
A Report of a Child with SEC31A-Related Neurodevelopmental Disorder

 , and
, and
Abstract

1. Introduction
2. Results
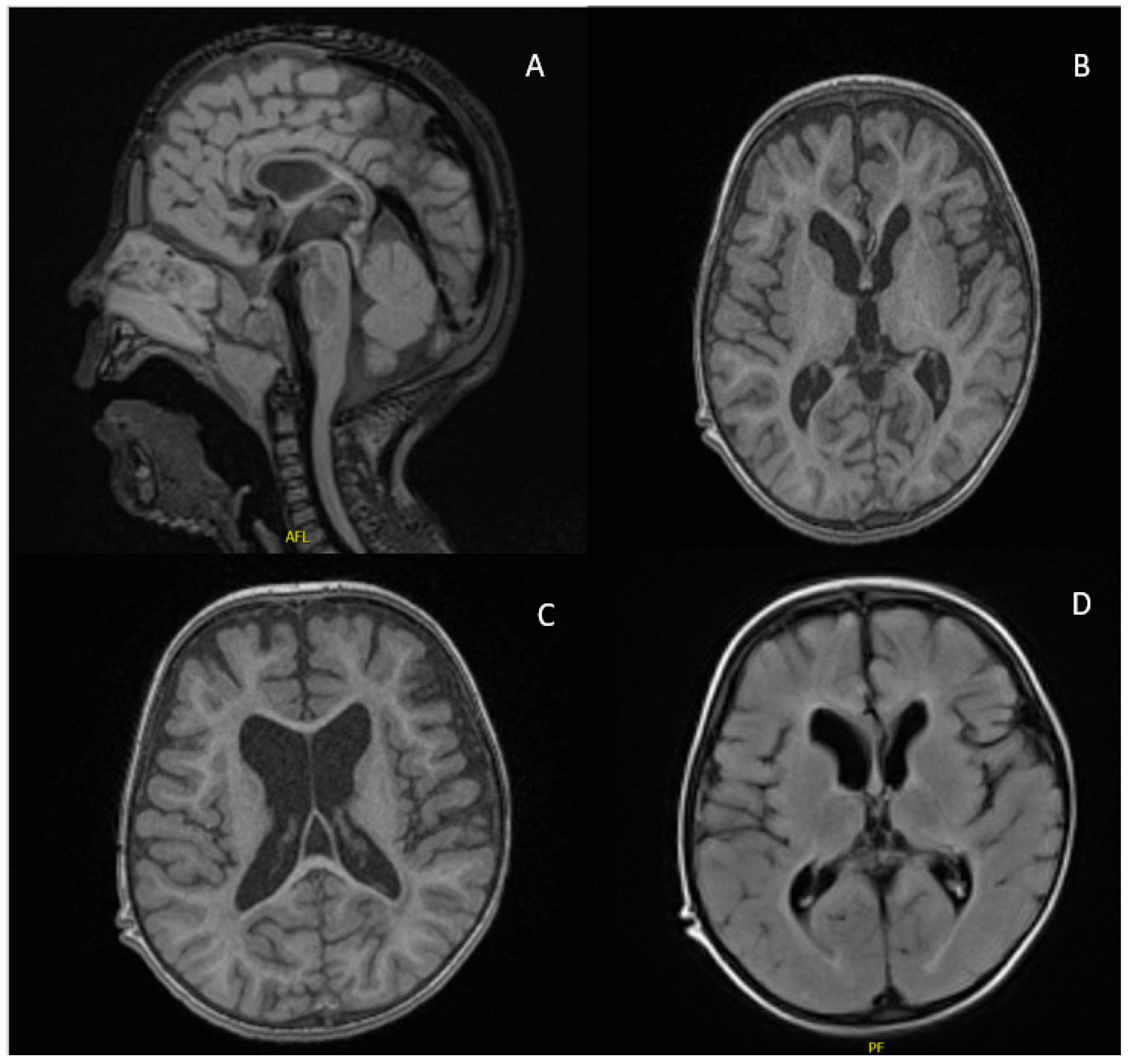
2.1. Case Report
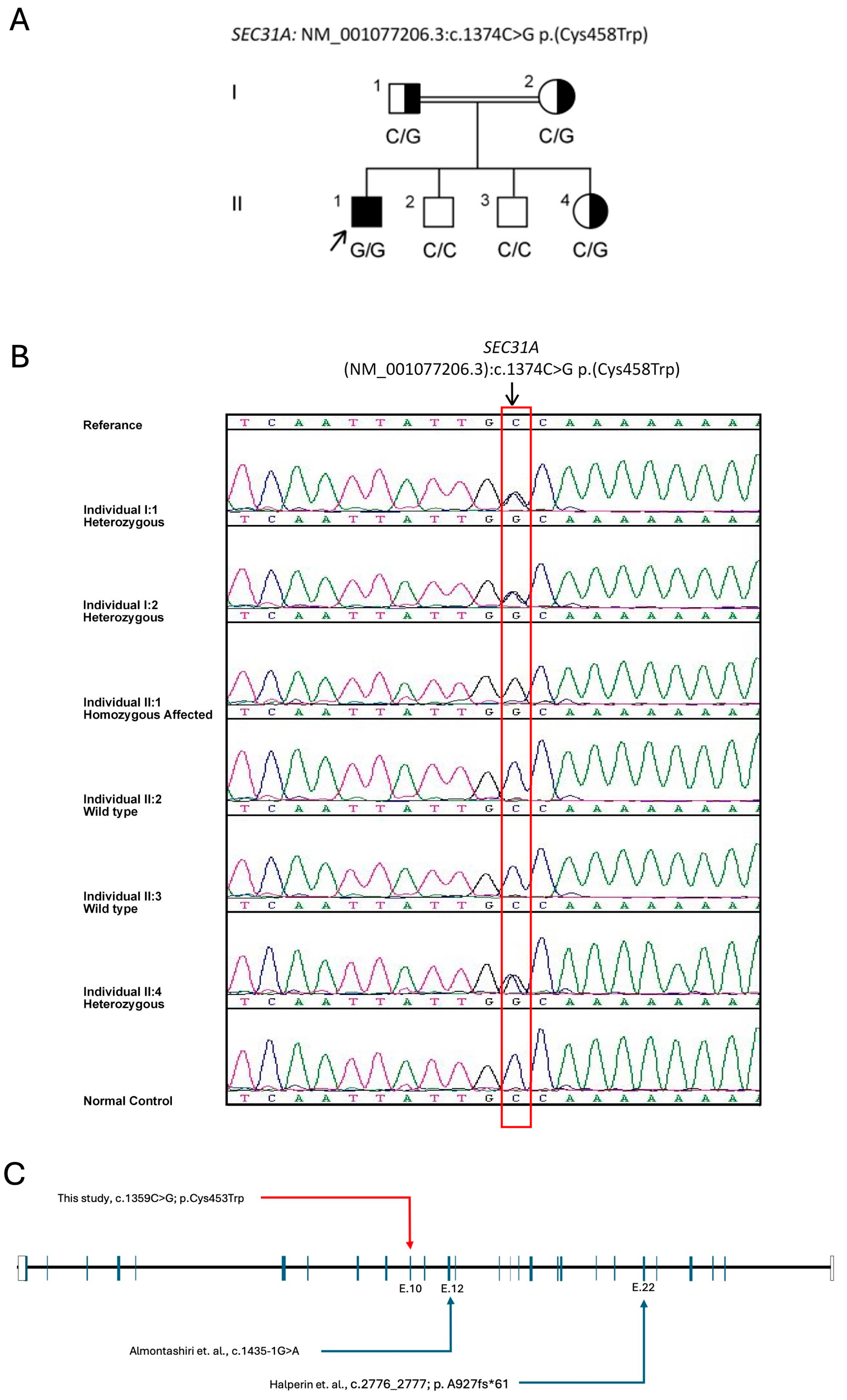
2.2. The Variant and in Silico Pathogenesis Analysis
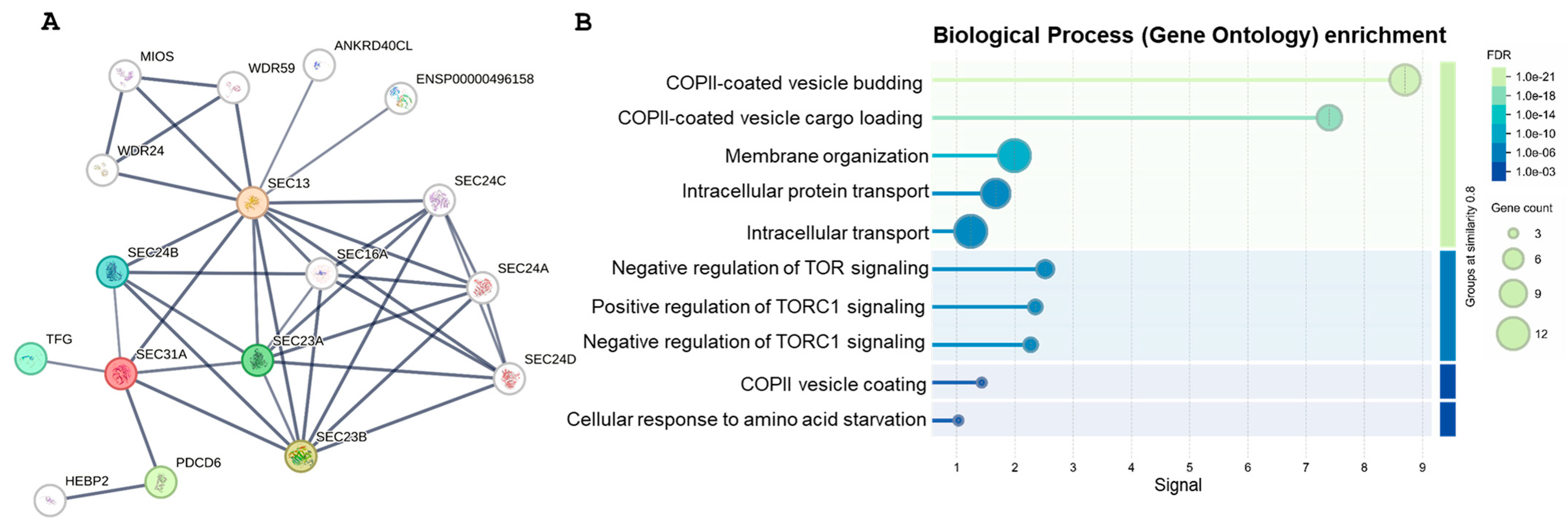
2.3. Gene Network Analysis (GNA)
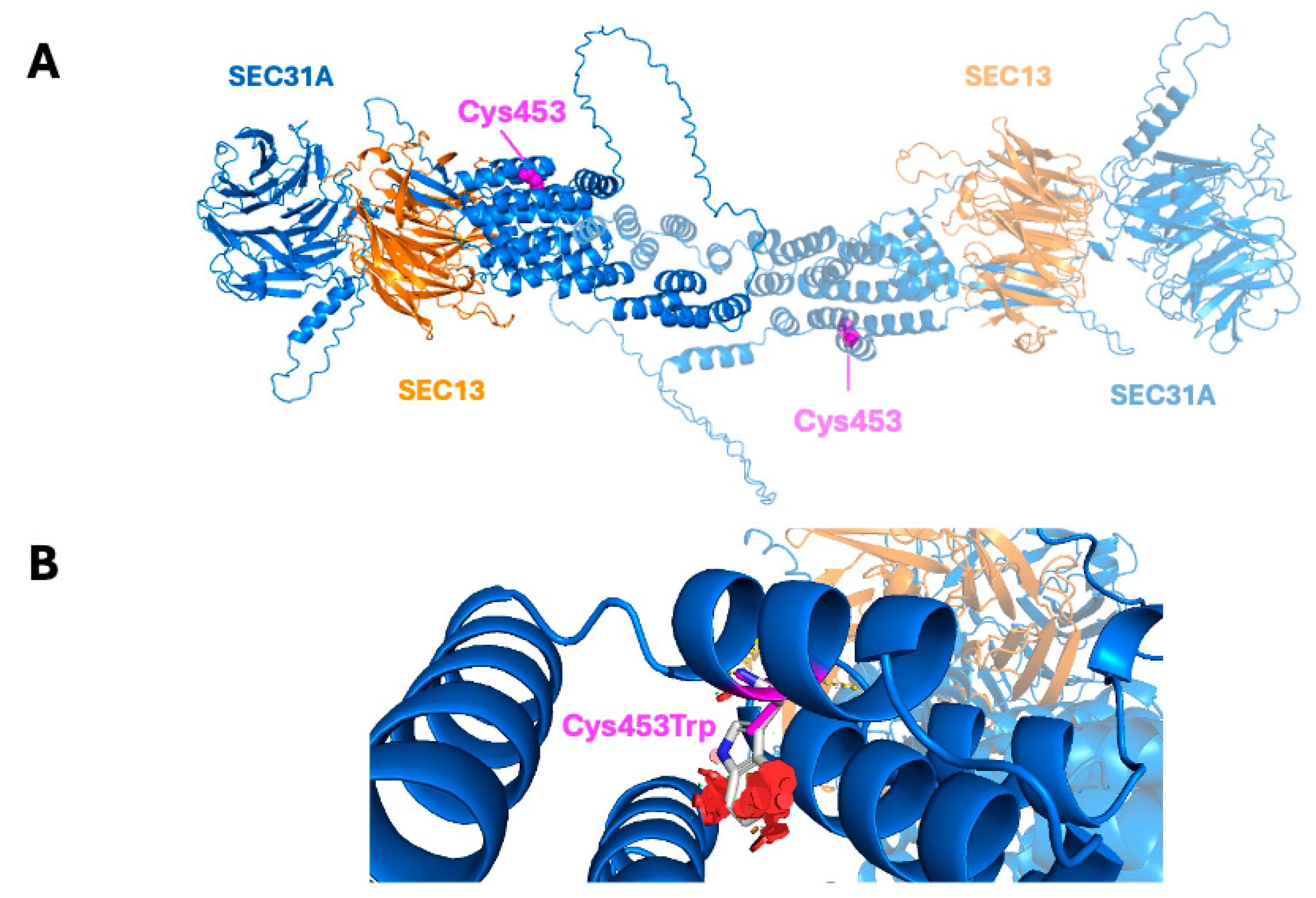
2.4. Structural Modeling of the Variant
2.5. ACMG Classification of c.1359C > G; p.Cys453Trp
3. Discussion
4. Materials and Methods
4.1. DNA Isolation, Polymerase Chain Reaction (PCR)
4.2. Whole Exome Sequencing (WES)
4.3. Whole Genome Sequencing (WGS)
4.4. Confirmatory Sanger Sequencing
4.5. Computational Structural Analysis of Mutants
4.6. Gene Network Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tang, B.L.; Zhang, T.; Low, D.Y.; Wong, E.T.; Horstmann, H.; Hong, W. Mammalian Homologues of Yeast Sec31p. J. Biol. Chem. 2000, 275, 13597–13604. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-L.; Kim, J. Consequences of mutations in the genes of the ER export machinery COPII in vertebrates. Cell Stress Chaperon- 2020, 25, 199–209. [Google Scholar] [CrossRef]
- Peotter, J.; Kasberg, W.; Pustova, I.; Audhya, A. COPII-mediated trafficking at the ER/ERGIC interface. Traffic 2019, 20, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Halperin, D.; Kadir, R.; Perez, Y.; Drabkin, M.; Yogev, Y.; Wormser, O.; Berman, E.M.; Eremenko, E.; Rotblat, B.; Shorer, Z.; et al. SEC31A mutation affects ER homeostasis, causing a neurological syndrome. J. Med Genet. 2019, 56, 139–148. [Google Scholar] [CrossRef]
- Russo, R.; Esposito, M.R.; Iolascon, A. Inherited hematological disorders due to defects in coat protein (COP)II complex. Am. J. Hematol. 2012, 88, 135–140. [Google Scholar] [CrossRef]
- Almontashiri, N.A.M.; Mushiba, A.; Alruqi, H.; Alharby, E.; Hashmi, J.A. Biallelic Loss of Function Variant in SEC31A Is Associated With Lethal Neurodevelopmental Disorder, Dysmorphic Features, and Skeletal Defects. Clin. Genet. 2024, 107, 477–479. [Google Scholar] [CrossRef]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef]
- Alirezaie, N.; Kernohan, K.D.; Hartley, T.; Majewski, J.; Hocking, T.D. ClinPred: Prediction Tool to Identify Disease-Relevant Nonsynonymous Single-Nucleotide Variants. Am. J. Hum. Genet. 2018, 103, 474–483. [Google Scholar] [CrossRef]
- Li, C.; Zhi, D.; Wang, K.; Liu, X. MetaRNN: Differentiating rare pathogenic and rare benign missense SNVs and InDels using deep learning. Genome Med. 2022, 14, 115. [Google Scholar] [CrossRef] [PubMed]
- Minton, K. Predicting variant pathogenicity with AlphaMissense. Nat. Rev. Genet. 2023, 24, 804. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Pesaran, T.; Chamberlin, A.; Fenwick, R.B.; Li, S.; Gau, C.-L.; Chao, E.C.; Lu, H.-M.; Black, M.H.; Qian, D. REVEL and BayesDel outperform other in silico meta-predictors for clinical variant classification. Sci. Rep. 2019, 9, 12752. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2014, 31, 761–763. [Google Scholar] [CrossRef]
- Raimondi, D.; Tanyalcin, I.; Ferté, J.; Gazzo, A.; Orlando, G.; Lenaerts, T.; Rooman, M.; Vranken, W. DEOGEN2: Prediction and interactive visualization of single amino acid variant deleteriousness in human proteins. Nucleic Acids Res. 2017, 45, W201–W206. [Google Scholar] [CrossRef]
- Lin, W.; Wells, J.; Wang, Z.; Orengo, C.; Martin, A.C.R. Enhancing missense variant pathogenicity prediction with protein language models using VariPred. Sci. Rep. 2024, 14, 8136. [Google Scholar] [CrossRef]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Day, I.N.M.; Gaunt, T.R. Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 2013, 29, 1504–1510. [Google Scholar] [CrossRef]
- Rogers, M.F.; A Shihab, H.; Mort, M.; Cooper, D.N.; Gaunt, T.R.; Campbell, C. FATHMM-XF: Accurate prediction of pathogenic point mutations via extended features. Bioinformatics 2017, 34, 511–513. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2015, 11, 1–9. [Google Scholar] [CrossRef]
- Fath, S.; Mancias, J.D.; Bi, X.; Goldberg, J. Structure and Organization of Coat Proteins in the COPII Cage. Cell 2007, 129, 1325–1336. [Google Scholar] [CrossRef]
- Anglès, F.; Gupta, V.; Wang, C.; Balch, W.E. COPII cage assembly factor Sec13 integrates information flow regulating endomembrane function in response to human variation. Sci. Rep. 2024, 14, 10160. [Google Scholar] [CrossRef]
- Wang, X.; Huang, R.; Wang, Y.; Zhou, W.; Hu, Y.; Yao, Y.; Cheng, K.; Li, X.; Xu, B.; Zhang, J.; et al. Manganese regulation of COPII condensation controls circulating lipid homeostasis. Nat. Cell Biol. 2023, 25, 1650–1663. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.; Jones, E.L.; Bonney, S.A.; Patel, H.N.; Mensenkamp, A.R.; Eichenbaum-Voline, S.; Rudling, M.; Myrdal, U.; Annesi, G.; Naik, S.; et al. Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat. Genet. 2003, 34, 29–31. [Google Scholar] [CrossRef] [PubMed]
- A Boyadjiev, S.; Fromme, J.C.; Ben, J.; Chong, S.S.; Nauta, C.; Hur, D.J.; Zhang, G.; Hamamoto, S.; Schekman, R.; Ravazzola, M.; et al. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat. Genet. 2006, 38, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Takeyari, S.; Kubota, T.; Miyata, K.; Yamamoto, K.; Nakayama, H.; Ohata, Y.; Kitaoka, T.; Yanagi, K.; Kaname, T.; Ozono, K. Japanese patient with Cole-carpenter syndrome with compound heterozygous variants of SEC24D. Am. J. Med Genet. Part A 2018, 176, 2882–2886. [Google Scholar] [CrossRef]
- Bögershausen, N.; Cavdarli, B.; Nagai, T.H.; Milev, M.P.; Wolff, A.; Mehranfar, M.; Schmidt, J.; Choudhary, D.; Gutiérrez-Gutiérrez, Ó.; Cyganek, L.; et al. SEC24C deficiency causes trafficking and glycosylation abnormalities in an epileptic encephalopathy with cataracts and dyserythropoeisis. J. Clin. Investig. 2025, 10, e173484. [Google Scholar] [CrossRef]
- Rosnoblet, C.; Peanne, R.; Legrand, D.; Foulquier, F. Glycosylation disorders of membrane trafficking. Glycoconj. J. 2012, 30, 23–31. [Google Scholar] [CrossRef]
- Bisnett, B.J.; Condon, B.M.; Lamb, C.H.; Georgiou, G.R.; Boyce, M. Export Control: Post-transcriptional Regulation of the COPII Trafficking Pathway. Front. Cell Dev. Biol. 2021, 8, 618652. [Google Scholar] [CrossRef]
- Helm, J.R.; Bentley, M.; Thorsen, K.D.; Wang, T.; Foltz, L.; Oorschot, V.; Klumperman, J.; Hay, J.C. Apoptosis-linked Gene-2 (ALG-2)/Sec31 Interactions Regulate Endoplasmic Reticulum (ER)-to-Golgi Transport. J. Biol. Chem. 2014, 289, 23609–23628. [Google Scholar] [CrossRef]
- AlBakheet, A.; AlQudairy, H.; Alkhalifah, J.; Almoaily, S.; Kaya, N.; Rahbeeni, Z. Detailed genetic and clinical analysis of a novel de novo variant in HPRT1: Case report of a female patient from Saudi Arabia with Lesch–Nyhan syndrome. Front. Genet. 2023, 13, 1044936. [Google Scholar] [CrossRef]
- AlHargan, A.; AlMuhaizea, M.A.; Almass, R.; Alwadei, A.H.; Daghestani, M.; Arold, S.T.; Kaya, N. SHQ1-associated neurodevelopmental disorder: Report of the first homozygous variant in unrelated patients and review of the literature. Hum. Genome Var. 2023, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Medico-Salsench, E.; Nikoncuk, A.; Ramakrishnan, R.; Lanko, K.; Kühn, N.A.; van der Linde, H.C.; Lor-Zade, S.; Albuainain, F.; Shi, Y.; et al. AMFR dysfunction causes autosomal recessive spastic paraplegia in human that is amenable to statin treatment in a preclinical model. Acta Neuropathol. 2023, 146, 353–368. [Google Scholar] [CrossRef]
- Al-Hassnan, Z.; AlDosary, M.; AlHargan, A.; AlQudairy, H.; Almass, R.; Alahmadi, K.O.; AlShahrani, S.; AlBakheet, A.; Almuhaizea, M.A.; Taylor, R.W.; et al. A novel missense mutation in ISCA2 causes aberrant splicing and leads to multiple mitochondrial dysfunctions syndrome 4. Front. Psychiatry 2024, 15, 1428175. [Google Scholar] [CrossRef] [PubMed]
- Altassan, R.; AlQudairy, H.; AlJebreen, S.; AlMuhaizea, M.; Al-Hindi, H.; Pena-Guerra, K.A.; Ghebeh, H.; Almzroua, A.; Albakheet, A.; AlDosary, M.; et al. Expanding the phenotypic and genotypic spectrum of GGPS1 related congenital muscular dystrophy. Am. J. Med. Genet. Part A 2023, 194, e63498. [Google Scholar] [CrossRef]
- Saudi Mendeliome Group Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases. Genome Biol. 2015, 16, 134. [CrossRef]
- Alazami, A.M.; Patel, N.; Shamseldin, H.E.; Anazi, S.; Al-Dosari, M.S.; Alzahrani, F.; Hijazi, H.; Alshammari, M.; Aldahmesh, M.A.; Salih, M.; et al. Accelerating Novel Candidate Gene Discovery in Neurogenetic Disorders via Whole-Exome Sequencing of Prescreened Multiplex Consanguineous Families. Cell Rep. 2015, 10, 148–161. [Google Scholar] [CrossRef] [PubMed]
- AlAbdi, L.; Maddirevula, S.; Aljamal, B.; Hamid, H.; Almulhim, A.; Hashem, M.O.; Algoos, Y.; Alqahtani, M.; Albaloshi, S.; Alghamdi, M.; et al. Arab founder variants: Contributions to clinical genomics and precision medicine. Med 2024, 6, 100528. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Guzmán-Vega, F.J.; González-Álvarez, A.C.; Peña-Guerra, K.A.; Cardona-Londoño, K.J.; Arold, S.T. Leveraging AI Advances and Online Tools for Structure-Based Variant Analysis. Curr. Protoc. 2023, 3, e857. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2022, 51, D638–D646. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prediction Tool | Score/Result | Pathogenicity Threshold | Interpretation * | Reference |
|---|---|---|---|---|
| AlphaMissense | 0.9915 | >0.564 | Pathogenic (top 5% of pathogenic variants, rankscore = 0.95066) | [14] |
| BayesDel_addAF | 0.3517 | >0.16 = Pathogenic | Pathogenic (supports deleteriousness) | [15] |
| BayesDel_noAF | 0.2674 (D) | >0.16 = Pathogenic | Pathogenic (independent of allele frequency) | [15] |
| CADD | 21.6 | >20 | Pathogenic (top 1% of deleterious variants genome-wide) | [16] |
| ClinPred | 0.9998 | >0.5 = Pathogenic | Pathogenic (near-maximal confidence) | [12] |
| DANN | 0.992 | >0.95 | Pathogenic (high-confidence prediction) | [17] |
| DEOGEN2 | 0.65427 | 0.89546 | Deleterious prediction | [18] |
| ESM1b/Variped | −18.109 | Lower = Deleterious | Deleterious (rankscore = 0.9965, “D” prediction) | [19] |
| Fathmm MKL | 0.9551 | >0.5 = Deleterious | Deleterious (group AEFGBI: likely functional impact) | [20] |
| Fathmm XF | 0.8938 | >0.5 = Deleterious | Deleterious (high confidence) | [21] |
| LRT | 0 | <0.05 = Deleterious | Deleterious (low conservation tolerance) | [22] |
| M_CAP | 0.1809 | >0.025 = Pathogenic | Pathogenic (moderate support) | [23] |
| MetaRNN | 0.9436 | >0.5 = Pathogenic | Pathogenic (high confidence) | [13] |
| MutPred | 0.767 | >0.5 | Pathogenic (rankscore = 0.89452; gain of MoRF binding, p = 0.0355) | [24] |
| MutationTaster | 1.0 | N/A (qualitative) | Disease-Causing (simple_aae model) | [9] |
| MutationAssessor | 3.575 | >3.5 = High Impact | High Impact (functional hotspot) | [25] |
| Polyphen2 HDIV | 1.0 | >0.85 = Probably Damaging | Damaging (maximal confidence) | [8] |
| Polyphen2 HVAR | 1.0 | >0.85 = Probably Damaging | Damaging (consistent with HDIV) | [8] |
| PROVEAN | −10.38 | ≤−2.5 = Deleterious | Deleterious (far below threshold) | [26] |
| REVEL | 0.498 | >0.5 | Uncertain significance (at border) | [27] |
| SIFT | 0 | <0.05 = Deleterious | Deleterious (strong evolutionary disruption) | [28] |
| SIFT4G | 0 | <0.05 = Deleterious | Deleterious (matches SIFT’s prediction) | [29] |
| This Study | Almontashiri et al. (2024) [6] | Halperin et al. (2019) [4] | |||
|---|---|---|---|---|---|
| Family | 1 | 1 | 1 | 1 | |
| Subjects | 1 | 1 | 1 | 2 | |
| Gender | Male | Female | Female | Male | |
| Age at presentation | 12 months | Antenatally | Birth | Birth | |
| Age/outcome | 5 years (alive) | 15 days (died) | 4 years (died) | 2 years (died) | |
| Consanguinity | (+) | (+) | (+) | (+) | |
| Ethnicity | Arab | Arab | Middle east Bedouin | ||
| Variant | c.DNA change | c.1359C > G | c.1435−1G > A | c.2776_2777 TA duplication | |
| Amino acid change | p.Cys453Trp | - | p.A927fs*61 | ||
| Zygosity | Homozygous | Homozygous | Homozygous | ||
| Neurological findings | Seizures (generalized tonic–clonic) | No seizure | Seizures (focal and generalized tonic–clonic) | ||
| Global developmental delay (cognition, motor, speech) Central hypotonia Hyperreflexia Spasticity | Hypotonia Hyperreflexia | Global developmental delay (cognition, motor, speech) Spastcic quadriplasia Hypotonia | |||
| Neuroimaging | Corpus callosum hypogenesis Hypomyelination Diffuse brain atrophy | Interhemispheric cyst Absent corpus callosum | Semilobar holoprosencephaly Enlargement of the subarachnoid space Corpus callosum agenesis Ventriculomegaly Colpocephaly | ||
| EEG | Abnormal electric activity | - | Disorganized background activity with an epileptic pattern of bilateral sharp waves and spikes discharges | ||
| Growth parameters | Microcephaly Short stature | Microcephaly Short stature | Microcephaly | ||
| Cardiac | Normal | Bradycardia | Peri-membranotic VSD | ||
| Genitourinary | Normal | Normal | Normal | ||
| GI problems | Constipation Dysphagia Recurrent aspiration Failure to thrive | Not reported | Pseudobulbar palsy Recurrent aspirations Congenital diaphragmatic hernia Umbilical and inguinal hernia Gastro-esophageal reflux Feeding difficulties Failure to thrive | ||
| Ophthalmological | Esotropia | Not reported | Nystagmus Lack of ocular fixation Bilateral nuclear cataracts | ||
| Dysmorphic features | Prominent nasal bridge, hypertelorism, epicanthal fold, frontal bossing, prominent ears | Wide anterior fontanelle, sloping forehead, hypertelorism, malformed ears, depressed nasal bridge, retrognathia, short neck Skeletal anomalies | Pointed triangular face, micrognathia and high-arched palate, thick lips, long eyelashes | ||
| Hearing | Bilateral hearing loss | Not reported | Bilateral hearing loss | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AlTassan, R.; AlQudairy, H.; Saydo, B.; Alammari, A.; Londoño, K.J.C.; Ramzan, K.; Colak, D.; Arold, S.T.; Kaya, N. A Report of a Child with SEC31A-Related Neurodevelopmental Disorder. Int. J. Mol. Sci. 2025, 26, 5296. https://doi.org/10.3390/ijms26115296
AlTassan R, AlQudairy H, Saydo B, Alammari A, Londoño KJC, Ramzan K, Colak D, Arold ST, Kaya N. A Report of a Child with SEC31A-Related Neurodevelopmental Disorder. International Journal of Molecular Sciences. 2025; 26(11):5296. https://doi.org/10.3390/ijms26115296
Chicago/Turabian StyleAlTassan, Ruqaiah, Hanan AlQudairy, Biam Saydo, Aseel Alammari, Kelly J. Cardona Londoño, Khushnooda Ramzan, Dilek Colak, Stefan T. Arold, and Namik Kaya. 2025. "A Report of a Child with SEC31A-Related Neurodevelopmental Disorder" International Journal of Molecular Sciences 26, no. 11: 5296. https://doi.org/10.3390/ijms26115296
APA StyleAlTassan, R., AlQudairy, H., Saydo, B., Alammari, A., Londoño, K. J. C., Ramzan, K., Colak, D., Arold, S. T., & Kaya, N. (2025). A Report of a Child with SEC31A-Related Neurodevelopmental Disorder. International Journal of Molecular Sciences, 26(11), 5296. https://doi.org/10.3390/ijms26115296