1. Introduction
Multiple myeloma (MM) is a cancer of plasma cells in the bone marrow (BM), which can manifest as bone pain, anaemia, BM failure, renal impairment, and a higher incidence of infections [
1]. The long-term remission and survival rates in MM patients have improved during the past two decades due to advances in treatment development, largely credited to the clinical approvals of targeted immunotherapies. These include monoclonal antibodies (mAbs), bispecific antibodies (bsAbs) redirecting T cells, and CAR T cell therapy. The affinity proteins used as the targeting moieties in these immunotherapies are typically based on antibodies or antibody fragments (mainly scFvs) [
1,
2,
3]. Additional antibody-based immunotherapies for MM treatment are currently in clinical trials with promising results [
4,
5,
6,
7]. However, MM is still considered an incurable disease, and further development of novel immunotherapies involving tumour target-recognition units is warranted [
4].
For such development, alternative classes of non-immunoglobulin affinity proteins may be considered. These proteins differ from antibody-based affinity proteins in e.g., their size, structural organisation, and engineering possibilities, which could be advantageous when used as target recognition units in biologicals [
8,
9]. Affibodies are a class of non-immunoglobulin affinity proteins with a small size (6.5–7 kDa) and a minimalist architecture consisting of three alpha helices [
10,
11]. Using combinatorial protein engineering principles, in which typically 13–15 surface-located positions are simultaneously randomised to create libraries for in vitro selection using e.g., phage or cell display, high-affinity affibody affinity proteins binding to diverse targets can be isolated [
11,
12]. Examples of clinically relevant targets addressed by affibodies include HER2, EGFR, PD-L1 and IL-17A, with demonstrated utility in in vivo tumour imaging, fluorescence-guided surgery, patient stratification in immuno-oncology, and autoimmune disease prevention, respectively [
13,
14,
15,
16].
B Cell Maturation Antigen (BCMA) is a target of considerable interest in MM [
17]. Overexpression of BCMA on malignant plasma cells is associated with the proliferation and survival of these cells, via activation by its natural ligands: A Proliferation Inducing Ligand (APRIL) and B cell Activating Factor (BAFF), of which APRIL is the preferred ligand [
18,
19,
20]. Several clinically approved immunotherapies are directed to BCMA, including the CAR T cell therapies idecabtagene vicleucel (Abecma) and ciltacabtagene autoleucel (Carvykti), and the BCMA x CD3 bsAbs teclistamab (Tecvayli) and elranatamab (Elrexfio) [
21].
In the present study, we describe the development of affibody affinity proteins targeting BCMA for potential application as a tumour-recognition unit in immunotherapeutic agents and as a diagnostic tool for patient stratification based on BCMA expression. We describe the use of phage display for the isolation of BCMA-targeting affibodies from naïve and second-generation libraries. The main candidate clone denoted 1-E6 was characterised in detail using binding kinetics analyses, circular dichroism spectroscopy and competitive binding measurements with the BCMA binding proteins APRIL and the clinically evaluated anti-BCMA mAb belantamab. Binding to BCMA-expressing cells was investigated using flow cytometry and fluorescence microscopy. Taken together, the results suggest that the developed anti-BCMA affibody 1-E6 has the potential to be used as a BCMA-targeting unit in reagents aimed for use in both therapeutic and diagnostic applications in MM.
3. Discussion
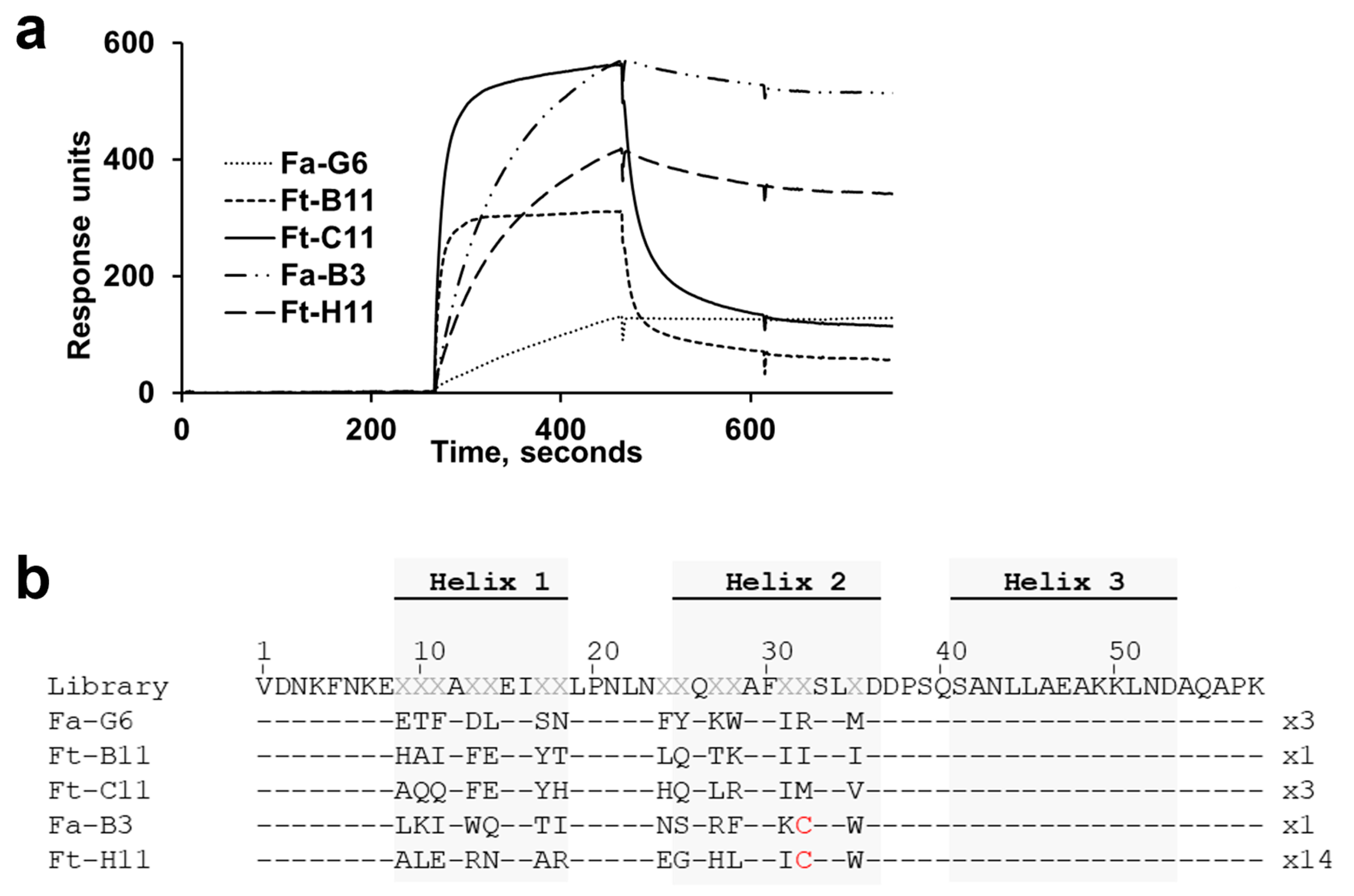
We describe the identification of novel affibody affinity proteins that bind BCMA, an important target for MM therapies. The BCMA ECD target protein used in this study is relatively small: 45 amino acid residues, i.e., smaller than the 58-residue affibody protein. It was therefore not obvious that it should contain any surfaces amenable for interaction with the affibody binding surface, which is a relatively flat surface and lacks protein loops. However, no less than 25 unique binders could be identified after screening 144 randomly picked clones from the fourth selection cycle output.
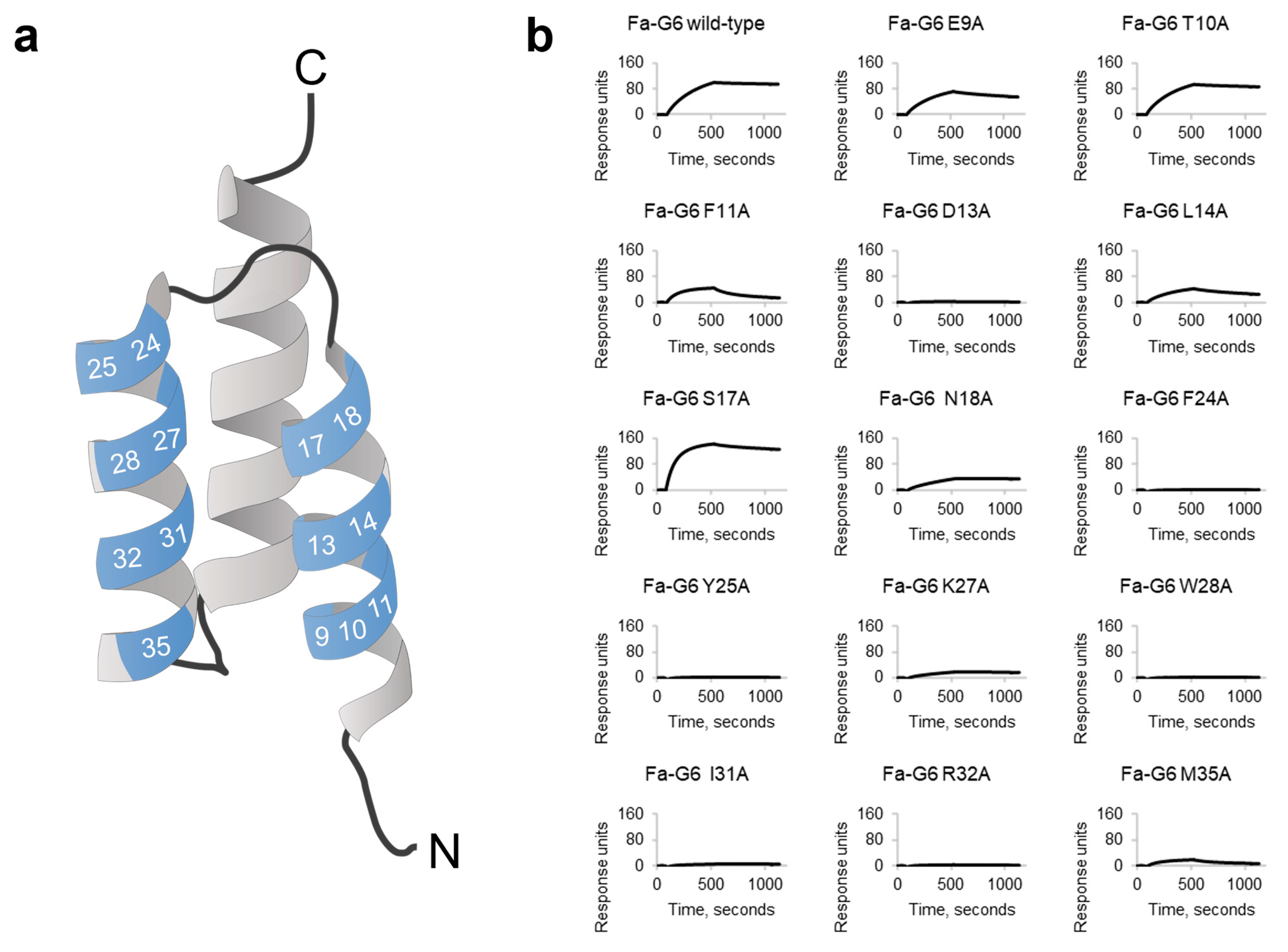
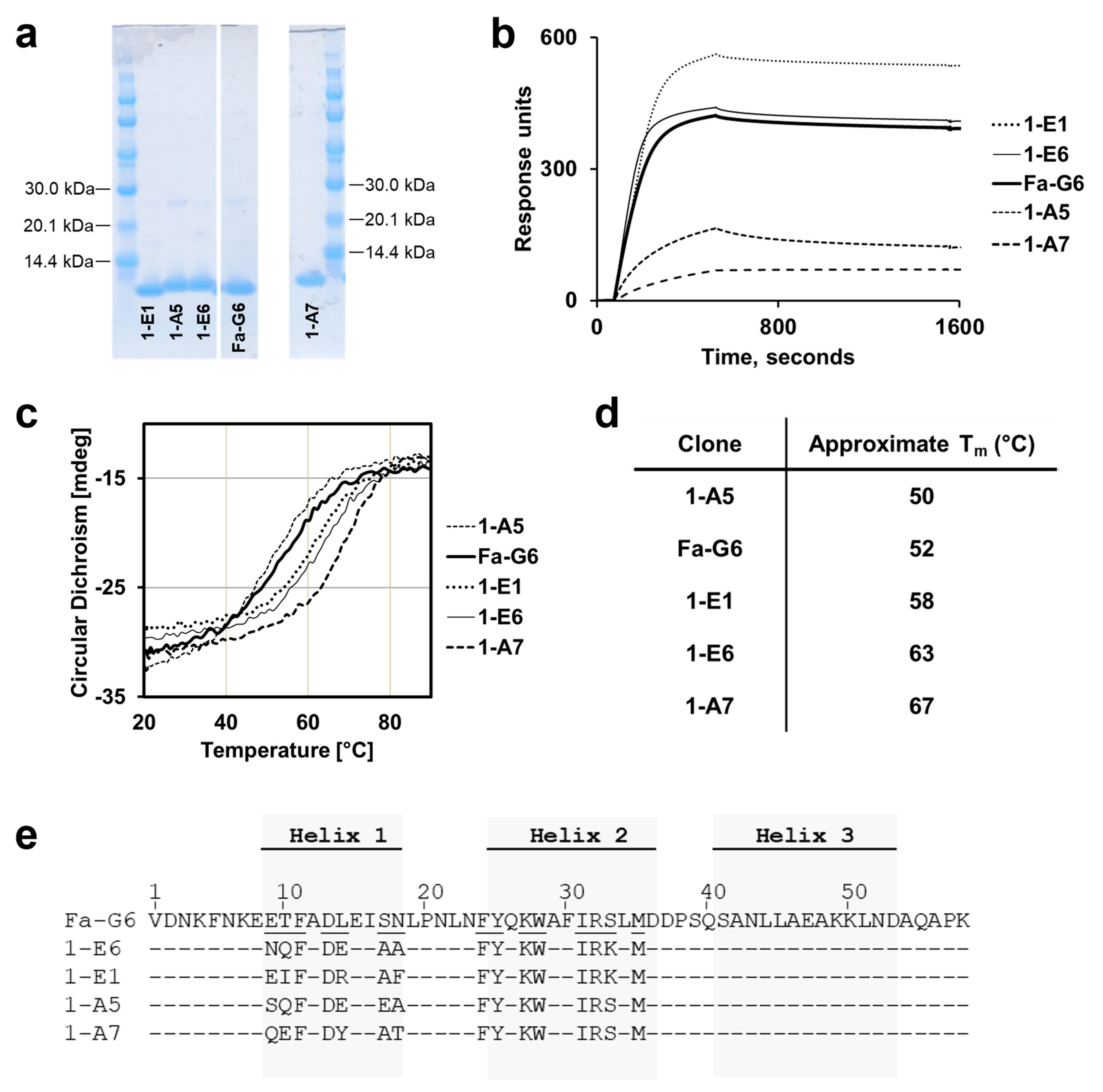
The identification of the top candidate 1-E6 affibody variant involved two consecutive library selection steps, including the construction of two second-generation libraries. The design of these was based on binding and secondary structure content data from a set of alanine mutants of a chosen first-generation variant, Fa-G6, showing the most preferable off-rate kinetics. While the assumption was that variants with significantly higher affinities would be possible to select from these second-generation libraries, the most prominent improvement in the output clones was instead seen in their structural stabilities, measured as higher Tm values. Intriguingly, the experimental set-up of the selections did not include any explicit measures where a higher thermostability would be favoured. Rather, gradually lower antigen concentrations and increasingly extensive washings were used and expected to promote high-affinity binding.
Although the alanine scanning experiment of the parental clone Fa-G6 showed that alanine substitutions at all seven positions in helix 2 severely affected binding, it is somewhat surprising that the output clones from the second-generation libraries almost exclusively contained the original Fa-G6 amino acid at six of these seven re-randomised positions, and that no replacement to chemically similar amino acids were observed. The output also showed a striking convergence to alanine in position 17. In the alanine scanning experiment of the Fa-G6 variant, it was observed that the S17A mutant showcased a high-affinity BCMA binding profile, with a faster complex formation rate than Fa-G6 wild-type, suggesting that the selection conditions to at least some extent promoted clones with improved binding.
Position 33 is not located in the binding surface of the affibody scaffold, but it is surface exposed and has been implied to be important for the scaffold stability [
23]. In both second-generation libraries this position, occupied by serine in the parental clone Fa-G6, was conservatively included in the randomisation and designed to be occupied by 90% serine and 10% lysine. However, the global analysis of the output demonstrated an increase of lysine to 40% occupancy, suggesting a favourable contribution during selections. CD spectroscopy showed that the second-generation clones 1-E1 and 1-E6, both containing lysine in this position, had improved Tm values by approximately 6 and 11 °C, respectively, supporting the notion of its importance for the structural stability. However, it is worth noting that the second-generation clone 1-A7, containing a serine at position 33, exhibited the most improved Tm, with a 15 °C increase compared to the parental Fa-G6 clone. However, this large increase in stability was not associated with a stronger binding affinity to BCMA.
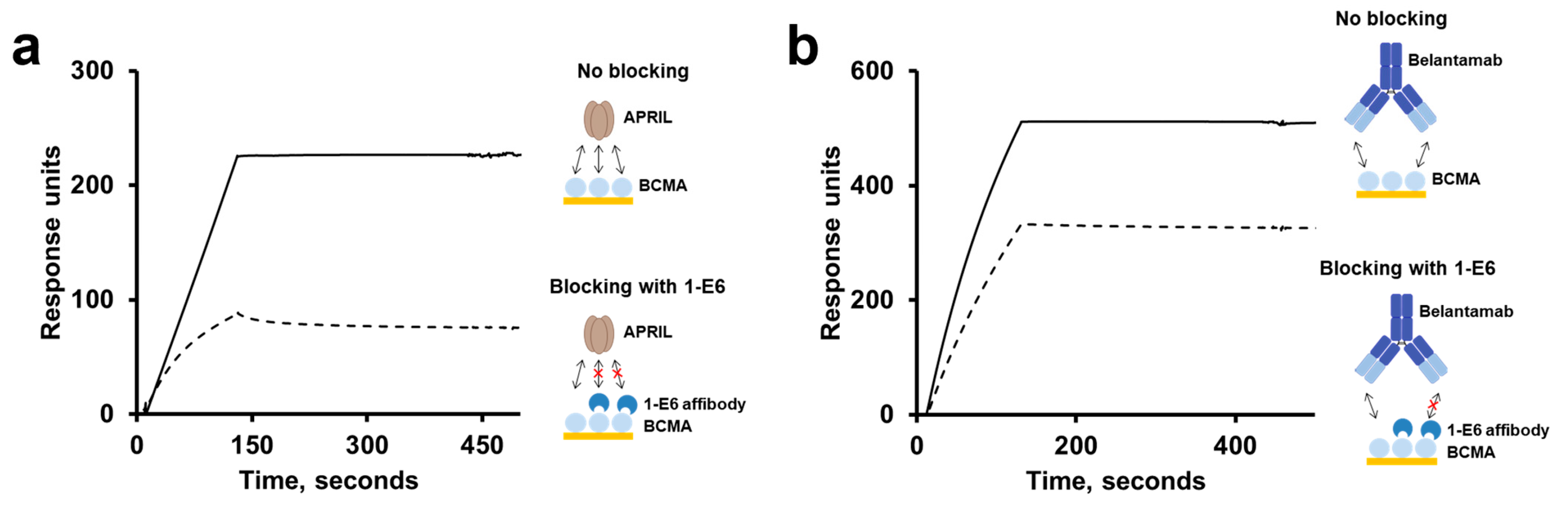
No binding was observed for the 1-E6 affibody to BCMA homologues from mouse, rhesus and marmoset, despite their high degrees of sequence identity to human BCMA in their ECDs. However, even if human and marmoset BCMA share 91% sequence identity, five amino acid positions and a small gap differ between the two proteins that could explain the findings. We found that the 1-E6 affibody was able to compete for binding to BCMA with APRIL, a native ligand to BCMA involved in the promotion of MM cell proliferation and survival [
18]. Similarly, the anti-BCMA monoclonal antibody belantamab has also been shown to be able to compete with APRIL for BCMA binding. This competition has been suggested to be one of belantamab’s mode of actions, when clinically investigated as an antibody-drug conjugate [
26]. Our data indicates that the respective epitopes of the 1-E6 affibody and belantamab are overlapping, or partially overlapping, and are coinciding with the binding site of APRIL on BCMA. This finding may be expected considering the small size of the BCMA ECD. It is appealing to theorise that the shared epitope space of the BCMA-binders APRIL, belantamab and the 1-E6 affibody may be a so-called “epitope hotspot” of the BCMA ECD. As suggested for belantamab, this may indicate that the 1-E6 affibody may inhibit BCMA-induced proliferation, via blocking of APRIL, as its therapeutic mode of action. On the other hand, it could also suggest that its therapeutic efficacy may be negatively affected by competition with soluble APRIL.
Resistance-driving mutations in BCMA have been described to negatively affect BCMA-binding reagents [
27]. However, in the same study APRIL was shown to be able to bind BCMA containing these resistance-driving mutations. Thus, since 1-E6 demonstrated overlapping binding epitopes with APRIL on BCMA, it is not impossible that 1-E6 may also recognise such BCMA variants.
An interesting observation was that two of the initially isolated binders with the highest binding responses (Fa-B3 and Ft-H11) each contained a single cysteine amino acid, and that substituting these cysteines for serines led to the loss of binding. In fact, a similar observation has earlier been described in the selection of affibodies against the Alzheimer’s disease-related amyloid β (Aβ) 1–40 peptide. Also here, homodimer formation of affibody monomers via a disulfide bridge between internal cysteines was found to be essential for efficient target binding. The Aβ binding of two cysteine-to-serine mutants was almost completely restored by homodimerisation through tandem head-to-tail fusion [
28]. Subsequent structural studies by NMR of the complex between a disulfide-bridged affibody homodimer and the Aβ 1–40 peptide showed a cooperative binding mode, involving each affibody unit interacting with a distinct epitope on a single Aβ 1–40 peptide monomer, providing an explanation for the observations [
29]. It is tempting to speculate that the cysteine-containing affibody variants selected against the small BCMA target in this work form similar affibody-target complexes of 2:1 stoichiometry. One advantage of the affibody scaffold is that it does not contain any native cysteine, thus allowing for introduction of cysteine in the coding sequence post-selection, to provide opportunities for site-directed conjugation of various functional groups, including fluorophores, biotin, cytotoxic drugs and chelators. Thus, affibody variants that contain additional cysteines of importance for homodimerisation and target binding are less attractive for many biotechnological or medical applications. However, as shown for the Aβ 1–40 peptide-binding affibodies, tandem head-to-tail fusion of cysteine-to-serine mutants could still be of interest, but this was not evaluated for the cysteine-containing anti-BCMA affibodies in this study.
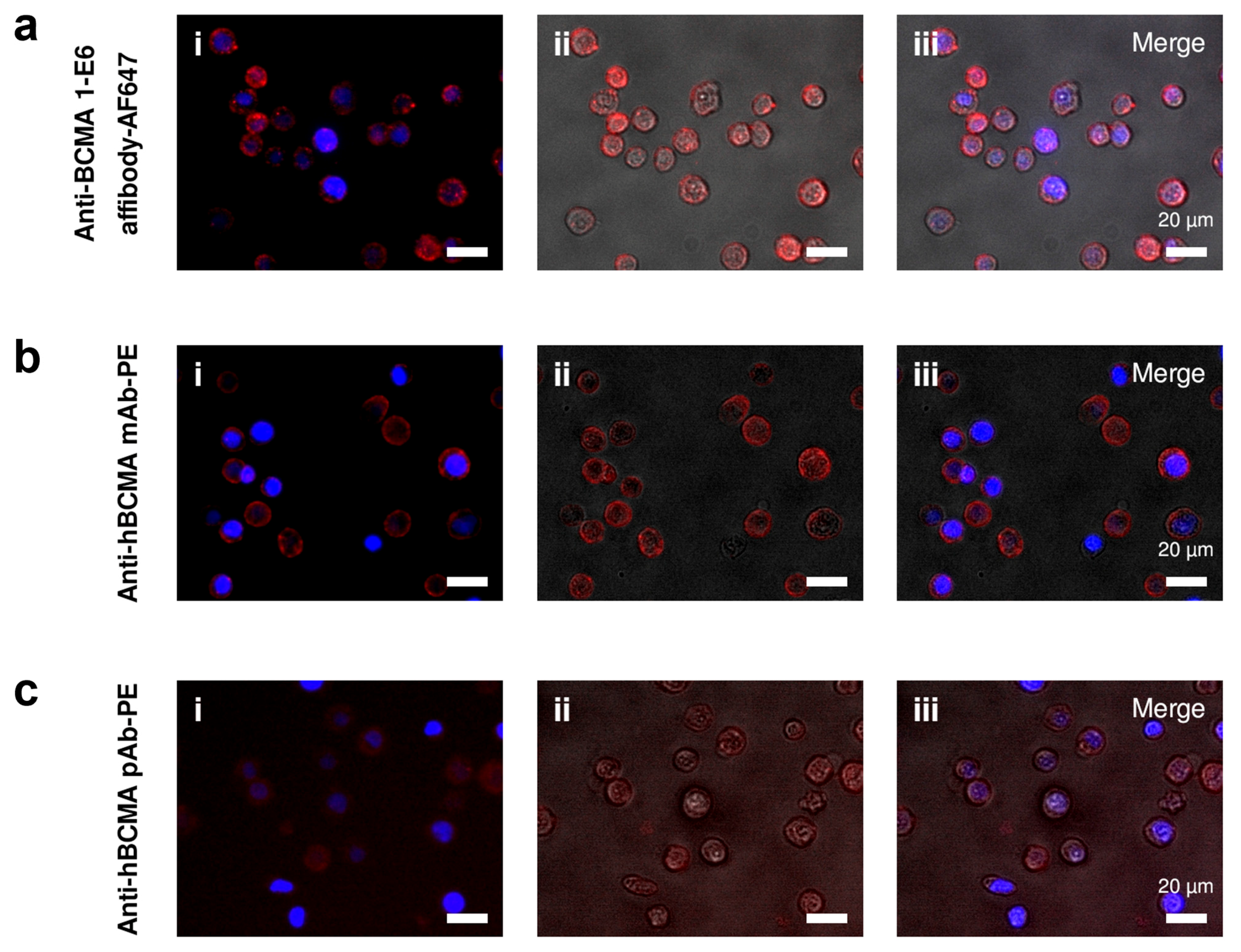
The second-generation 1-E6 affibody (1-E6-1-E6-His6) clearly demonstrated specific binding to BCMA expressed on MM.1s cells in flow cytometry and in fluorescence microscopy. Although not directly comparable due to the different fluorochromes used, the binding activity was analogous to that of BCMA-binding antibodies. In addition, no binding was observed for BCMA− SKBR3 cells. In these assays, it is inconclusive if any internalisation has occurred. If the 1-E6 homodimer induces BCMA internalisation it could affect its usage in therapeutic constructs, depending on the intended mode of action, e.g., internalisation could be beneficial for its use in drug conjugates, but negatively affect its use for immune cell activation.
The 1-E6 homodimer preparation used was randomly labelled using primary amines present in the protein sequence, meaning that the prepared sample was of a heterogeneous nature and that the binding to BCMA could have been affected. This was especially a risk considering the lysine residue in position 27, which was observed to be essential for binding in the alanine scanning. We did, however, not observe any diminished BCMA binding by labelled 1-E6 homodimer in SPR, but future studies with labelled protein could potentially be done with affibodies labelled in a site-specific manner as mentioned above, e.g., via introduction of a unique C-terminal cysteine, for coupling via maleimide chemistry. Nevertheless, our results show the feasibility of using a BCMA-binding affibody in both flow cytometry and microscopy assay settings, for the detection of BCMA-expressing cells. It should be noted that since affibodies are devoid of any Fc regions, the use of so-called Fc blockers, commonly used to compete out unwanted interactions between diagnostic antibodies and cell surface Fc receptors, are not needed.
A BCMA-directed CAR T cell therapy, anitocabtagene autoleucel (anito-cel; CART-ddBCMA), has been assessed in a Phase I clinical trial (NCT04155749) and shown an impressive 100% overall response rate (ORR) in heavily pretreated relapsed or refractory MM (RRMM) patients [
30]. The CAR component constitutes an 8 kDa BCMA-targeting D-domain, based on a
de novo-designed three-helix bundle scaffold. The D-domain scaffold does not share sequence similarity with affibodies, but both scaffolds appear similar in their overall topology [
31,
32]. Although its functional success may be due to several different factors, it cannot be ruled out that the structural characteristics of the CAR component plays an important role, including its small size and compact structure, which consequently allow for a higher CAR expression level during manufacturing and improved functional efficacy compared to a scFv CAR [
31]. Thus, the encouraging clinical data presented for anito-cel provides a compelling motivation for the further development and use of alternative, non-immunoglobulin scaffolds, including affibodies, in therapeutic applications. In fact, an initial study on the use of the anti-BCMA 1-E6 affibody—developed in the present work—as the tumour antigen-binding unit in a BCMA × CD16a dual engager for NK cell-mediated killing of MM.1s cells in vitro, has yielded encouraging results [
22].
Anti-drug antibodies (ADAs) may be elicited against proteins drugs, including humanised or even fully human monoclonal antibodies [
33]. Such antibodies may affect both safety and efficacy. The non-human origin of the affibody scaffold may therefore raise immunogenicity concerns. In two clinical studies (52 weeks and up to three years, respectively) of the affibody-based drug Izokibep (an IL-17A inhibitor), ADAs were detected in some individuals. However, these responses were reported not to affect pharmacokinetic (PK) parameters, safety, or efficacy [
34,
35]. This holds promise that the described anti-BCMA affibody could be included as the targeting unit in drugs aimed for MM therapy in humans.
In summary, this work demonstrates the successful isolation and development of anti-BCMA affibodies, using phage display technology. Characterisation of the second-generation anti-BCMA clone 1-E6 demonstrated its high thermostability and specific, high-affinity binding to BCMA, highlighting the potential for broad applicability of this small, non-immunoglobulin reagent in e.g., diagnostic and medical applications.
4. Materials and Methods
4.1. First-Generation Phage Library and Stock Preparation
Preparation of an M13 phage library of affibodies and the subsequent production of a naïve affibody-displaying phage stock based on the pAffi-1 phagemid [
28] were done as described in detail in [
22]. The corresponding phage library preparation and phage stock production using the pAffi-100 phagemid were performed similarly. pAffi-100 differs from the pAffi-1 phagemid in that it contains a trypsin protease cleavage site before the amber stop codon, which is followed by full-length M13 phage coat protein III.
4.2. Selection of First-Generation Binders to BCMA Using Phage Display
Four selection cycles were performed in three parallel tracks, with different selection conditions for each track. Biotinylated human BCMA-rFc (human TNFRSF17/BCMA/CD269 Protein (His & human IgG1 Fc tag, Biotinylated), cat. no. 10620-H03H-B, Sino Biological, Eschborn, Germany), corresponding to residues 1–54 of Uniprot entry Q02223, was used as the target antigen. Biotinylated human BCMA-rFc (150 nM (cycle 1), 100 nM (cycle 2) and 50 nM (cycles 3–4)) was immobilised on 1.5 mg streptavidin (SA)-coated paramagnetic beads (Dynabeads M-280 Streptavidin, cat. no. 11205D, Invitrogen, Waltham, MA, USA) for 1 h at room temperature (RT) and under constant end-over-end (eoe) rotation. Affibody-displaying phage stocks generated from the libraries based on pAffi-1 and pAffi-100 phagemids were added to the immobilised biotinylated human BCMA-rFc (pre-blocked with 1% (w/v) bovine serum albumin (BSA) in PBS-T (PBS (150 mM NaCl, 8 mM Na2HPO4, 2 mM NaH2PO4) supplemented with 0.05% (v/v) Tween-20, pH 7.4)) for selection incubation for 3 h (cycle 1), 4 h (cycle 2), 2 h (cycle 3) or 1 h and 20 min (cycle 4), at RT with eoe rotation. Prior to the selection incubation step in cycles 2–4, negative selection was performed to remove phage carrying binders against SA and Fc, by pre-incubating the phage stocks in PBS-T with 0.1% (w/v) BSA and SA-beads containing biotinylated trastuzumab mAb (contains Fc region) (Herceptin, Roche, Basel, Germany) for 1 h at RT with eoe rotation.
After several washes with PBS-T (eoe at RT), bound phage was eluted with 0.3 M acetic acid, pH 2.8, for 15 min eoe at RT, followed by neutralisation with an equal volume of 1 M Tris-HCl, pH 8; or eluted with 0.25 mg/mL trypsin (Gibco Life Technologies, Carlsbad, CA, USA) for 30 min, eoe at RT. After selection cycles 1–3, helper phage M13K07 for pAffi-1 or KM13 for pAffi-100 were used to amplify new phage stocks in E. coli XL-1 Blue cells (Agilent, Santa Clara, CA, USA), for use as the input phage in the subsequent selection cycle. Phage stock titres were measured by infecting E. coli XL-1 Blue and performing spot titration on carbenicillin plates (30 g/L blood agar, 100 µg/mL carbenicillin). Colony PCR was used to analyse the percentage of infected colonies carrying phagemids with the correct affibody insert size.
4.3. Surface Plasmon Resonance Characterisation of First-Generation Candidate BCMA-Binding Clones
ELISA-positive and unique clones (
Supplementary Materials and Methods) were subcloned for soluble expression in
E. coli as fusion proteins with an N-terminal His
6 tag and a C-terminal ABD (His
6-affibody-ABD format). DNA fragments encoding the clones of interest were first amplified by PCR from the library phagemids with specific primers introducing restriction recognition sites for Xhol and AscI restriction enzymes. The fragments were digested with XhoI and AscI and then ligated to a T7 promoter-based
E. coli expression vector digested with the same restriction enzymes. The resulting DNA constructs were sequence verified using Sanger sequencing (Microsynth) and transformed into
E. coli BL21(DE3) for expression. Cells were chemically lysed, and His
6-tagged fusion proteins were purified under denaturing conditions using HisPur Cobalt IMAC Resin (cat. no. 89966, Thermo Scientific, Waltham, MA, USA) for IMAC purification. Following purification, the protein buffer was exchanged to PBS using PD-10 desalting columns (cat. no. 17085101, Cytiva, Uppsala, Sweden). Concentrations were estimated based on absorbances measured at 280 nm and using molecular weights (MW) and extinction coefficients calculated from the amino acid sequences. SDS-PAGE analysis under reducing conditions was performed to confirm the purities and concentrations.
A Biacore T200 instrument (Cytiva, Uppsala, Sweden) was used to analyse the real-time interactions of the fusion proteins with BCMA, by SPR. The protein ligand human BCMA-rFc (cat. no. 10620-H15H, Sino Biological, Eschborn, Germany) was immobilised on a Series S CM5 sensor chip (Cytiva, Uppsala, Sweden) by amine coupling, using the manufacturer’s instructions. One flow cell was activated and deactivated to be used as a reference cell. The affibody fusion proteins were injected at 200 nM over the flow cells. After each run cycle, the flow cells were regenerated with 10 mM HCl. PBS-T was used as the running buffer and sample buffer.
4.4. Alanine Scanning of the First-Generation BCMA-Binding Clone Fa-G6
Fa-G6 was subcloned into a T7 promoter-based E. coli expression vector for soluble expression of proteins with an N-terminal His6 tag (His6-affibody format), similarly as described above. The expression plasmid containing the Fa-G6 construct was subsequently used as the DNA template for alanine scanning mutagenesis. A total of 14 individual mutagenesis PCR reactions were performed to substitute the original codons in the 14 variable positions (residues 9, 10, 11, 13, 14, 17, 18, 24, 25, 27, 28, 31, 32, and 35) to alanine codons, using primers designed for each position. The 14 alanine substitution variants were sequence verified using Sanger sequencing (Microsynth, Göttingen, Germany). E. coli BL21(DE3) cells were separately transformed with individual plasmids encoding the N-terminally His6-tagged alanine variants, as well as the Fa-G6 wild-type construct. Production, purification under denaturing conditions, buffer exchange, absorbance measurement, and SDS-PAGE analysis were performed as described above.
A Biacore T200 instrument (Cytiva, Uppsala, Sweden) was used to analyse the interactions of the Fa-G6 wild-type and the 14 alanine variants with BCMA in real time. Immobilisation of the protein ligand human BCMA-rFc (cat. no. 10620-H15H, Sino Biological, Eschborn, Germany), generation of a reference cell, binding analysis and regeneration of flow cells were performed as described above. PBS-T was used as the running buffer and sample buffer.
4.5. Second-Generation Library Construction, Selection and Monoclonal Phage ELISA
Two M13 phage display second-generation library (Library A and Library B) genes were designed, based the Fa-G6 clone, and synthesised as oligonucleotides (121 bases) encoding amino acids 3–41 (reverse complementary strand) of the Z domain [
36]. The oligonucleotides were synthesised by ELLA Biotech (Furstenfeldbruck, Germany) using custom mixtures of trinucleotide codon building blocks in the 14 variable positions (residues 9, 10, 11, 13, 14, 17, 18, 24, 25, 27, 28, 31, 32 and 35). Validation of the library gene designs was done by sequencing (Microsynth, Göttingen, Germany) of 96 individually obtained colonies following a small-scale test ligation: Library A and Library B oligonucleotides were PCR-amplified using specific primers to introduce restriction recognition sites for XhoI and NheI restriction enzymes, then XhoI/NheI digested and ligated to XhoI/NheI digested pAffi-1 (Library A) or pAffi-100 (Library B), and finally transformed into electrocompetent ER2738
E. coli cells (F’,
glnV amber suppressor) (Lucigen, San Antonio, TX, USA) by electroporation. Following library validation, full-scale libraries were generated as described above, using ligation reaction mixes of 5.4 µg of XhoI/NheI-digested PCR products of Library A or Library B with 30 µg of XhoI/NheI-digested pAffi-1 or pAffi-100, respectively. Recovered post-electroporation transformants were analysed by titration via spreading of dilution series on ampicillin plates (30 g/L blood agar, 100 µg/mL ampicillin) and cultivated overnight in TSB+Y supplemented with 1.5% (
w/
v) glucose and 100 µg/mL ampicillin. Overnight cultures were pelleted by centrifugation and resuspended in cold 40% (
v/
v) glycerol, followed by freezing at -80 °C. Library size (diversity) was calculated from the post-electroporation titrations, which for Library A in pAffi-1 was approximately 3.4 × 10
7 and for Library B in pAffi-100 was approximately 5.9 × 10
7.
Second-generation affibody displaying phage stocks based on Library A in pAffi-1 and Library B in pAffi-100 were prepared as described previously [
22]. Recombinant, biotinylated human BCMA (human TNFRS/BCMA/CD269 Protein (His & Fc tag), Biotinylated, cat. no. 10620-H03H-B, Sino Biological, Eschborn, Germany), corresponding to residues 1–54 of Uniprot entry Q02223, was used as the target antigen. Three selection cycles were performed in nine parallel tracks (three using Library A and six using Library B), with different selection conditions for each track (
Supplementary Materials and Methods).
Following selection cycle 3, 20–21 bacterial colonies generating the correct affibody insert size in colony PCR and equally representing the nine selection tracks were individually grown for monoclonal phage ELISA. MaxiSorp ELISA plates (Clear Flat-Bottom Immuno Nonsterile 384-Well Plates, cat. no. 464718, Thermo Fisher Scientific, Waltham, MA, USA) were coated with 1 µg/mL target antigen biotinylated human BCMA-Fc or control antigens: 20 µg/mL HSA, 10 µg/ul SA or 10 µg/mL trastuzumab (Fc control) (Herceptin, Roche, Basel, Switzerland); all in 100 mM sodium carbonate buffer, pH 9.6, for 16–18 h at 4 °C with slow shaking. The amplified monoclonal phage stocks were subjected to ELISA, as described following the naïve selection (
Supplementary Materials and Methods). Candidates that were considered ELISA-positive clones had high HSA signals and relatively high signals to BCMA, compared to signals observed for SA and trastuzumab (Fc control) and the unrelated antigen control. Following ELISA screening, a total of 120 ELISA-positive clones were sent for DNA sequencing by Sanger sequencing (Microsynth).
4.6. Characterisation of Second-Generation Candidate BCMA-Binding Clones
A subset of 18 s-generation BCMA-binding clones, equally representing the nine selection tracks, were chosen for expression as soluble proteins with a C-terminal His6 tag (affibody-His6 format). DNA fragments encoding the 18 clones and the Fa-G6 wild-type clone were amplified with specific primers, designed to introduce overhangs complementary to ends of a linearised T7 promoter-based E. coli expression vector for cloning using In-Fusion HD Cloning Kit (TakaraBio, Gothenburg, Sweden). The resulting constructs were sequence-verified using Sanger sequencing (Microsynth). Production of the 18 constructs and the Fa-G6 wild-type clone, purification under denaturing conditions, buffer exchange, absorbance measurement and SDS-PAGE analysis were performed as described above.
A Biacore T200 instrument (Cytiva, Uppsala, Sweden) was used to analyse the interactions of the second-generation binding clones and the Fa-G6 wild-type clone with BCMA in real time. Immobilisation of the protein ligand human BCMA-rFc (cat. no. 10620-H15H, Sino Biological, Eschborn, Germany), generation of a reference cell, binding analysis, and regeneration of surfaces were performed as described above, but with binding clones diluted to 100 nM for binding analysis. PBS-T was used as the running buffer and sample buffer.
Secondary structure contents and thermal denaturation profiles were analysed as described for the first-generation clones (
Supplementary Materials and Methods), but using samples diluted to 0.2 mg/mL.
4.7. Determination of Binding Kinetics Using SPR
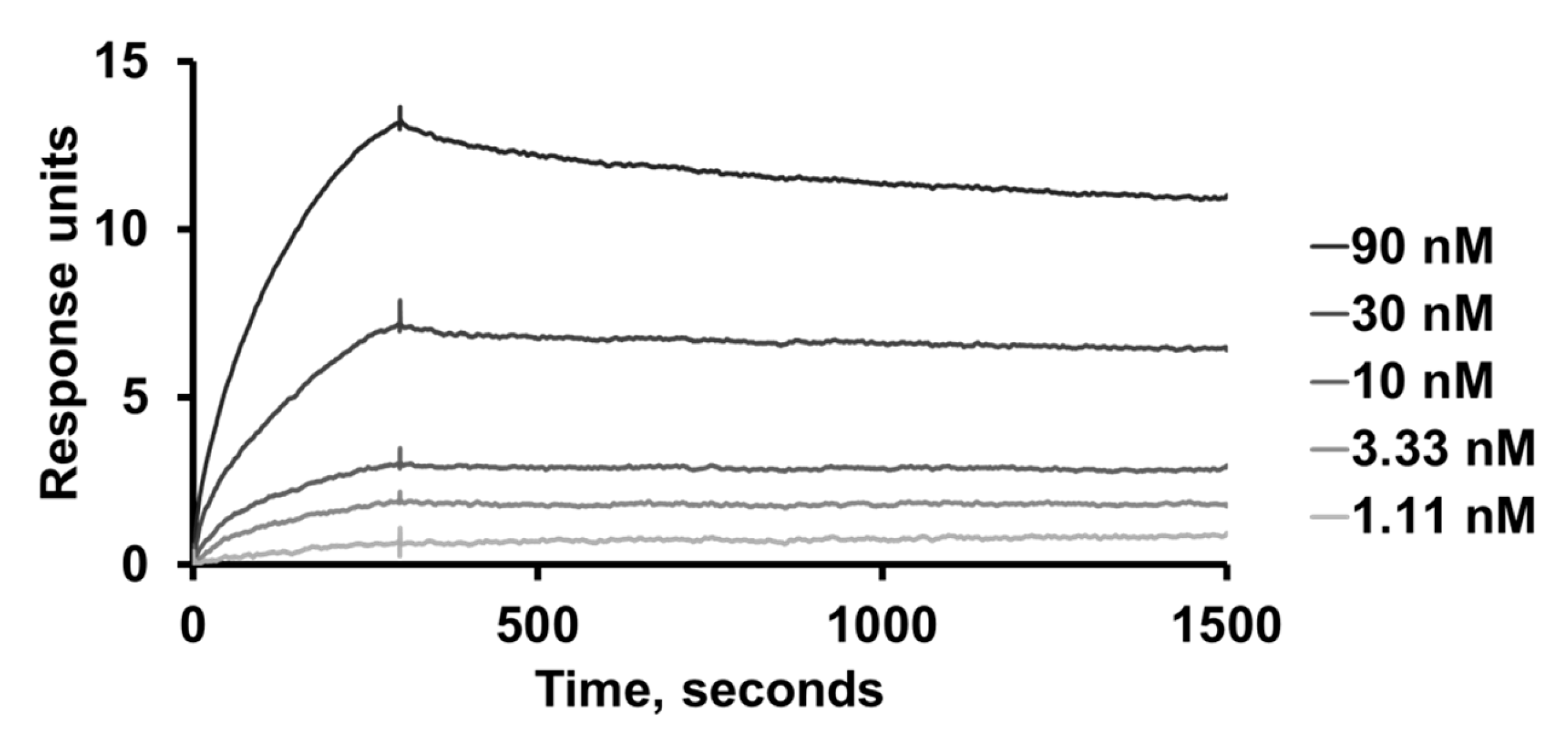
Kinetic analysis of the second-generation clone 1-E6 was conducted using SPR and a Biacore T200 instrument (Cytiva, Uppsala, Sweden). Immobilisation of the protein ligand human BCMA-rFc (cat. no. 10620-H15H, Sino Biological, Eschborn, Germany), generation of a reference cell, and regeneration of flow cells were performed as described above, but with a lower surface density. PBS-T was used as the running buffer and sample buffer. Duplicate 1:3 serial dilutions of 1-E6 (affibody-His6 format) ranging 1.11–90 nM, were injected over the surfaces. Kinetic constants were estimated from the resulting sensorgrams using BIAevaluation software (Cytiva, Uppsala, Sweden) and assuming 1:1 binding.
4.8. Epitope Binning Analyses by SPR
For epitope binning analyses of the 1-E6 affibody, blocking assays were set up using a Biacore T200 instrument (Cytiva, Uppsala, Sweden). Immobilisation of the protein ligand human BCMA-rFc (cat. no. 10620-H15H, Sino Biological, Eschborn, Germany), generation of a reference cell, and regeneration of flow cells were performed as described above. PBS-T was used as the running buffer and sample buffer. 1 µM of 1-E6 (affibody-His6 format) was injected over immobilised human BCMA-rFc, followed by an injection of running buffer, 100 nM of APRIL (R&D systems, Minneapolis, MN, USA), or 25 nM belantamab (Belantamab Biosimilar—Research Grade, cat. no. ICH5011, ichorbio, Wantage, UK). Reference runs were performed where running buffer was injected, followed by either 100 nM APRIL or 25 nM belantamab.
4.9. Flow Cytometry Analyses of BCMA-Binding 1-E6 Homodimer
Cultured MM.1s and SKBR3 cells were detached from cultivation flasks with Versene solution (cat. no. 150400-066, Gibco, Thermo Scientific, Waltham, MA, USA). Cells were washed with PBS, and approximately 105 cells were seeded per well in a 96-well plate (TC treated V-bottom, Corning, Thermo Scientific, Waltham, MA, USA). The cells were stained for viability (LIVE/DEAD Fixable Aqua Dead Cell Stain Kit, cat. no. L34957, Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) for 10 min at 4 °C in the dark. Cells were washed twice with Flow staining buffer (eBioscience™ Flow Cytometry Staining Buffer, cat. no. 00-4222-26, Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA), then blocked with Fc Receptor Blocking Reagent (Human Seroblock, cat. no. BUF070B, Bio-Rad). 200 nM AF647-labelled 1-E6 affibody (1-E6-1-E6-His6) or control was added to the cells and incubated for 20 min at 4 °C in the dark. The controls used were AF647-labelled affibody control (Alexa Fluor 647 NHS Ester (Succinimidyl Ester), cat. no. A20006, Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) (binding unrelated targets HER2 and CD16a), PE-labelled anti-BCMA monoclonal antibody (PE anti-human CD269/BCMA Antibody, cat. no. 357503, Biolegend, San Diego, CA, USA), anti-BCMA polyclonal antibody (Human BCMA/TNFRSF17 PE-conjugated Antibody, cat. no. FAB193P, R&D Systems, Minneapolis, MN, USA) or corresponding isotype control antibodies (PE Mouse IgG2a, κ Isotype Ctrl (FC) Antibody, cat. no. 400213, Biolegend, San Diego, CA, USA; Goat IgG PE-conjugated Antibody, cat. no. IC108P, R&D Systems, Minneapolis, MN, USA). Cells were washed twice with Flow staining buffer, fixed with Fixation buffer (Flow Cytometry Fixation Buffer, cat. no. FC004, R&D Systems, Minneapolis, MN, USA) for 10 min at RT, then washed again and resuspended in Flow staining buffer for flow cytometric measurement (Sony ID7000 Spectral Cell Analyzer). Mean fluorescence intensity (MFI) was plotted and analysed using FlowJo Software (BD Life Sciences, Ashland, OR, USA).
4.10. Fluorescence and Brightfield Microscopy
SKBR3 cells were prepared by enzymatic detachment (Gibco™ TrypLE™ Express Enzyme (1X), phenol red, cat. no. 12605-028, Gibco, Thermo Scientific, Waltham, MA, USA), seeding at approximately 2 × 104 per well a 96-well plate (Thermo Scientific™ Nunc™ MicroWell™ 96-Well, Nunclon Delta-Treated, Flat-Bottom Microplate, cat. no. 10212811, Thermo Scientific, Waltham, MA, USA), and allowing the cells to adhere and grow in the plate for 7 days. Cultured MM.1s cells were detached from cultivation flasks with Versene solution (cat. no. 150400-066, Gibco, Thermo Scientific, Waltham, MA, USA). Approximately 105 cells per well were transferred to a 96-well conical bottom plate (Nunc™ 96-Well Polystyrene Conical Bottom MicroWell™ Plates, cat. no. 249935, Thermo Scientific, Waltham, MA, USA).
The cells were washed twice with PBS, then blocked with 1% (v/v) goat serum (cat. no. G9023, Sigma-Aldrich, Saint Louis, MO, USA) in PBS for 5 min at RT. 1 µM AF647-labelled 1-E6 affibody (1-E6-1-E6-His6) or control was added to the cells and incubated for 20 min at 4 °C in the dark. The controls were 1 µM control affibody (AF647-labelled anti-HER2/CD16a affibody), or anti-BCMA antibody (PE anti-human CD269/BCMA Antibody, cat. no. 357503, Biolegend, San Diego, CA, USA), anti-BCMA polyclonal antibody (Human BCMA/TNFRSF17 PE-conjugated Antibody, cat. no. FAB193P, R&D Systems, Minneapolis, MN, USA) or corresponding isotype control (PE Mouse IgG2a, κ Isotype Ctrl (FC) Antibody, cat. no. 400213, Biolegend, San Diego, CA, USA; Goat IgG PE-conjugated Antibody, cat. no. IC108P, R&D Systems, Minneapolis, MN, USA). Anti-BCMA antibodies and isotype controls were added at 5x the recommended reagent volume per cell according to the manufacturers. Cells were washed twice with PBS, then fixed with 4% paraformaldehyde (cat. no. 15424389, Thermo Scientific, Waltham, MA, USA) for 10 min at RT, then washed twice with PBS again. Cell nuclei were stained with DAPI (cat. no. D9542, Sigma-Aldrich, Saint Louis, MO, USA) for 10 min at RT. Cells were washed with PBS for 5 min. Lastly, the MM.1s cells were transferred to a 96-well flat-bottom plate (Thermo Scientific™ Nunc™ MicroWell™ 96-Well, Nunclon Delta-Treated, Flat-Bottom Microplate, cat. no. 10212811, Thermo Scientific, Waltham, MA, USA). Cell imaging was done using widefield microscopy (DMI6000 B microscope, Leica Microsystems, Wetzlar, Germany).


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}