
bTRM Control of Murine Cytomegalovirus CNS Reactivation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
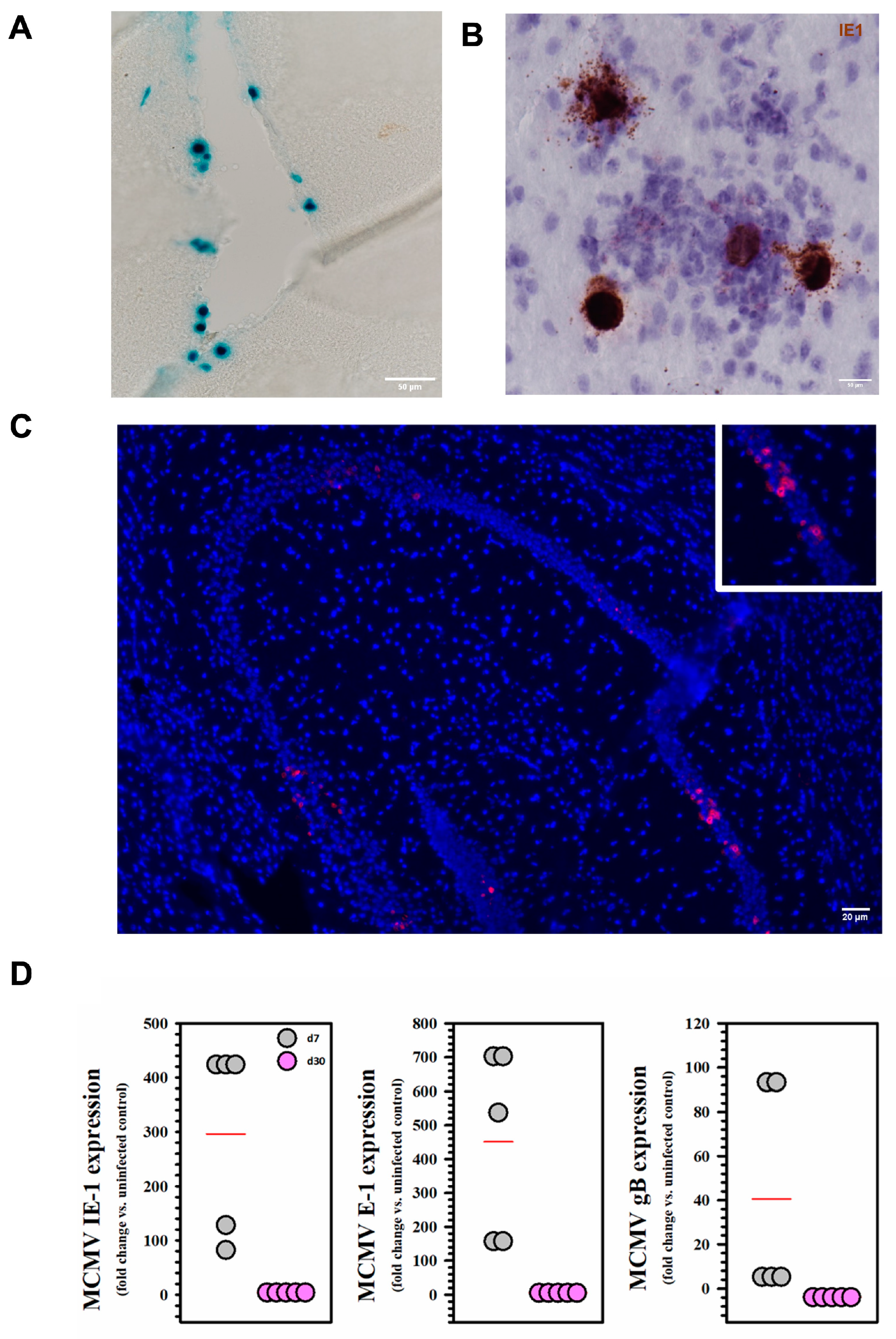
2.1. Viral Expression and Establishment of Latency Following MCMV Infection
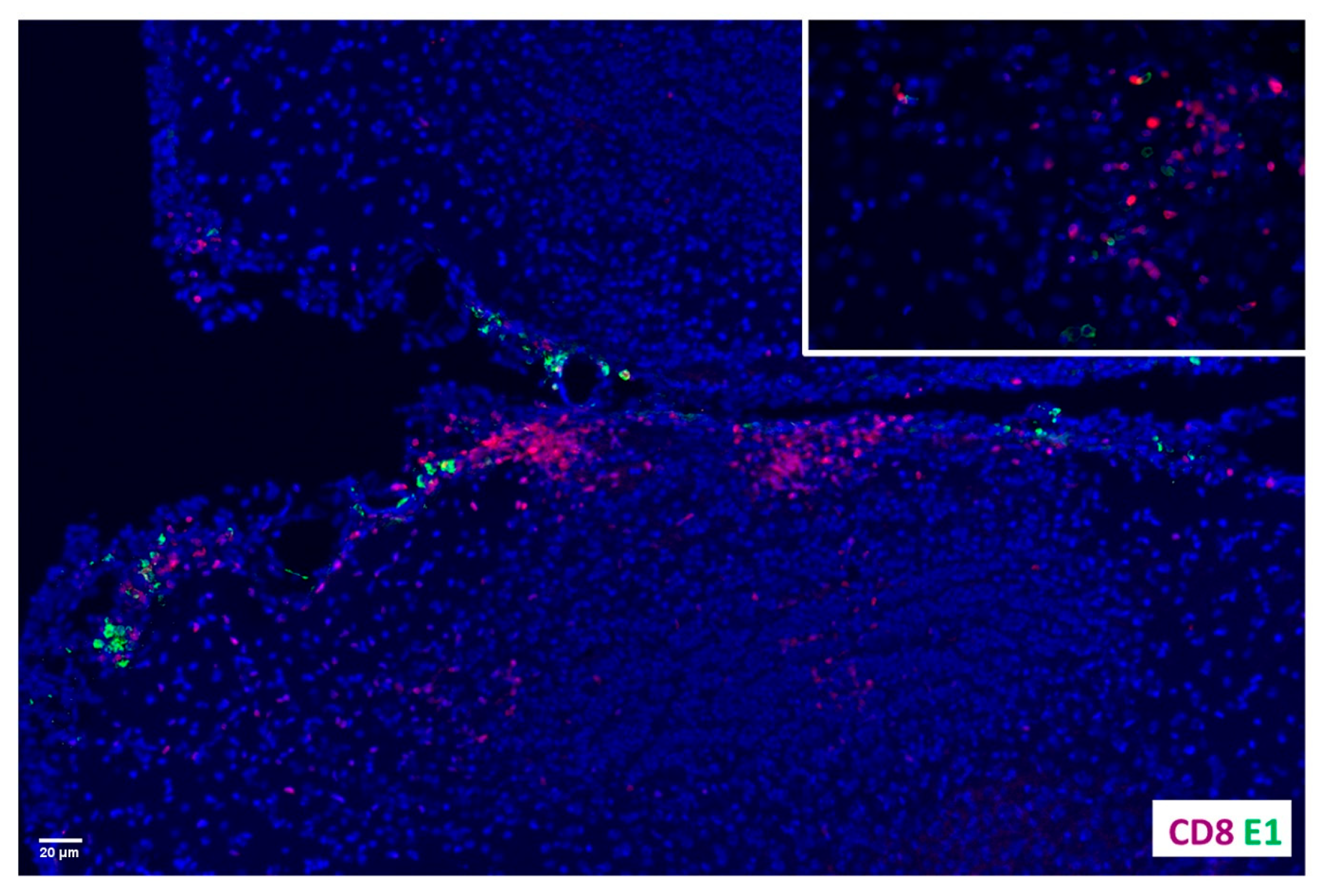
2.2. Co-Localization of CD8+ T-Cells and MCMV E1-Retaining Cells
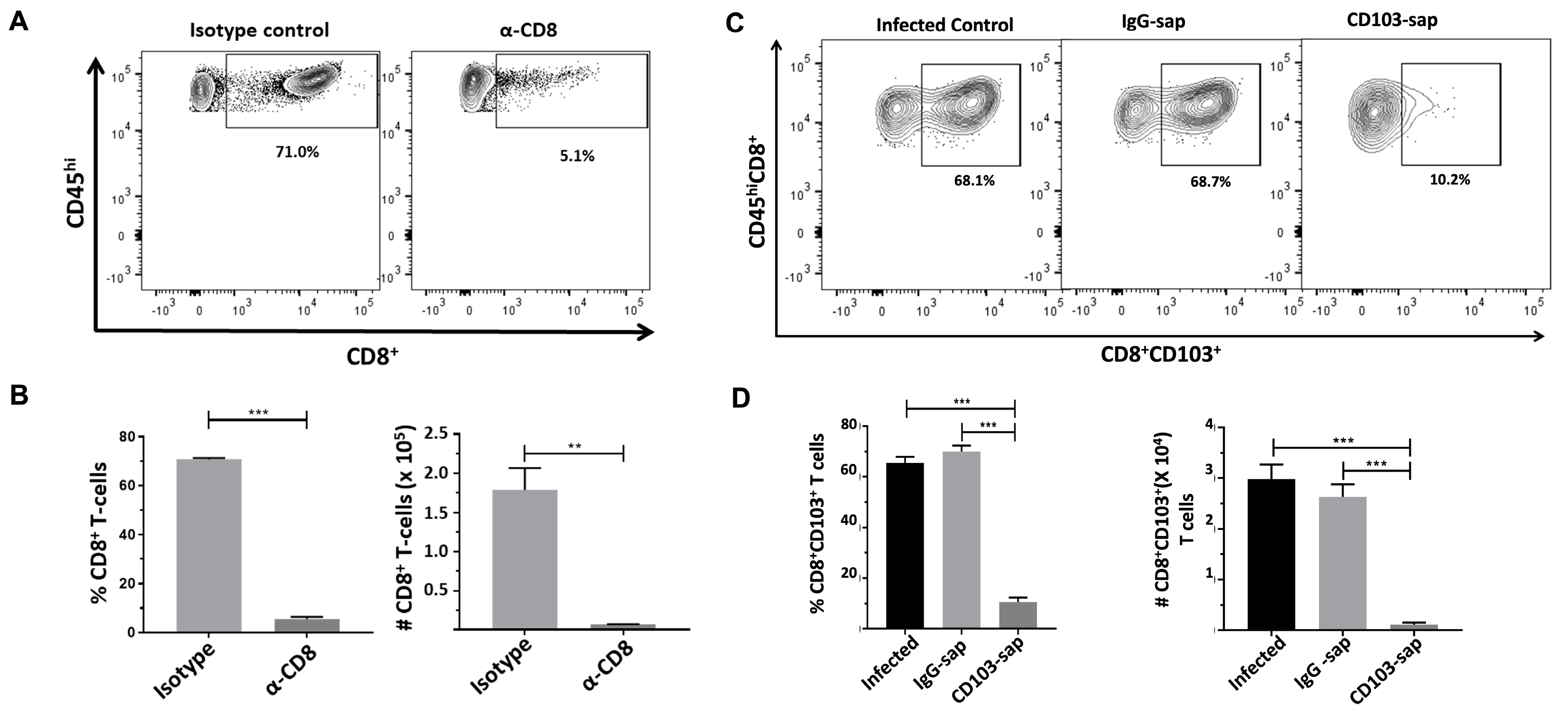
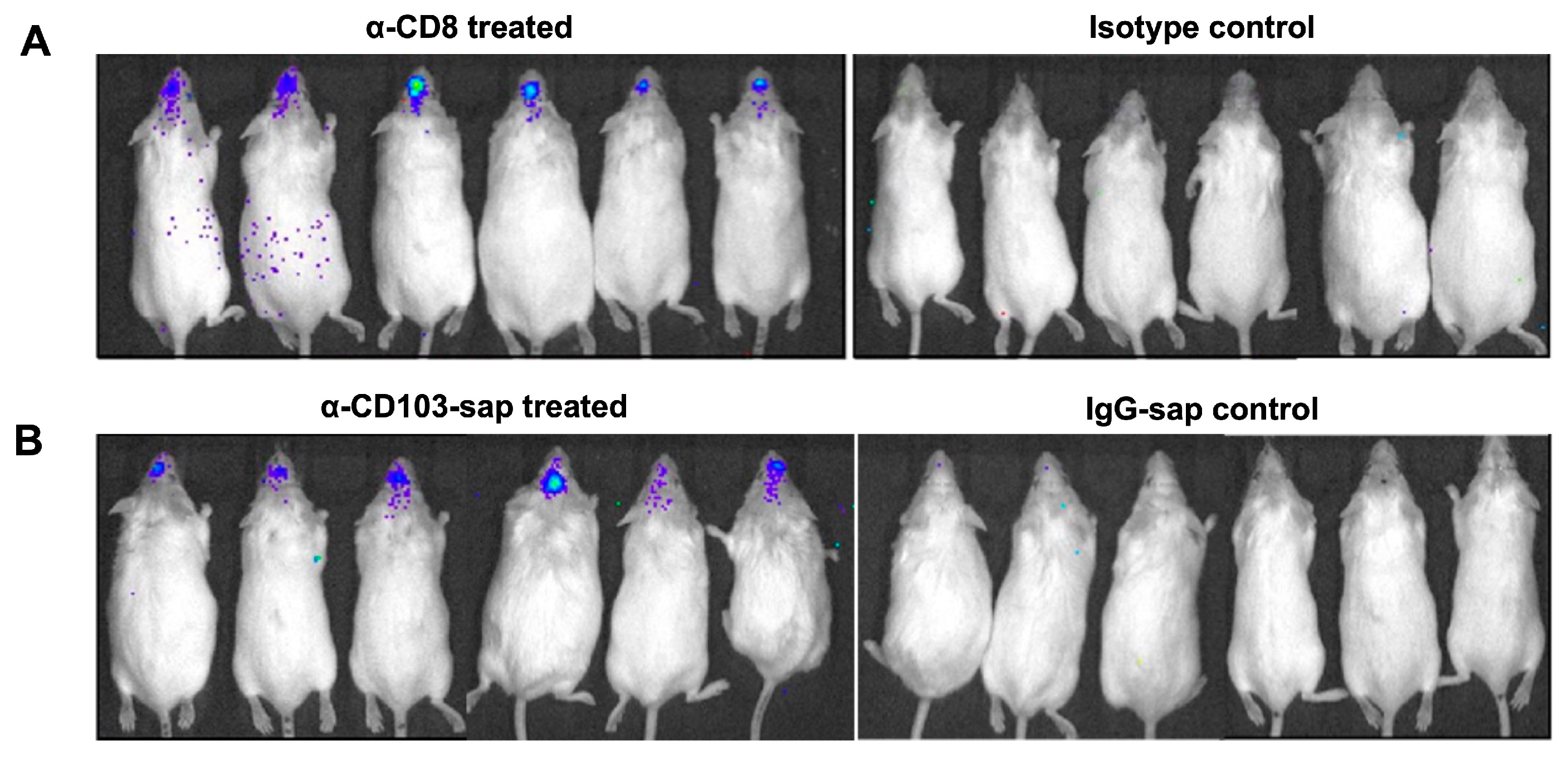
2.3. Depletion of CD8+ and CD8+CD103+ T-Cells from the Brains of MCMV-Infected Animals
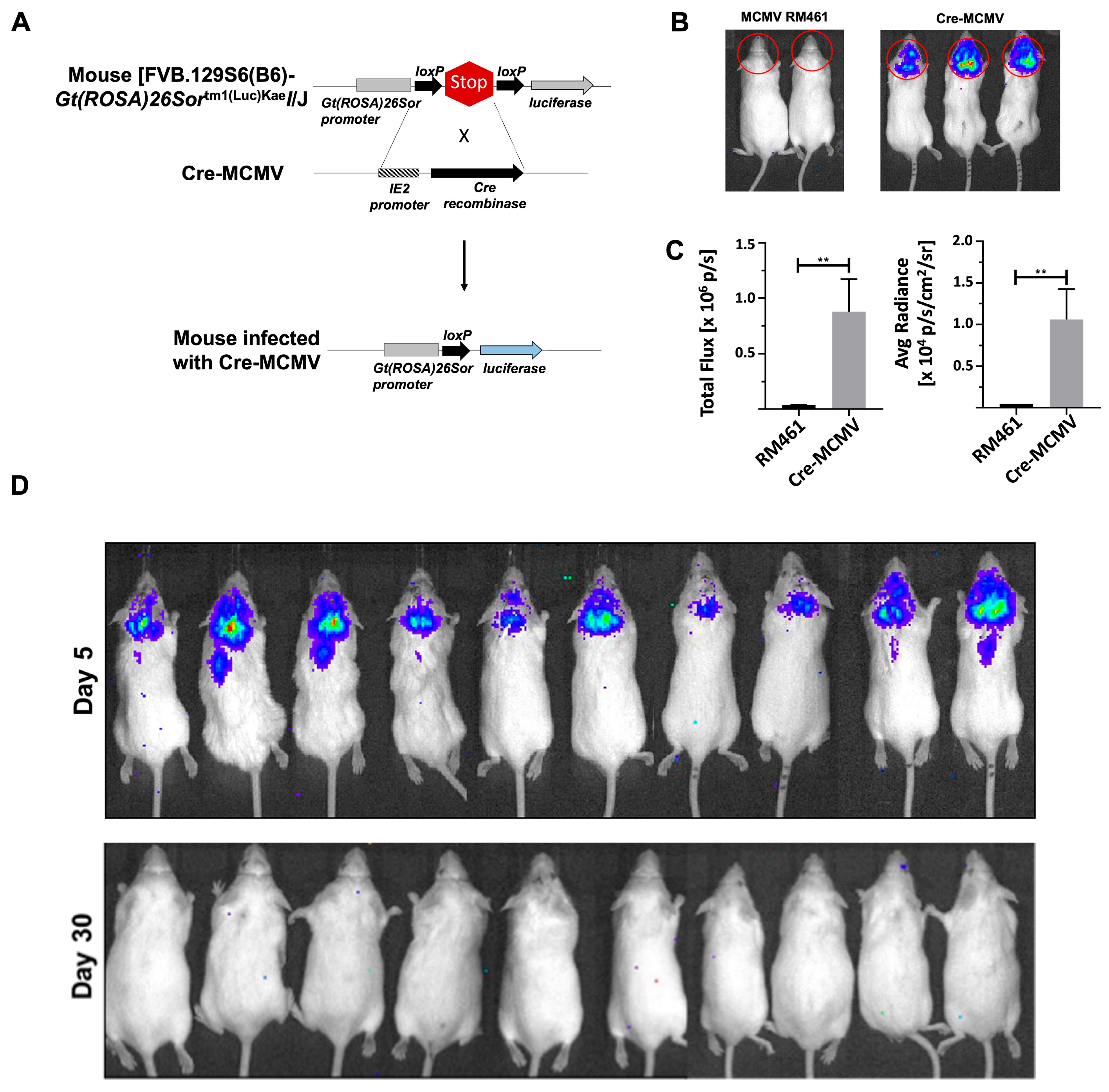
2.4. Establishment of Latent cre-MCMV Infection in FVB Transgenic Mice Using Bioluminescent Imaging
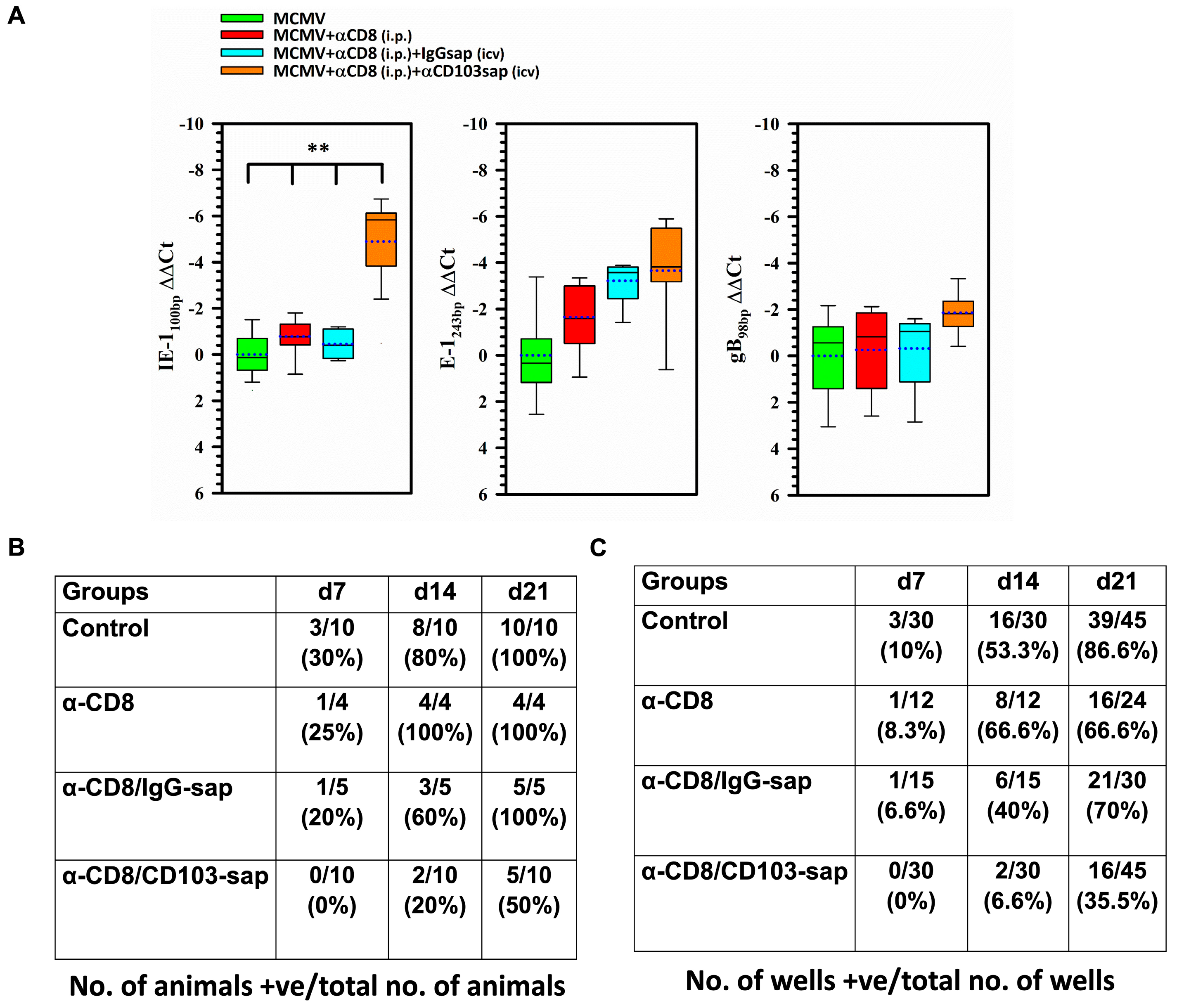
2.5. Role of CD8 or CD103 in Controlling Viral Reactivation Within the Brain
2.6. Recovery of Reactivated Infectious Virus from the Brains of Latently-Infected Animals Depleted of bTRM
2.7. Profiling Brain Microenvironments
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Experimental Animals
4.3. Virus and Growth Conditions
4.4. Intracerebroventricular (icv) Infection of Mice
4.5. Isolation of Brain Leukocytes and Flow Cytometric Analysis
4.6. Bioluminescence Imaging
4.7. Semi-Quantitative RT-PCR
4.8. Immunohistochemistry
4.9. RNAscope™ ISH
4.10. Explant Assay
4.11. Nanostring Analysis
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MCMV | Murine cytomegalovirus |
| bTRMs | Brain tissue-resident memory T-cells |
| LgalS3 | Galectin 3 |
| gpnmb | Glycoprotein nonmetastatic melanoma protein B |
| Hmox1 | Heme oxygenase I |
| TREM2 | Triggering receptor on myeloid cells 2 |
| DAM | Disease-associated Microglia |
| MGnD | Neurodegenerative Microglia |
| NSC | Neural Stem cells |
| SVZ | Sub-ventricular zone |
| CTL | Cytotoxic T-cell |
| CNS | Central Nervous system |
| d.p.i | Days post infection |
| ICV | Intracerebroventricular |
| Ab | Antibody |
| Ag | Antigen |
| BMNCs | Brain mononuclear cells |
| HSV | Herpes Simplex virus |
| IFN | Interferon |
| FBS | Fetal bovine serum |
| TCID50 | 50% Tissue culture infective doses |
| CPE | Cytopathic effect |
| HPRT | Hypoxanthine phosphoribosyl transferase |
| PBS | Phosphate-buffered saline |
| RT | Room temperature |
References
- Hu, S.; Rotschafer, J.H.; Lokensgard, J.R.; Cheeran, M.C. Activated CD8+ T lymphocytes inhibit neural stem/progenitor cell proliferation: Role of interferon-gamma. PLoS ONE 2014, 9, e105219. [Google Scholar] [CrossRef] [PubMed]
- Cheeran, M.C.; Jiang, Z.; Hu, S.; Ni, H.T.; Palmquist, J.M.; Lokensgard, J.R. Cytomegalovirus infection and interferon-gamma modulate major histocompatibility complex class I expression on neural stem cells. J. Neurovirol. 2008, 14, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Mutnal, M.B.; Cheeran, M.C.; Hu, S.; Lokensgard, J.R. Murine cytomegalovirus infection of neural stem cells alters neurogenesis in the developing brain. PLoS ONE 2011, 6, e16211. [Google Scholar] [CrossRef] [PubMed]
- Mutnal, M.B.; Hu, S.; Lokensgard, J.R. Persistent humoral immune responses in the CNS limit recovery of reactivated murine cytomegalovirus. PLoS ONE 2012, 7, e33143. [Google Scholar] [CrossRef]
- Prasad, S.; Hu, S.; Sheng, W.S.; Singh, A.; Lokensgard, J.R. Tregs Modulate Lymphocyte Proliferation, Activation, and Resident-Memory T-Cell Accumulation within the Brain during MCMV Infection. PLoS ONE 2015, 10, e0145457. [Google Scholar] [CrossRef]
- Prasad, S.; Hu, S.; Sheng, W.S.; Chauhan, P.; Lokensgard, J.R. Reactive glia promote development of CD103(+) CD69(+) CD8(+) T-cells through programmed cell death-ligand 1 (PD-L1). Immun. Inflamm. Dis. 2018, 6, 332–344. [Google Scholar] [CrossRef]
- Brizic, I.; Susak, B.; Arapovic, M.; Huszthy, P.C.; Hirsl, L.; Kvestak, D.; Juranic Lisnic, V.; Golemac, M.; Pernjak Pugel, E.; Tomac, J.; et al. Brain-resident memory CD8(+) T cells induced by congenital CMV infection prevent brain pathology and virus reactivation. Eur. J. Immunol. 2018, 48, 950–964. [Google Scholar] [CrossRef]
- Yeh, H.; Ikezu, T. Transcriptional and Epigenetic Regulation of Microglia in Health and Disease. Trends Mol. Med. 2019, 25, 96–111. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Afridi, R.; Lee, W.H.; Suk, K. Microglia Gone Awry: Linking Immunometabolism to Neurodegeneration. Front. Cell. Neurosci. 2020, 14, 246. [Google Scholar] [CrossRef]
- Shinmura, Y.; Aiba-Masago, S.; Kosugi, I.; Li, R.Y.; Baba, S.; Tsutsui, Y. Differential expression of the immediate-early and early antigens in neuronal and glial cells of developing mouse brains infected with murine cytomegalovirus. Am. J. Pathol. 1997, 151, 1331–1340. [Google Scholar] [PubMed]
- Tsutsui, Y.; Kashiwai, A.; Kawamura, N.; Aiba-Masago, S.; Kosugi, I. Prolonged infection of mouse brain neurons with murine cytomegalovirus after pre- and perinatal infection. Arch. Virol. 1995, 140, 1725–1736. [Google Scholar] [CrossRef] [PubMed]
- Proenca, J.T.; Coleman, H.M.; Connor, V.; Winton, D.J.; Efstathiou, S. A historical analysis of herpes simplex virus promoter activation in vivo reveals distinct populations of latently infected neurones. J. Gen. Virol. 2008, 89, 2965–2974. [Google Scholar] [CrossRef] [PubMed]
- Dutia, B.M.; Reid, S.J.; Drummond, D.D.; Ligertwood, Y.; Bennet, I.; Rietberg, W.; Silvia, O.; Jarvis, M.A.; Nash, A.A. A novel Cre recombinase imaging system for tracking lymphotropic virus infection in vivo. PLoS ONE 2009, 4, e6492. [Google Scholar] [CrossRef]
- Simon, C.O.; Holtappels, R.; Tervo, H.M.; Bohm, V.; Daubner, T.; Oehrlein-Karpi, S.A.; Kuhnapfel, B.; Renzaho, A.; Strand, D.; Podlech, J.; et al. CD8 T cells control cytomegalovirus latency by epitope-specific sensing of transcriptional reactivation. J. Virol. 2006, 80, 10436–10456. [Google Scholar] [CrossRef]
- Reddehase, M.J.; Simon, C.O.; Seckert, C.K.; Lemmermann, N.; Grzimek, N.K. Murine model of cytomegalovirus latency and reactivation. Curr. Top. Microbiol. Immunol. 2008, 325, 315–331. [Google Scholar]
- Seckert, C.K.; Griessl, M.; Buttner, J.K.; Scheller, S.; Simon, C.O.; Kropp, K.A.; Renzaho, A.; Kuhnapfel, B.; Grzimek, N.K.; Reddehase, M.J. Viral latency drives ‘memory inflation’: A unifying hypothesis linking two hallmarks of cytomegalovirus infection. Med. Microbiol. Immunol. 2012, 201, 551–566. [Google Scholar] [CrossRef]
- Di Liberto, G.; Egervari, K.; Kreutzfeldt, M.; Schurch, C.M.; Hewer, E.; Wagner, I.; Du Pasquier, R.; Merkler, D. Neurodegenerative phagocytes mediate synaptic stripping in Neuro-HIV. Brain 2022, 145, 2730–2741. [Google Scholar] [CrossRef]
- Thammahakin, P.; Maezono, K.; Maekawa, N.; Kariwa, H.; Kobayashi, S. Detection of disease-associated microglia among various microglia phenotypes induced by West Nile virus infection in mice. J. Neurovirol. 2023, 29, 367–375. [Google Scholar] [CrossRef]
- Manet, C.; Mansuroglu, Z.; Conquet, L.; Bortolin, V.; Comptdaer, T.; Segrt, H.; Bourdon, M.; Menidjel, R.; Stadler, N.; Tian, G.; et al. Zika virus infection of mature neurons from immunocompetent mice generates a disease-associated microglia and a tauopathy-like phenotype in link with a delayed interferon beta response. J. Neuroinflamm. 2022, 19, 307. [Google Scholar] [CrossRef]
- Lee, K.H.; Kwon, D.E.; Do Han, K.; La, Y.; Han, S.H. Association between cytomegalovirus end-organ diseases and moderate-to-severe dementia: A population-based cohort study. BMC Neurol. 2020, 20, 216. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, G.; Piacentini, R.; Fabiani, M.; Mastrodonato, A.; Marcocci, M.E.; Limongi, D.; Napoletani, G.; Protto, V.; Coluccio, P.; Celestino, I.; et al. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog. 2019, 15, e1007617. [Google Scholar] [CrossRef]
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Li Puma, D.D.; Lomonte, P.; Grassi, C.; Palamara, A.T.; De Chiara, G. Herpes Simplex Virus-1 in the Brain: The Dark Side of a Sneaky Infection. Trends Microbiol. 2020, 28, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Readhead, B.; Haure-Mirande, J.V.; Funk, C.C.; Richards, M.A.; Shannon, P.; Haroutunian, V.; Sano, M.; Liang, W.S.; Beckmann, N.D.; Price, N.D.; et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron 2018, 99, 64–82.e7. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.A.; Harris, E.A. Molecular Mechanisms for Herpes Simplex Virus Type 1 Pathogenesis in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 48. [Google Scholar] [CrossRef]
- Rizzo, R. Controversial role of herpesviruses in Alzheimer’s disease. PLoS Pathog. 2020, 16, e1008575. [Google Scholar] [CrossRef]
- Romeo, M.A.; Gilardini Montani, M.S.; Gaeta, A.; D’Orazi, G.; Faggioni, A.; Cirone, M. HHV-6A infection dysregulates autophagy/UPR interplay increasing beta amyloid production and tau phosphorylation in astrocytoma cells as well as in primary neurons, possible molecular mechanisms linking viral infection to Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165647. [Google Scholar] [CrossRef]
- Romeo, M.A.; Faggioni, A.; Cirone, M. Could autophagy dysregulation link neurotropic viruses to Alzheimer’s disease? Neural Regen. Res. 2019, 14, 1503–1506. [Google Scholar]
- Barnes, L.L.; Capuano, A.W.; Aiello, A.E.; Turner, A.D.; Yolken, R.H.; Torrey, E.F.; Bennett, D.A. Cytomegalovirus infection and risk of Alzheimer disease in older black and white individuals. J. Infect. Dis. 2015, 211, 230–237. [Google Scholar] [CrossRef]
- Lin, W.R.; Wozniak, M.A.; Wilcock, G.K.; Itzhaki, R.F. Cytomegalovirus is present in a very high proportion of brains from vascular dementia patients. Neurobiol. Dis. 2002, 9, 82–87. [Google Scholar] [CrossRef]
- McMaster, S.R.; Wilson, J.J.; Wang, H.; Kohlmeier, J.E. Airway-Resident Memory CD8 T Cells Provide Antigen-Specific Protection against Respiratory Virus Challenge through Rapid IFN-gamma Production. J. Immunol. 2015, 195, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Ariotti, S.; Hogenbirk, M.A.; Dijkgraaf, F.E.; Visser, L.L.; Hoekstra, M.E.; Song, J.Y.; Jacobs, H.; Haanen, J.B.; Schumacher, T.N. T cell memory. Skin-resident memory CD8(+) T cells trigger a state of tissue-wide pathogen alert. Science 2014, 346, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Hu, S.; Sheng, W.S.; Chauhan, P.; Lokensgard, J.R. Recall Responses from Brain-Resident Memory CD8(+) T Cells (bT(RM)) Induce Reactive Gliosis. iScience 2019, 20, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 2014, 346, 98–101. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Cicin-Sain, L.; Podlech, J.; Messerle, M.; Reddehase, M.J.; Koszinowski, U.H. Frequent coinfection of cells explains functional in vivo complementation between cytomegalovirus variants in the multiply infected host. J. Virol. 2005, 79, 9492–9502. [Google Scholar] [CrossRef]
- Cheeran, M.C.; Hu, S.; Palmquist, J.M.; Bakken, T.; Gekker, G.; Lokensgard, J.R. Dysregulated interferon-gamma responses during lethal cytomegalovirus brain infection of IL-10-deficient mice. Virus Res. 2007, 130, 96–102. [Google Scholar] [CrossRef]
- Ford, A.L.; Goodsall, A.L.; Hickey, W.F.; Sedgwick, J.D. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J. Immunol. 1995, 154, 4309–4321. [Google Scholar] [CrossRef]
- Marten, N.W.; Stohlman, S.A.; Zhou, J.; Bergmann, C.C. Kinetics of virus-specific CD8+ -T-cell expansion and trafficking following central nervous system infection. J. Virol. 2003, 77, 2775–2778. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chauhan, P.; Hu, S.; Sheng, W.S.; Prasad, S.; Lokensgard, J.R. bTRM Control of Murine Cytomegalovirus CNS Reactivation. Int. J. Mol. Sci. 2025, 26, 5275. https://doi.org/10.3390/ijms26115275
Chauhan P, Hu S, Sheng WS, Prasad S, Lokensgard JR. bTRM Control of Murine Cytomegalovirus CNS Reactivation. International Journal of Molecular Sciences. 2025; 26(11):5275. https://doi.org/10.3390/ijms26115275
Chicago/Turabian StyleChauhan, Priyanka, Shuxian Hu, Wen S. Sheng, Sujata Prasad, and James R. Lokensgard. 2025. "bTRM Control of Murine Cytomegalovirus CNS Reactivation" International Journal of Molecular Sciences 26, no. 11: 5275. https://doi.org/10.3390/ijms26115275
APA StyleChauhan, P., Hu, S., Sheng, W. S., Prasad, S., & Lokensgard, J. R. (2025). bTRM Control of Murine Cytomegalovirus CNS Reactivation. International Journal of Molecular Sciences, 26(11), 5275. https://doi.org/10.3390/ijms26115275