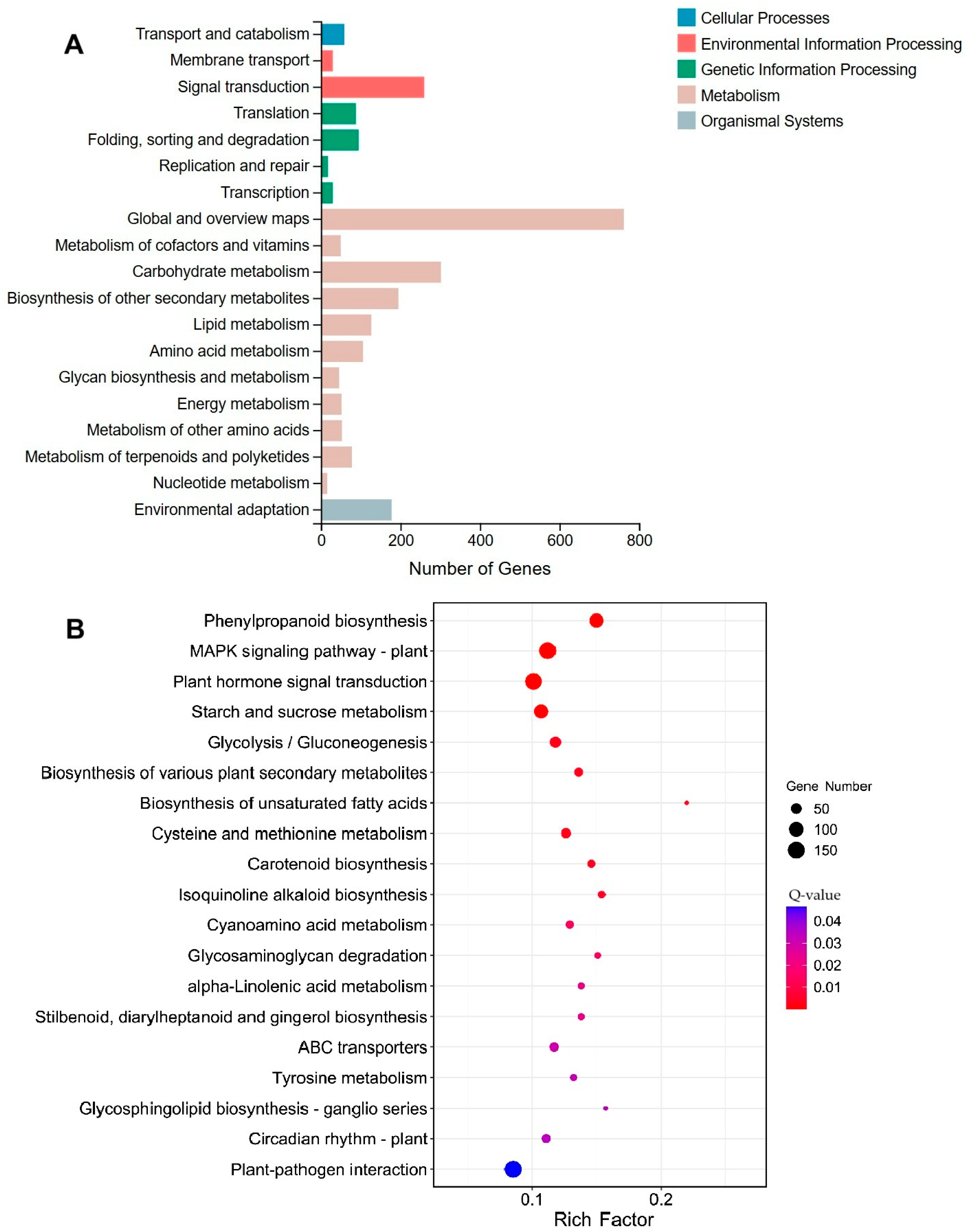
2.4. Functional Annotation of Differentially Expressed Genes
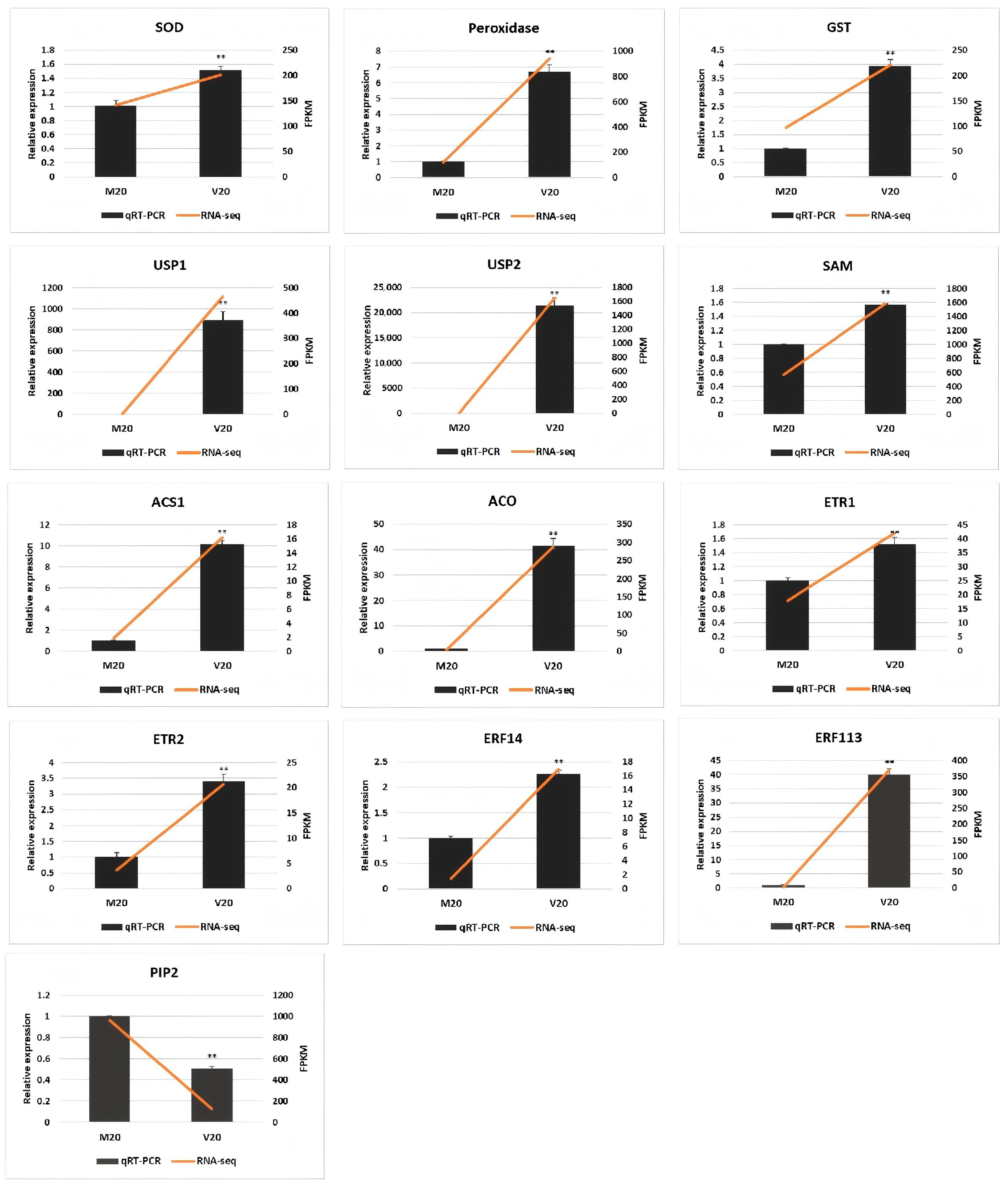
2.5. Quantitative Real-Time PCR Analysis
2.6. Determination of the Endogenous Hormone Content
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction, cDNA Library Preparation, and Sequencing
4.3. Sequencing Data Analysis
4.4. qRT-PCR Validation
4.5. Measurement of the Endogenous Hormone Content
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Polivanova, O.B.; Bedarev, V.A. Hyperhydricity in Plant Tissue Culture. Plants 2022, 11, 3313. [Google Scholar] [CrossRef] [PubMed]
- van den Dries, N.; Giannì, S.; Czerednik, A.; Krens, F.A.; de Klerk, G.-J.M. Flooding of the apoplast is a key factor in the development of hyperhydricity. J. Exp. Bot. 2013, 64, 5221–5230. [Google Scholar] [CrossRef]
- Gantait, S.; Mahanta, M. Hyperhydricity-induced changes among in vitro regenerants of gerbera. S. Afr. J. Bot. 2022, 149, 496–501. [Google Scholar] [CrossRef]
- Chakrabarty, D.; Park, S.Y.; Ali, M.B.; Shin, K.S.; Paek, K.Y. Hyperhydricity in apple: Ultrastructural and physiological aspects. Tree Physiol. 2006, 26, 377–388. [Google Scholar] [CrossRef]
- Gao, H.; Li, J.; Ji, H.; An, L.; Xia, X. Hyperhydricity-induced ultrastructural and physiological changes in blueberry (Vaccinium spp.). Plant Cell Tissue Organ Cult. 2018, 133, 65–76. [Google Scholar] [CrossRef]
- Tabart, J.; Franck, T.; Kevers, C.; Dommes, J. Effect of polyamines and polyamine precursors on hyperhydricity in micropropagated apple shoots. Plant Cell Tissue Organ Cult. 2015, 120, 11–18. [Google Scholar] [CrossRef]
- Muneer, S.; Park, Y.G.; Jeong, B.R. Red and Blue Light Emitting Diodes (LEDs) Participate in Mitigation of Hyperhydricity in In Vitro-Grown Carnation Genotypes (Dianthus caryophyllus). J. Plant Growth Regul. 2018, 37, 370–379. [Google Scholar] [CrossRef]
- Ivanova, M.; Van Staden, J. Natural ventilation effectively reduces hyperhydricity in shoot cultures of Aloe polyphylla Schönland ex Pillans. Plant Growth Regul. 2010, 60, 143–150. [Google Scholar] [CrossRef]
- Lai, C.C.; Lin, H.M.; Nalawade, S.M.; Fang, W.; Tsay, H.-S. Hyperhydricity in shoot cultures of Scrophularia yoshimurae can be effectively reduced by ventilation of culture vessels. J. Plant Physiol. 2005, 162, 355–361. [Google Scholar] [CrossRef]
- Ivanova, M.; van Staden, J. Effect of ammonium ions and cytokinins on hyperhydricity and multiplication rate of in vitro regenerated shoots of Aloe polyphylla. Plant Cell Tissue Organ Cult. 2008, 92, 227–231. [Google Scholar] [CrossRef]
- Ivanova, M.; Van Staden, J. Influence of gelling agent and cytokinins on the control of hyperhydricity in Aloe polyphylla. Plant Cell Tissue Organ Cult. 2011, 104, 13–21. [Google Scholar] [CrossRef]
- Kadota, M.; Niimi, Y. Effects of cytokinin types and their concentrations on shoot proliferation and hyperhydricity in in vitro pear cultivar shoots. Plant Cell Tissue Organ Cult. 2003, 72, 261–265. [Google Scholar] [CrossRef]
- Hassannejad, S.; Bernard, F.; Mirzajani, F.; Gholami, M. SA improvement of hyperhydricity reversion in Thymus daenensis shoots culture may be associated with polyamines changes. Plant Physiol. Biochem. PPB 2012, 51, 40–46. [Google Scholar] [CrossRef]
- Liu, M.; Jiang, F.; Kong, X.; Tian, J.; Wu, Z.; Wu, Z. Effects of multiple factors on hyperhydricity of Allium sativum L. Sci. Hortic. 2017, 217, 285–296. [Google Scholar] [CrossRef]
- Gao, H.; Xia, X.; An, L.; Xin, X.; Liang, Y. Reversion of hyperhydricity in pink (Dianthus chinensis L.) plantlets by AgNO3 and its associated mechanism during in vitro culture. Plant Sci. 2017, 254, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Xu, P.; Li, J.; Ji, H.; An, L.; Xia, X. AgNO3 prevents the occurrence of hyperhydricity in Dianthus chinensis L. by enhancing water loss and antioxidant capacity. In Vitro Cell. Dev. Biol.-Plant 2017, 53, 561–570. [Google Scholar] [CrossRef]
- Maurya, R.L.; Kumar, M.; Sirohi, U.; Priya; Chaudhary, V.; Sharma, V.R.; Yadav, D.; Yadav, M.K. Effect of Silver Nitrate and Thidiazuron on Shoot Proliferation, Hyperhydricity and Assessment of Genetic Fidelity of Microplants in Carnation (Dianthus caryophyllus L.). Cytol. Genet. 2023, 57, 87–94. [Google Scholar] [CrossRef]
- Bakir, Y.; Eldem, V.; Zararsiz, G.; Unver, T. Global Transcriptome Analysis Reveals Differences in Gene Expression Patterns Between Nonhyperhydric and Hyperhydric Peach Leaves. Plant Genome 2016, 9, 1–9. [Google Scholar] [CrossRef]
- Muneer, S.; Soundararajan, P.; Jeong, B.R. Proteomic and Antioxidant Analysis Elucidates the Underlying Mechanism of Tolerance to Hyperhydricity Stress in In Vitro Shoot Cultures of Dianthus caryophyllus. J. Plant Growth Regul. 2016, 35, 667–679. [Google Scholar] [CrossRef]
- Gao, H.; Xia, X.; An, L. Critical roles of the activation of ethylene pathway genes mediated by DNA demethylation in Arabidopsis hyperhydricity. Plant Genome 2022, 15, e20202. [Google Scholar] [CrossRef]
- Zhang, H.; Shi, M.; Su, S.; Zheng, S.; Wang, M.; Lv, J.; Wang, X.; Yang, Q.; Gao, H. Whole-genome methylation analysis reveals epigenetic differences in the occurrence and recovery of hyperhydricity in Dendrobium officinale plantlets. In Vitro Cell. Dev. Biol.-Plant 2022, 58, 290–301. [Google Scholar] [CrossRef]
- Lelis, T.M.; Santos, I.R.; Silva-Cardoso, I.M.A.; de Souza, A.L.X.; Gomes, A.C.M.M.; Mehta, A.; Scherwinski-Pereira, J.E. Unraveling the occurrence of hyperhydricity in oil palm somatic embryos during somatic embryogenesis process. Protoplasma 2025, 262, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Muneer, S.; Wei, H.; Park, Y.G.; Jeong, H.K.; Jeong, B.R. Proteomic Analysis Reveals the Dynamic Role of Silicon in Alleviation of Hyperhydricity in Carnation Grown In Vitro. Int. J. Mol. Sci. 2017, 19, 50. [Google Scholar] [CrossRef]
- Saher, S.; Piqueras, A.; Hellin, E.; Olmos, E. Hyperhydricity in micropropagated carnation shoots: The role of oxidative stress. Physiol. Plant. 2004, 120, 152–161. [Google Scholar] [CrossRef]
- Dewir, Y.H.; Chakrabarty, D.; Ali, M.B.; Hahn, E.; Paek, K. Lipid peroxidation and antioxidant enzyme activities of Euphorbia millii hyperhydric shoots. Environ. Exp. Bot. 2006, 58, 93–99. [Google Scholar] [CrossRef]
- Franck, T.; Kevers, C.; Gaspar, T. Protective enzymatic systems against activated oxygen species compared in normal and vitrified shoots of Prunus avium L. L. raised in vitro. Plant Growth Regul. 1995, 16, 253–256. [Google Scholar] [CrossRef]
- Dhanyalakshmi, K.H.; Nataraja, K.N. Universal stress protein-like gene from mulberry enhances abiotic stress tolerance in Escherichia coli and transgenic tobacco cells. Plant Biol. 2021, 23, 1190–1194. [Google Scholar] [CrossRef]
- Luo, D.; Wu, Z.; Bai, Q.; Zhang, Y.; Huang, M.; Huang, Y.; Li, X. Universal Stress Proteins: From Gene to Function. Int. J. Mol. Sci. 2023, 24, 4725. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Che, S.; Zhang, Y.; Wang, H.; Wei, T.; Yan, G.; Song, W.; Yu, W. Universal stress protein in Malus sieversii confers enhanced drought tolerance. J. Plant Res. 2019, 132, 825–837. [Google Scholar] [CrossRef]
- Kataeva, N.V.; Alexandrova, I.G.; Butenko, R.G.; Dragavtceva, E.V. Effect of applied and internal hormones on vitrification and apical necrosis of different plants cultured in vitro. Plant Cell Tissue Organ Cult. 1991, 27, 149–154. [Google Scholar] [CrossRef]
- Jain, A.; Husain, S.; Kothari, S.L. Micropropagation of Dianthus caryophyllus L.—Control of Vitrification. J. Plant Biochem. Biotechnol. 1997, 6, 35–37. [Google Scholar] [CrossRef]
- Ding, L.; Uehlein, N.; Kaldenhoff, R.; Guo, S.; Zhu, Y.; Kai, L. Aquaporin PIP2;1 affects water transport and root growth in rice (Oryza sativa L.). Plant Physiol. Biochem. PPB 2019, 139, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Yaneff, A.; Vitali, V.; Amodeo, G. PIP1 aquaporins: Intrinsic water channels or PIP2 aquaporin modulators? FEBS Lett. 2015, 589, 3508–3515. [Google Scholar] [CrossRef] [PubMed]
- Kemat, N.; Visser, R.G.F.; Krens, F.A. Hypolignification: A Decisive Factor in the Development of Hyperhydricity. Plants 2021, 10, 2625. [Google Scholar] [CrossRef]
- Berthet, S.; Demont-Caulet, N.; Pollet, B.; Bidzinski, P.; Cézard, L.; Le Bris, P.; Borrega, N.; Hervé, J.; Blondet, E.; Balzergue, S.; et al. Disruption of LACCASE4 and 17 results in tissue-specific alterations to lignification of Arabidopsis thaliana stems. Plant Cell 2011, 23, 1124–1137. [Google Scholar] [CrossRef]
- Caparrós-Ruiz, D.; Fornalé, S.; Civardi, L.; Puigdomènech, P.; Rigau, J. Isolation and characterisation of a family of laccases in maize. Plant Sci. 2006, 171, 217–225. [Google Scholar] [CrossRef]
- Liu, D.; Yuan, H.; Yao, Y.; Cheng, L.; Tang, L.; Kang, Q.; Song, X.; Chen, S.; Wu, G. Genome-Wide Identification and Expression Pattern Analysis of the F5H Gene Family in Flax (Linum usitatissimum L.). Agronomy 2023, 13, 1108. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads (M) | Clean Reads (M) | Clean Bases (Gb) | Q20 (%) | Q30 (%) | Mapping Ratio (%) |
|---|---|---|---|---|---|---|
| M20a | 47.19 | 43.32 | 6.5 | 97.9 | 92.92 | 96.35 |
| M20b | 43.69 | 40.7 | 6.11 | 98.47 | 94.33 | 96.51 |
| M20c | 45.44 | 42.55 | 6.38 | 97.83 | 92.61 | 96.76 |
| V20a | 42.7 | 40.12 | 6.02 | 98.38 | 94.02 | 96.72 |
| V20b | 47.19 | 43.3 | 6.5 | 98.21 | 93.59 | 96.43 |
| V20c | 48.93 | 44.2 | 6.63 | 98.02 | 93.21 | 96.08 |
| Group | Total | Upregulated Genes | Downregulated Genes |
|---|---|---|---|
| M20 vs. V20 | 2698 | 1667 | 1031 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.; Cheng, L.; Tang, L.; Yang, L.; Jiang, Z.; Song, X.; Kang, Q.; Yao, D.; Chen, S.; Ru, J.; et al. Transcriptome and Endogenous Hormone Analysis Reveals the Molecular Mechanism of Callus Hyperhydricity in Flax (Linum usitatissimum L.). Int. J. Mol. Sci. 2025, 26, 5360. https://doi.org/10.3390/ijms26115360
Liu D, Cheng L, Tang L, Yang L, Jiang Z, Song X, Kang Q, Yao D, Chen S, Ru J, et al. Transcriptome and Endogenous Hormone Analysis Reveals the Molecular Mechanism of Callus Hyperhydricity in Flax (Linum usitatissimum L.). International Journal of Molecular Sciences. 2025; 26(11):5360. https://doi.org/10.3390/ijms26115360
Chicago/Turabian StyleLiu, Dandan, Lili Cheng, Lili Tang, Lie Yang, Zhongjuan Jiang, Xixia Song, Qinghua Kang, Dandan Yao, Si Chen, Jiarong Ru, and et al. 2025. "Transcriptome and Endogenous Hormone Analysis Reveals the Molecular Mechanism of Callus Hyperhydricity in Flax (Linum usitatissimum L.)" International Journal of Molecular Sciences 26, no. 11: 5360. https://doi.org/10.3390/ijms26115360
APA StyleLiu, D., Cheng, L., Tang, L., Yang, L., Jiang, Z., Song, X., Kang, Q., Yao, D., Chen, S., Ru, J., Zhang, L., Wu, G., & Yuan, H. (2025). Transcriptome and Endogenous Hormone Analysis Reveals the Molecular Mechanism of Callus Hyperhydricity in Flax (Linum usitatissimum L.). International Journal of Molecular Sciences, 26(11), 5360. https://doi.org/10.3390/ijms26115360