
Transcriptome and Co-Expression Network Analyses of Resistant and Susceptible Rice Cultivars in Response to Meloidogyne graminicola

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
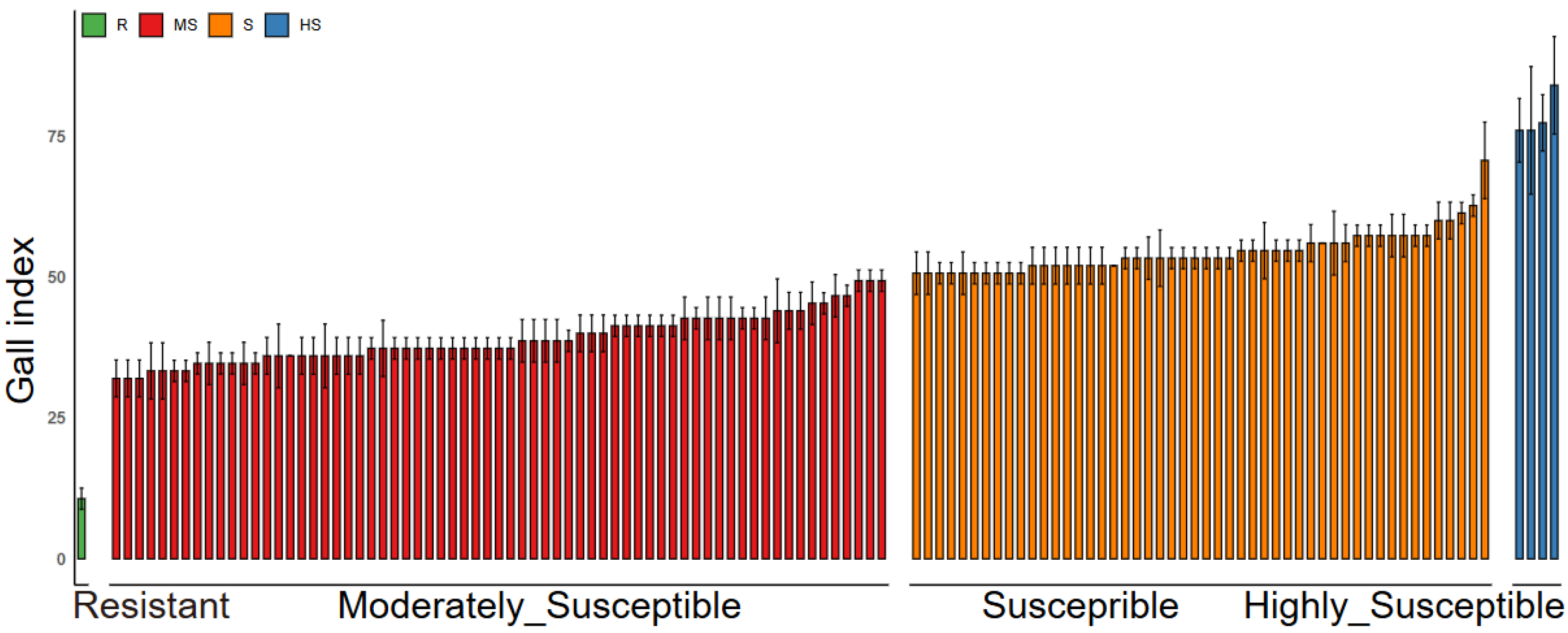
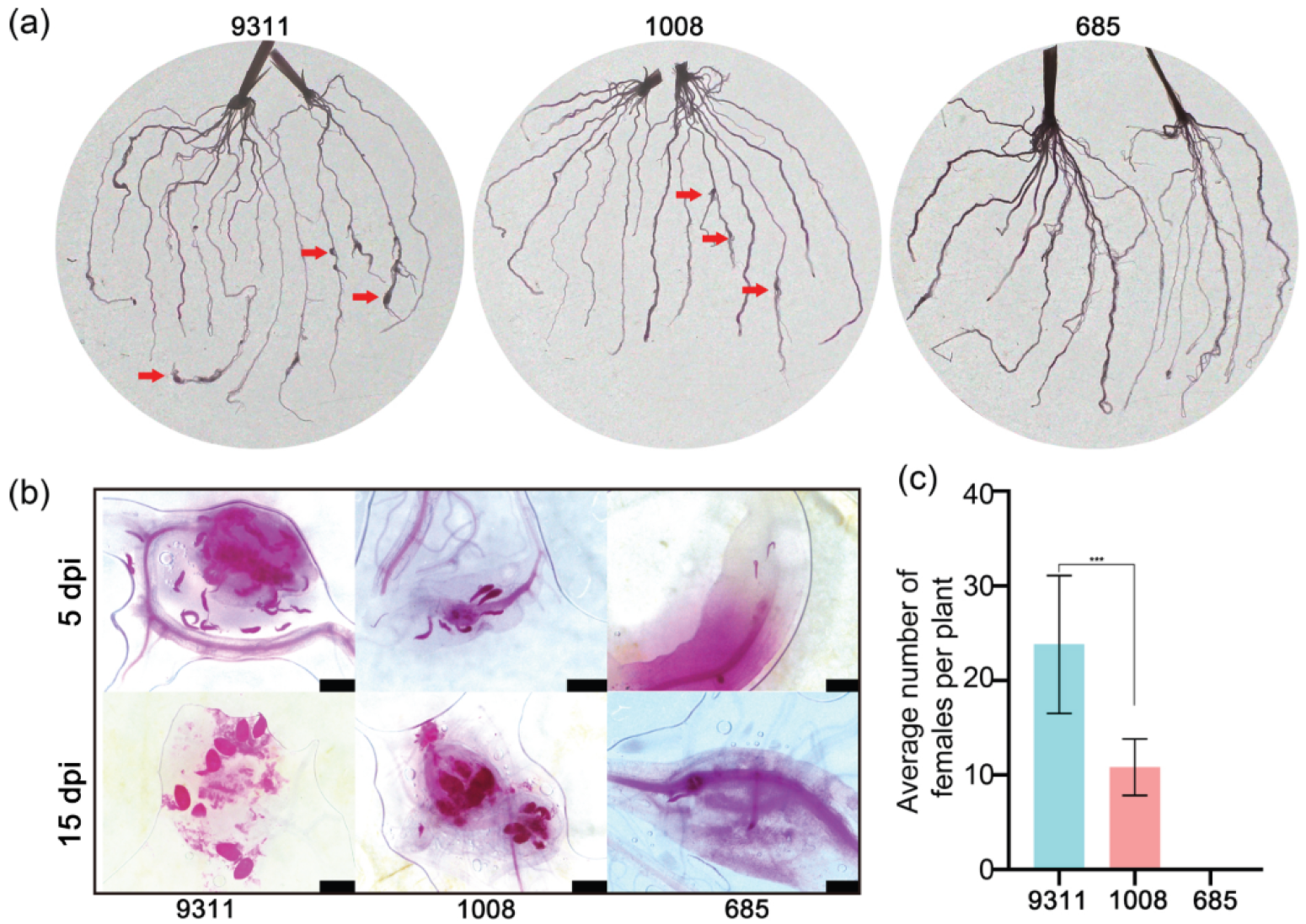
2.1. Identification of Resistance to M. graminicola in Indica Rice Cultivars
2.2. RNA-Seq Data Alignment to the Reference Genome
2.3. Comparison of the DEGs Between Resistant and Susceptible Rice Cultivars in Response to M. graminicola Infection
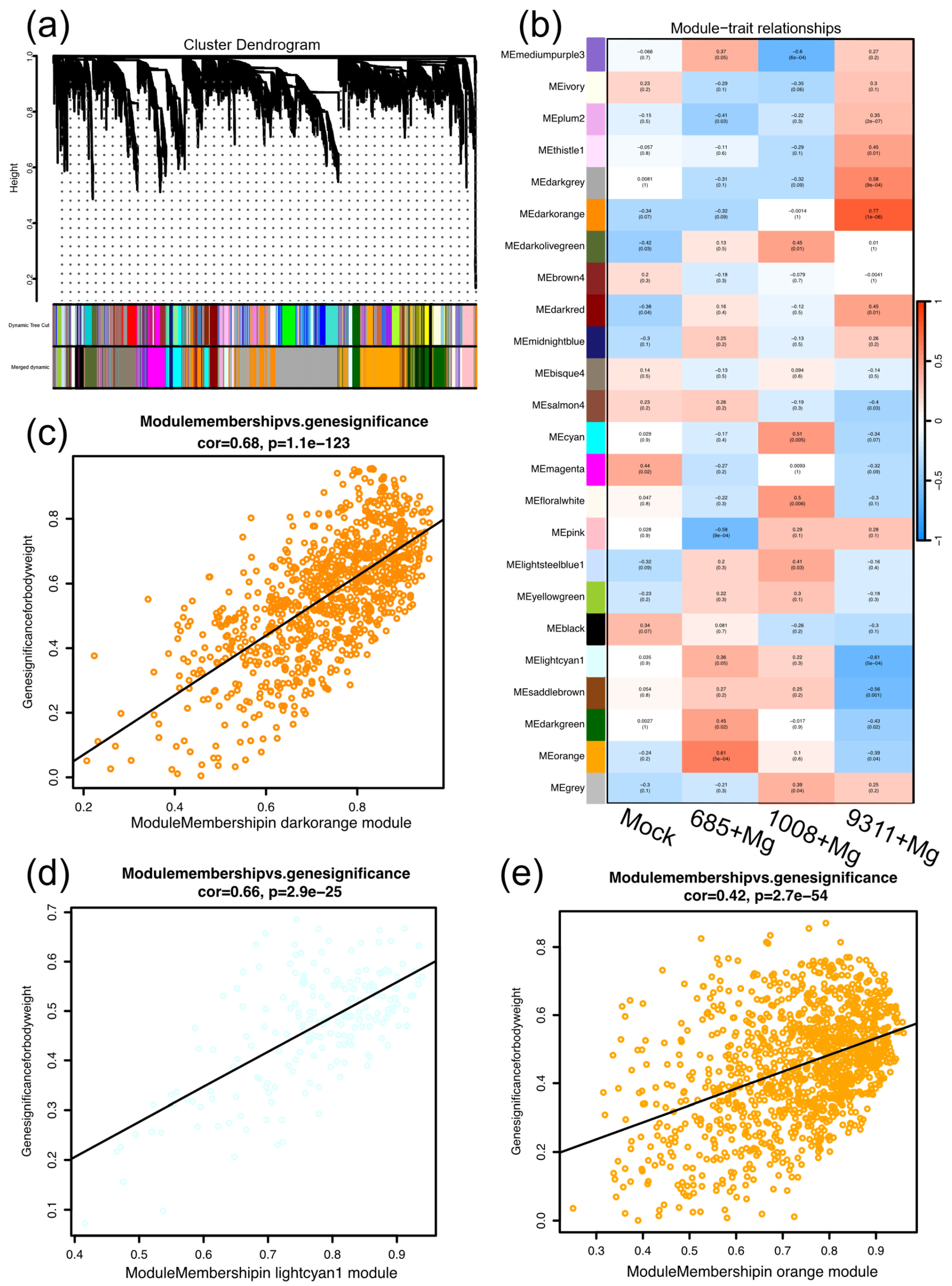
2.4. Weighted Gene Co-Correlation Network Analysis
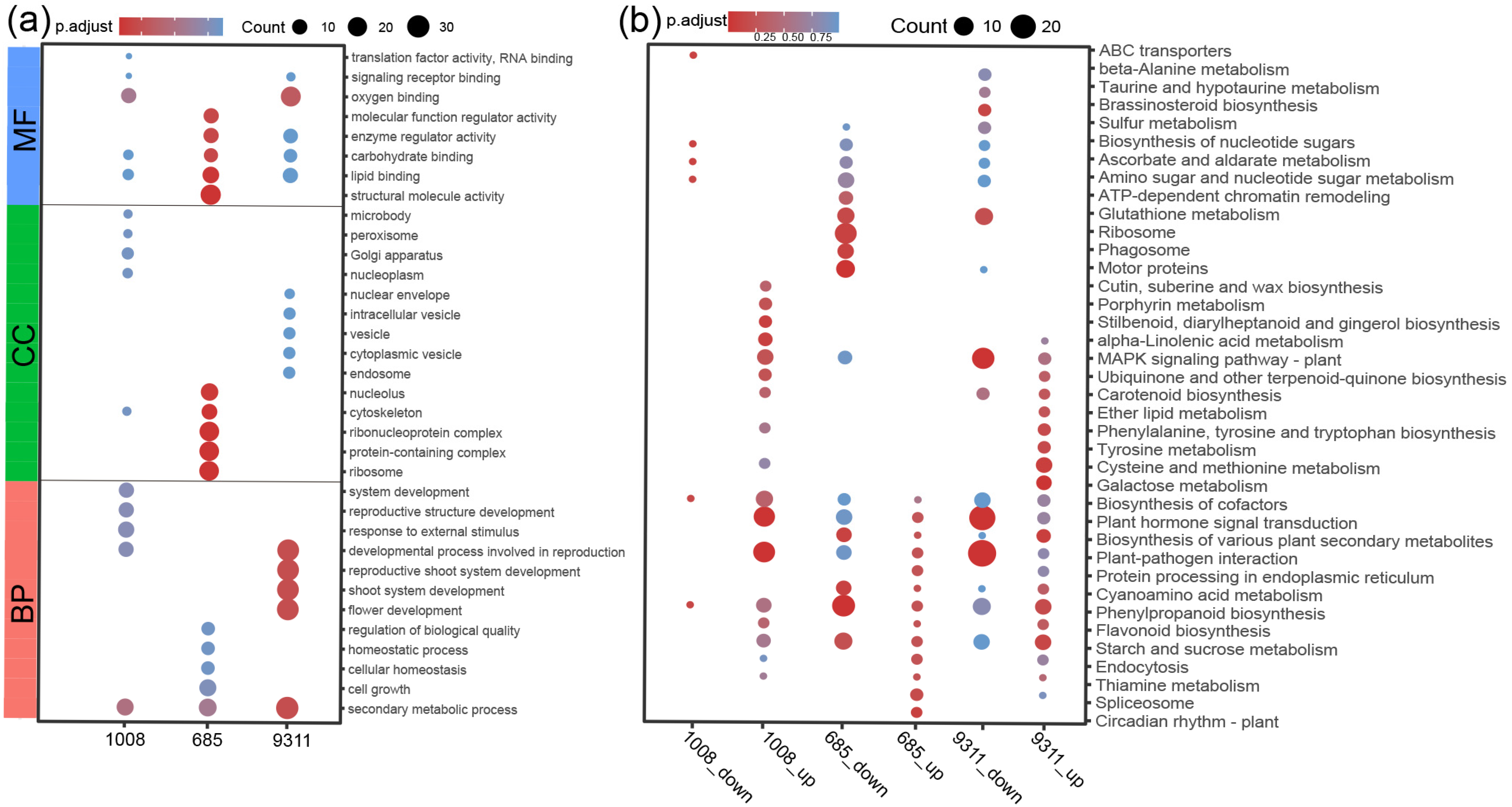
2.5. Functional Analysis of Gene Modules Associated with Susceptibility and Resistance to M. graminicola
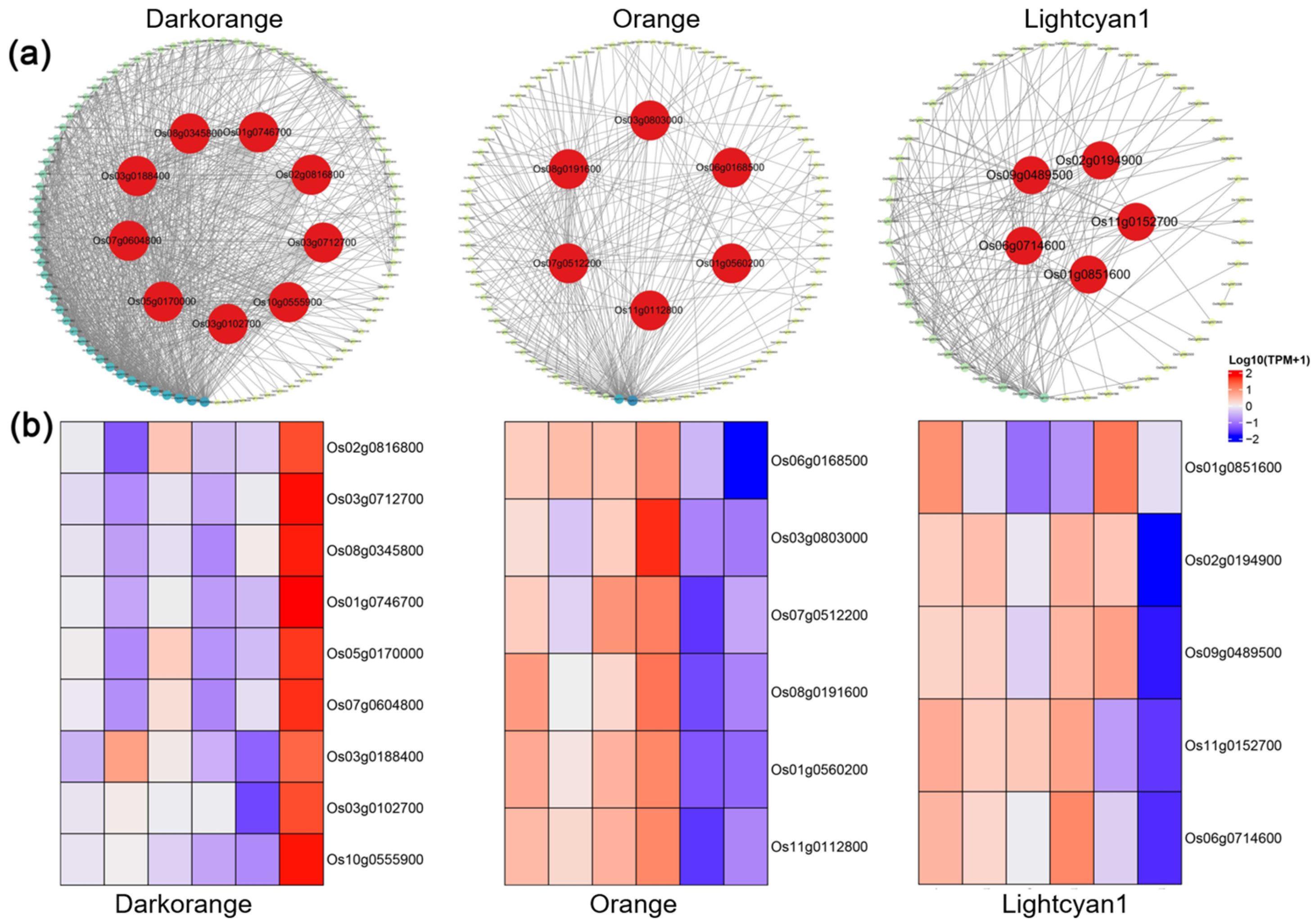
2.6. Identification of Hub Genes Correlated with Susceptibility and Resistance to M. graminicola
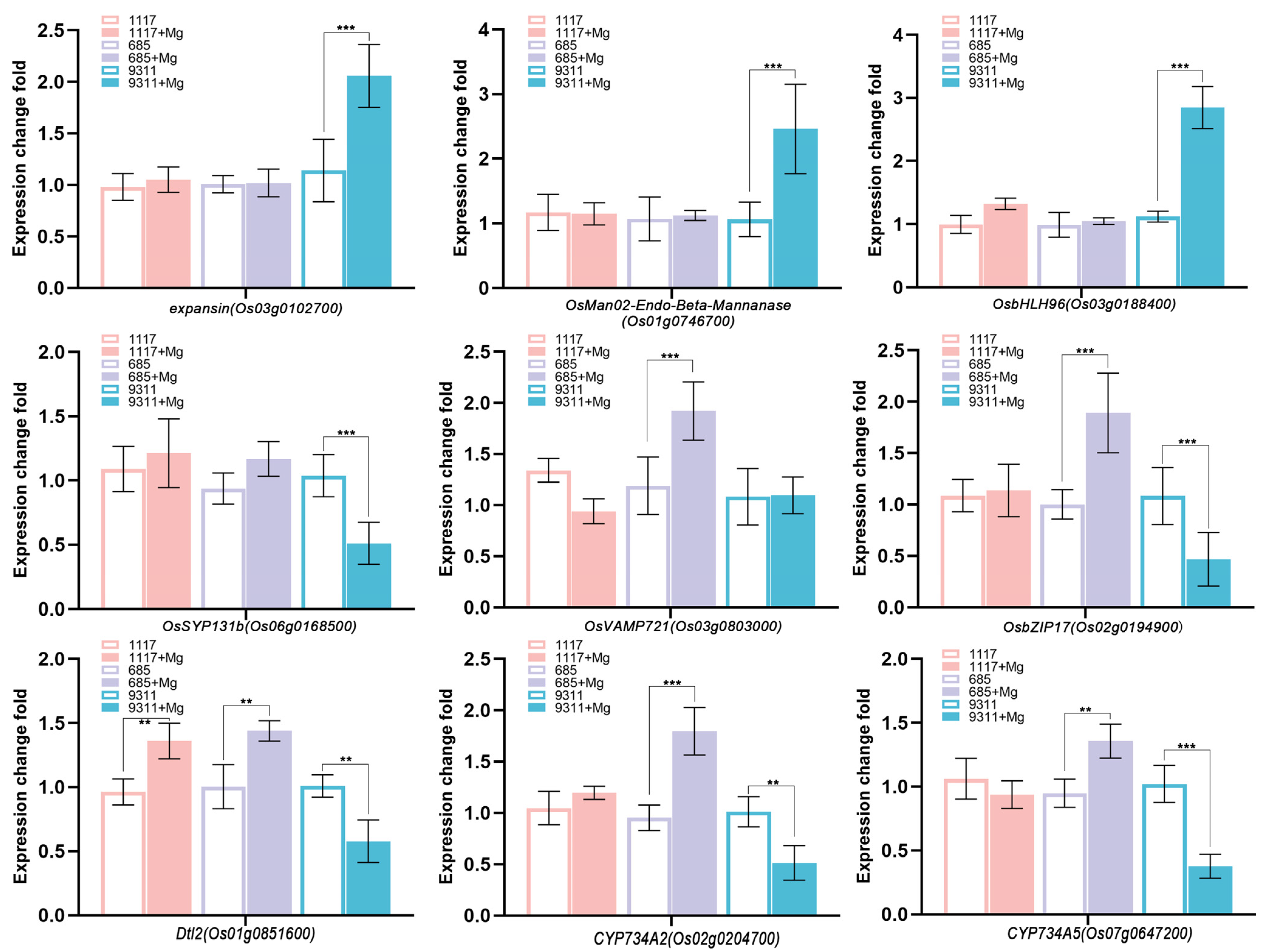
2.7. Validation of Hub Genes by RT-qPCR
3. Discussion
3.1. Mining of M. graminicola Resistance Resources and Molecular Mechanism
3.2. Sugar Metabolism Has an Important Role in Nematode–Host Interactions
3.3. Autophagic Degradation Pathway Regulates Plant-Nematode Immune Interactions
3.4. M. graminicola Regulate the Salicylic Acid (SA) and Brassinosteroid (BR) Signaling Pathway to Promote Nematode Parasitism
4. Materials and Methods
4.1. Plant Material and Nematode Inoculation
4.2. Nematode Infection Experiments
4.3. RNA-Seq and Differentially Expressed Gene Analysis
4.4. Weighted Gene Co-Expression Network Analysis
4.5. Differential Gene Expression Analysis and Gene Enrichment Analysis
4.6. Validation of Genes by qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
Abbreviations
| DEGs | Differential expressed genes |
| GCs | Giant cells |
| RNK | Root-knot nematode |
| WGCNA | Weighted gene co-expression network analysis |
| SCN | Soybean cyst nematode |
| GO | Gene ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| HR | Hypersensitive response |
References
- Fukagawa, N.K.; Ziska, L.H. Rice: Importance for global nutrition. J. Nutr. Sci. Vitaminol. 2019, 65, S2–S3. [Google Scholar] [CrossRef]
- Savary, S.; Willocquet, L.; Pethybridge, S.J.; Esker, P.; McRoberts, N.; Nelson, A. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 2019, 3, 430–439. [Google Scholar] [CrossRef]
- Abad, P.; Gouzy, J.; Aury, J.-M.; Castagnone-Sereno, P.; Danchin, E.G.J.; Deleury, E.; Perfus-Barbeoch, L.; Anthouard, V.; Artiguenave, F.; Blok, V.C.; et al. Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nat. Biotechnol. 2008, 26, 909–915. [Google Scholar] [CrossRef]
- Hassan, M.A.; Pham, T.H.; Shi, H.; Zheng, J. Nematodes threats to global food security. Acta Agric. Scand. Sect. B-Soil Plant Sci. 2013, 63, 420–425. [Google Scholar] [CrossRef]
- Arun, A.; Shanthi, A.; Raveendran, M.; Seenivasan, N.; Pushpam, R.; Shandeep, G. An insight into occurrence, biology, and pathogenesis of rice root-knot nematode Meloidogyne graminicola. Biology 2023, 12, 987. [Google Scholar] [CrossRef]
- Sacchi, S.; Torrini, G.; Marianelli, L.; Mazza, G.; Fumagalli, A.; Cavagna, B.; Ciampitti, M.; Roversi, P.F. Control of Meloidogyne graminicola a root-knot nematode using rice plants as trap crops: Preliminary results. Agriculture 2021, 11, 37. [Google Scholar] [CrossRef]
- Ravindra, H.; Sehgal, M.; Narasimhamurthy, H.B.; Jayalakshmi, K.; Imran Khan, H.S. Rice root-knot nematode (Meloidogyne graminicola) an emerging problem. Int. J. Curr. Microbiol. App. Sci. 2017, 6, 3143–3171. [Google Scholar] [CrossRef]
- Bellafiore, S.; Jougla, C.; Chapuis, É.; Besnard, G.; Suong, M.; Vu, P.N.; De Waele, D.; Gantet, P.; Thi, X.N. Intraspecific variability of the facultative meiotic parthenogenetic root-knot nematode (Meloidogyne graminicola) from rice fields in vietnam. Comptes Rendus Biol. 2015, 338, 471–483. [Google Scholar] [CrossRef]
- Davis, E. Getting to the roots of parasitism by nematodes. Trends Parasitol. 2004, 20, 134–141. [Google Scholar] [CrossRef]
- Chen, J.; Lin, B.; Huang, Q.; Hu, L.; Zhuo, K.; Liao, J. A novel Meloidogyne graminicola effector, MgGPP, is secreted into host cells and undergoes glycosylation in concert with proteolysis to suppress plant defenses and promote parasitism. PLoS Pathog. 2017, 13, e1006301. [Google Scholar] [CrossRef]
- Favery, B.; Quentin, M.; Jaubert-Possamai, S.; Abad, P. Gall-forming root-knot nematodes hijack key plant cellular functions to induce multinucleate and hypertrophied feeding cells. J. Insect Physiol. 2016, 84, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef]
- Wang, X.; Cheng, R.; Xu, D.; Huang, R.; Li, H.; Jin, L.; Wu, Y.; Tang, J.; Sun, C.; Peng, D.; et al. MG1 interacts with a protease inhibitor and confers resistance to rice root-knot nematode. Nat. Commun. 2023, 14, 3354. [Google Scholar] [CrossRef]
- Milligan, S.B.; Bodeau, J.; Yaghoobi, J.; Kaloshian, I.; Zabel, P.; Williamson, V.M. The root knot nematode resistance gene Mi from tomato is a member of the leucine zipper, nucleotide binding, leucine-rich repeat family of plant genes. Plant Cell 1998, 10, 1307–1319. [Google Scholar] [CrossRef]
- Chen, R.; Li, H.; Zhang, L.; Zhang, J.; Xiao, J.; Ye, Z. CaMi, a root-knot nematode resistance gene from hot pepper (Capsium Annuum L.) confers nematode resistance in tomato. Plant Cell Rep. 2007, 26, 895–905. [Google Scholar] [CrossRef]
- Claverie, M.; Dirlewanger, E.; Bosselut, N.; Van Ghelder, C.; Voisin, R.; Kleinhentz, M.; Lafargue, B.; Abad, P.; Rosso, M.-N.; Chalhoub, B.; et al. The Ma gene for complete-spectrum resistance to Meloidogyne species in prunus is a TNL with a huge repeated C-terminal post-LRR region. Plant Physiol. 2011, 156, 779–792. [Google Scholar] [CrossRef]
- Huang, Z.; Jiang, M.; Sun, L.; Zhang, J.; Jiang, C.; Lu, A.; Li, D.; Nan, N.; Sun, W. Exploiting susceptibility genes in rice: From molecular mechanism to application. Phytopathol. Res. 2024, 6, 67. [Google Scholar] [CrossRef]
- Büschges, R.; Hollricher, K.; Panstruga, R.; Simons, G.; Wolter, M.; Frijters, A.; Van Daelen, R.; Van Der Lee, T.; Diergaarde, P.; Groenendijk, J.; et al. The barley Mlo gene: A novel control element of plant pathogen resistance. Cell 1997, 88, 695–705. [Google Scholar] [CrossRef]
- Radakovic, Z.S.; Anjam, M.S.; Escobar, E.; Chopra, D.; Cabrera, J.; Silva, A.C.; Escobar, C.; Sobczak, M.; Grundler, F.M.W.; Siddique, S. Arabidopsis HIPP27 is a host susceptibility gene for the beet cyst nematode Heterodera schachtii. Mol. Plant Pathol. 2018, 19, 1917–1928. [Google Scholar] [CrossRef]
- Andersen, E.J.; Ali, S.; Byamukama, E.; Yen, Y.; Nepal, M.P. Disease resistance mechanisms in plants. Genes 2018, 9, 339. [Google Scholar] [CrossRef]
- Xing, X.; Li, X.; Zhang, M.; Wang, Y.; Liu, B.; Xi, Q.; Zhao, K.; Wu, Y.; Yang, T. Transcriptome analysis of resistant and susceptible tobacco (Nicotiana tabacum) in response to root-knot nematode Meloidogyne incognita infection. Biochem. Biophys. Res. Commun. 2017, 482, 1114–1121. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Li, H.; Wang, Q.; Zhao, P.; Zhu, G.; Fang, W.; Chen, C.; Wang, X.; Wang, L. Comparative transcriptome analysis of genes involved in the response of resistant and susceptible peach cultivars to nematode infection. Sci. Hortic. 2017, 215, 20–27. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Sun, B.; Zhou, X.; Chen, C.; Chen, C.; Chen, K.; Chen, M.; Liu, S.; Chen, G.; Cao, B.; Cao, F.; et al. Coexpression network analysis reveals an MYB transcriptional activator involved in capsaicinoid biosynthesis in hot peppers. Hortic. Res. 2020, 7, 162. [Google Scholar] [CrossRef]
- Jiang, H.; Zhou, C.; Ma, J.; Qu, S.; Liu, F.; Sun, H.; Zhao, X.; Han, Y. Weighted gene co-expression network analysis identifies genes related to HG Type 0 resistance and verification of hub gene GmHg1. Front. Plant Sci. 2022, 13, 1118503. [Google Scholar] [CrossRef]
- Jones, J.T.; Haegeman, A.; Danchin, E.G.J.; Gaur, H.S.; Helder, J.; Jones, M.G.K.; Kikuchi, T.; Manzanilla-López, R.; Palomares-Rius, J.E.; Wesemael, W.M.L.; et al. Top 10 plant-parasitic nematodes in molecular plant pathology. Mol. Plant Pathol. 2013, 14, 946–961. [Google Scholar] [CrossRef]
- Caromel, B.; Gebhardt, C. Breeding for nematode resistance: Use of genomic information. In Genomics and Molecular Genetics of Plant-Nematode Interactions; Jones, J., Gheysen, G., Fenoll, C., Eds.; Springer: Dordrecht, The Netherlands, 2011; pp. 465–492. [Google Scholar] [CrossRef]
- Zhan, L.; Ding, Z.; Peng, D.; Peng, H.; Kong, L.; Liu, S.; Liu, Y.; Li, Z.; Huag, W. Evaluation of Chinese rice varieties resistant to the root-knot nematode Meloidogyne graminicola. J. Integr. Agric. 2018, 17, 621–630. [Google Scholar] [CrossRef]
- Soriano, I.R.; Schmit, V.; Brar, D.S.; Prot, J.-C.; Reversat, G. Resistance to rice root-knot nematode Meloidogyne graminicola identified in Oryza longistaminata and O. Glaberrima. Nematology 1999, 1, 395–398. [Google Scholar] [CrossRef]
- Hofmann, J.; Wieczorek, K.; Blochl, A.; Grundler, F.M.W. Sucrose supply to nematode-induced syncytia depends on the apoplasmic and symplasmic pathways. J. Exp. Bot. 2007, 58, 1591–1601. [Google Scholar] [CrossRef]
- Hofmann, J.; Szakasits, D.; Blöchl, A.; Sobczak, M.; Daxböck-Horvath, S.; Golinowski, W.; Bohlmann, H.; Grundler, F.M.W. Starch serves as carbohydrate storage in nematode-induced syncytia. Plant Physiol. 2008, 146, 228–235. [Google Scholar] [CrossRef]
- Baldacci-Cresp, F.; Chang, C.; Maucourt, M.; Deborde, C.; Hopkins, J.; Lecomte, P.; Bernillon, S.; Brouquisse, R.; Moing, A.; Abad, P.; et al. (Homo)Glutathione deficiency impairs root-knot nematode development in Medicago truncatula. PLoS Pathog. 2012, 8, e1002471. [Google Scholar] [CrossRef] [PubMed]
- Kyndt, T.; Vieira, P.; Gheysen, G.; De Almeida-Engler, J. Nematode feeding sites: Unique organs in plant roots. Planta 2013, 238, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Koch, K. Sucrose metabolism: Regulatory mechanisms and pivotal roles in sugar sensing and plant development. Curr. Opin. Plant Biol. 2004, 7, 235–246. [Google Scholar] [CrossRef]
- Wu, Y.; Fang, W.; Peng, W.; Jiang, M.; Chen, G.; Xiong, F. Sucrose transporter in rice. Plant Signal Behav. 2021, 16, 1952373. [Google Scholar] [CrossRef]
- Zeng, X.; Luo, Y.; Vu, N.T.Q.; Shen, S.; Xia, K.; Zhang, M. CRISPR/Cas9-mediated mutation of OsSWEET14 in rice cv. Zhonghua11 confers resistance to Xanthomonas oryzae Pv. Oryzae without yield penalty. BMC Plant Biol. 2020, 20, 313. [Google Scholar] [CrossRef]
- Kim, P.; Xue, C.Y.; Song, H.D.; Gao, Y.; Feng, L.; Li, Y.; Xuan, Y.H. Tissue-specific activation of promotes rice resistance to sheath blight disease and increases grain weight. Plant Biotechnol. J. 2021, 19, 409–411. [Google Scholar] [CrossRef]
- Streubel, J.; Pesce, C.; Hutin, M.; Koebnik, R.; Boch, J.; Szurek, B. Five phylogenetically close rice SWEET genes confer TAL. New Phytol. 2013, 200, 808–819. [Google Scholar] [CrossRef]
- Zhou, J.; Peng, Z.; Long, J.; Sosso, D.; Liu, B.; Eom, J.; Huang, S.; Liu, S.; Vera Cruz, C.; Frommer, W.B.; et al. Gene targeting by the effector PthXo2 reveals cryptic resistance gene for bacterial blight of rice. Plant J. 2015, 82, 632–643. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, D.; Duan, Y.; Chen, L.; Fan, H.; Wang, Y.; Liu, X.; Chen, L.-Q.; Xuan, Y.; Zhu, X. AtSWEET1 negatively regulates plant susceptibility to root-knot nematode disease. Front. Plant Sci. 2023, 14, 1010348. [Google Scholar] [CrossRef]
- Xiao, L.; Gheysen, G.; Yang, M.; Xiao, X.; Xu, L.; Guo, X.; Yang, L.; Liu, W.; He, Y.; Peng, D.; et al. Brown planthopper infestation on rice reduces plant susceptibility to Meloidogyne graminicola by reducing root sugar allocation. New Phytol. 2024, 242, 262–277. [Google Scholar] [CrossRef]
- Matuszkiewicz, M.; Sobczak, M.; Cabrera, J.; Escobar, C.; Karpiński, S.; Filipecki, M. The role of programmed cell death regulator LSD1 in nematode-induced syncytium formation. Front. Plant Sci. 2018, 9, 314. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, S.; Xu, C.; Yang, H.; Achom, M.; Wang, X. A key virulence effector from cyst nematodes targets host autophagy to promote nematode parasitism. New Phytol. 2023, 237, 1374–1390. [Google Scholar] [CrossRef]
- Zou, J.; Chen, X.; Liu, C.; Guo, M.; Kanwar, M.K.; Qi, Z.; Yang, P.; Wang, G.; Bao, Y.; Bassham, D.C.; et al. Autophagy promotes jasmonate-mediated defense against nematodes. Nat. Commun. 2023, 14, 4769. [Google Scholar] [CrossRef]
- Kwon, C.; Lee, J.-H.; Yun, H.S. SNAREs in Plant biotic and abiotic stress responses. Mol. Cells 2020, 43, 501–508. [Google Scholar] [CrossRef]
- Liu, Y.-N.; Liu, C.-C.; Guo, R.; Tian, L.; Cheng, J.-F.; Wu, Y.-N.; Wang, D.; Wang, B. The rice Qa-SNAREs in SYP13 Subfamily are involved in regulating arbuscular mycorrhizal symbiosis and seed rertility. Front. Plant Sci. 2022, 13, 898286. [Google Scholar] [CrossRef]
- Kyndt, T.; Denil, S.; Haegeman, A.; Trooskens, G.; Bauters, L.; Van Criekinge, W.; De Meyer, T.; Gheysen, G. Transcriptional reprogramming by root knot and migratory nematode infection in rice. New Phytol. 2012, 196, 887–900. [Google Scholar] [CrossRef]
- Lu, C.; Liu, X.; Tang, Y.; Fu, Y.; Zhang, J.; Yang, L.; Li, P.; Zhu, Z.; Dong, P. A Comprehensive review of TGA transcription factors in plant growth, stress responses, and beyond. Int. J. Biol. Macromol. 2024, 258, 128880. [Google Scholar] [CrossRef]
- Ekengren, S.K.; Liu, Y.; Schiff, M.; Dinesh-Kumar, S.P.; Martin, G.B. Two MAPK cascades, NPR1, and TGA transcription f actors play a role in Pto-mediated disease resistance in tomato. Plant J. 2003, 36, 905–917. [Google Scholar] [CrossRef]
- Pascual, S.; Emiliozzi, M.; Nombela, G. Role of Two Transcription factors (TGA 1a and TGA 2.1) in the Mi-1-mediated resistance of tomato to the root-knot nematode Meloidogyne javanica. Horticulturae 2024, 10, 134. [Google Scholar] [CrossRef]
- Kim, Y.-W.; Youn, J.-H.; Roh, J.; Kim, J.-M.; Kim, S.-K.; Kim, T.-W. Brassinosteroids enhance salicylic acid-mediated immune responses by inhibiting BIN2 phosphorylation of clade I TGA transcription factors in Arabidopsis. Mol. Plant. 2022, 15, 991–1007. [Google Scholar] [CrossRef]
- Sakamoto, T.; Kawabe, A.; Tokida-Segawa, A.; Shimizu, B.-I.; Takatsuto, S.; Shimada, Y.; Fujioka, S.; Mizutani, M. Rice CYP734As function as multisubstrate and multifunctional enzymes in brassinosteroid catabolism. Plant J. 2011, 67, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yimer, H.Z.; Nahar, K.; Kyndt, T.; Haeck, A.; Van Meulebroek, L.; Vanhaecke, L.; Demeestere, K.; Höfte, M.; Gheysen, G. Gibberellin antagonizes jasmonate-induced defense against Meloidogyne graminicola in rice. New Phytol. 2018, 218, 646–660. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Xiao, Z.; Shen, K.; Lai, W.; Du, S.; Zhou, L.; Chen, J. Transcriptome and Co-Expression Network Analyses of Resistant and Susceptible Rice Cultivars in Response to Meloidogyne graminicola. Int. J. Mol. Sci. 2025, 26, 5315. https://doi.org/10.3390/ijms26115315
Zhang S, Xiao Z, Shen K, Lai W, Du S, Zhou L, Chen J. Transcriptome and Co-Expression Network Analyses of Resistant and Susceptible Rice Cultivars in Response to Meloidogyne graminicola. International Journal of Molecular Sciences. 2025; 26(11):5315. https://doi.org/10.3390/ijms26115315
Chicago/Turabian StyleZhang, Shirui, Zitong Xiao, Kexiang Shen, Wentao Lai, Shunbiao Du, Lingyan Zhou, and Jiansong Chen. 2025. "Transcriptome and Co-Expression Network Analyses of Resistant and Susceptible Rice Cultivars in Response to Meloidogyne graminicola" International Journal of Molecular Sciences 26, no. 11: 5315. https://doi.org/10.3390/ijms26115315
APA StyleZhang, S., Xiao, Z., Shen, K., Lai, W., Du, S., Zhou, L., & Chen, J. (2025). Transcriptome and Co-Expression Network Analyses of Resistant and Susceptible Rice Cultivars in Response to Meloidogyne graminicola. International Journal of Molecular Sciences, 26(11), 5315. https://doi.org/10.3390/ijms26115315