

Review of Four Refined Clinical Entities in Hereditary Retinal Disorders from Japan
Abstract
1. Background
2. Introduction
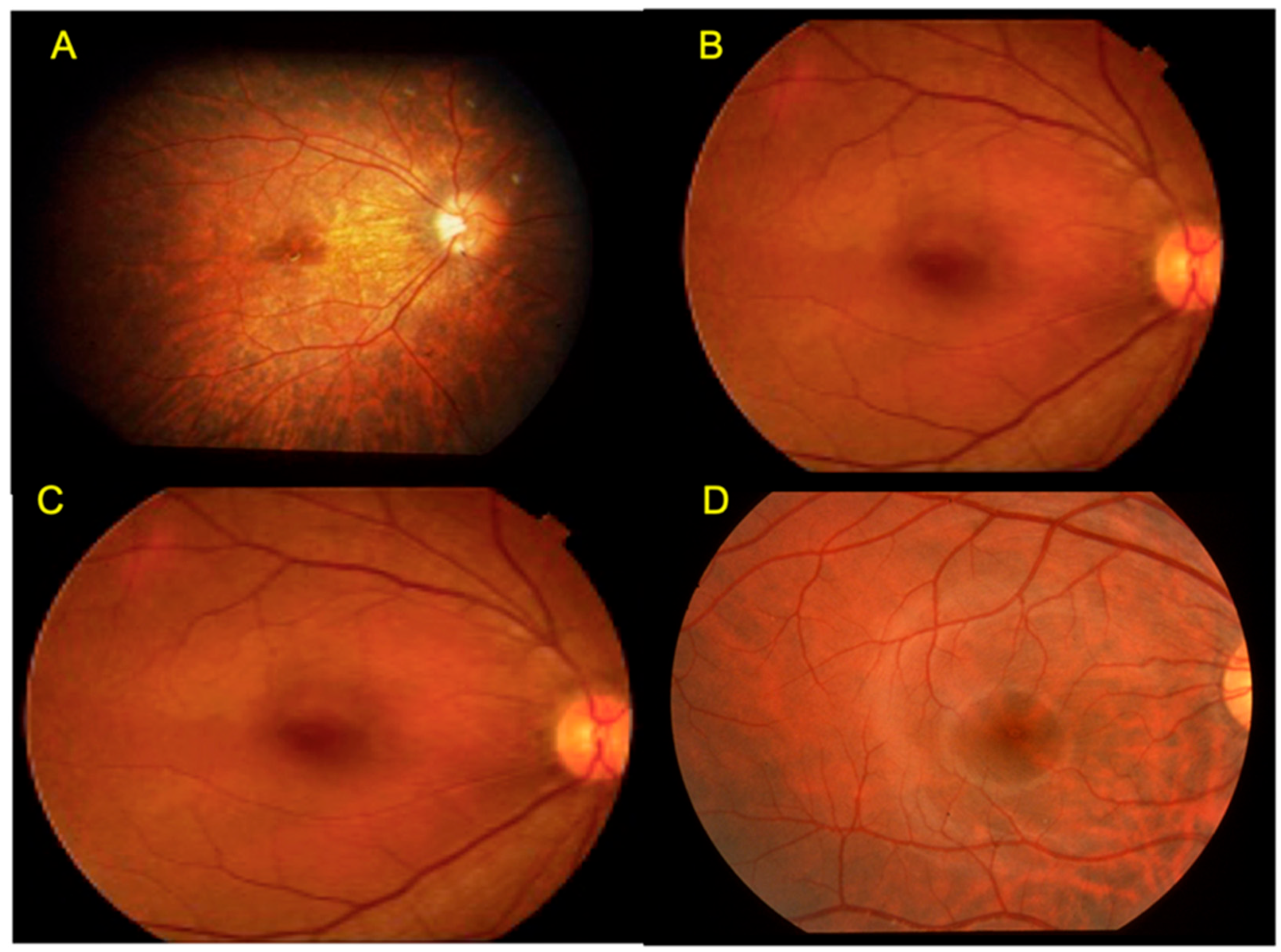
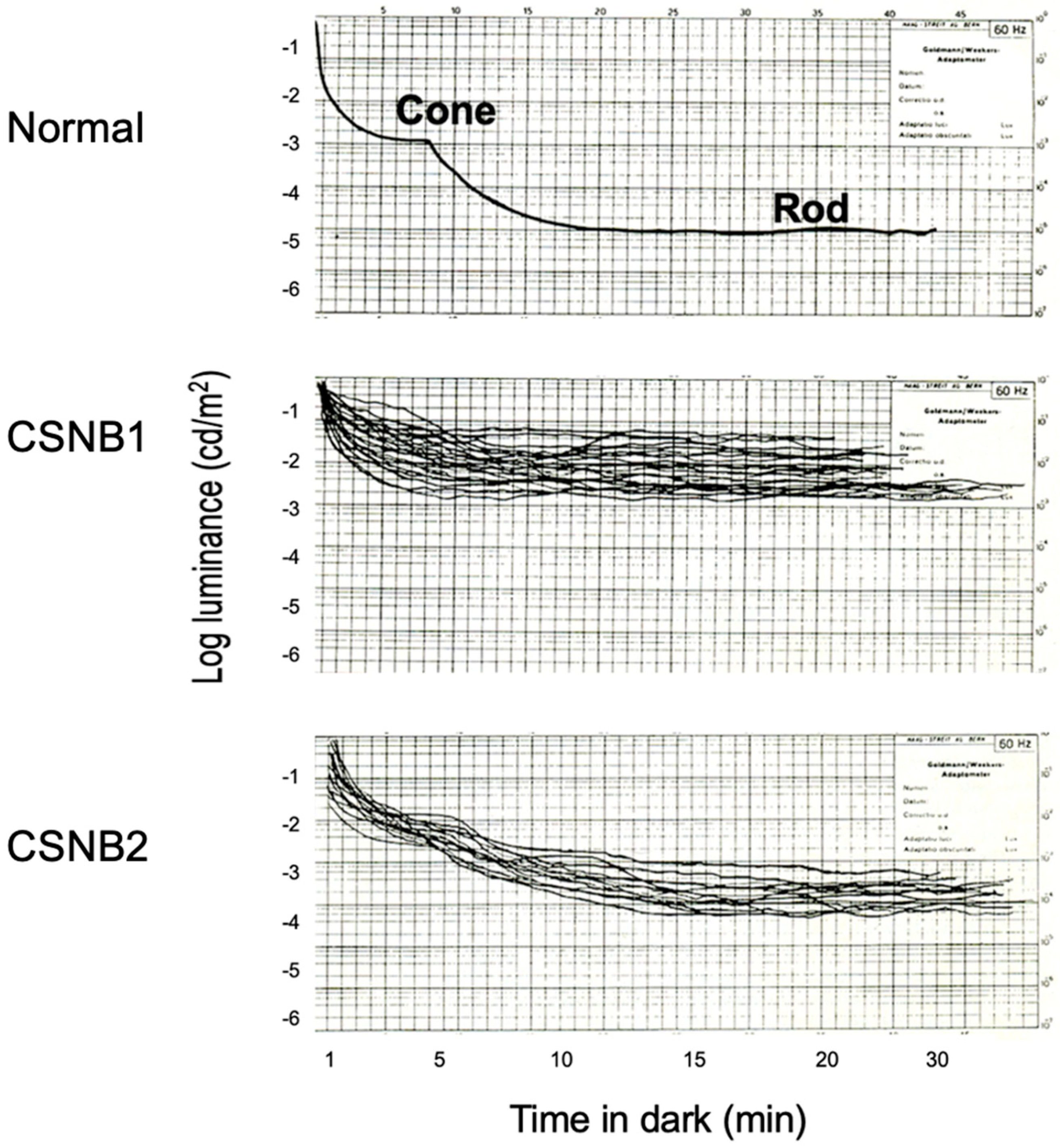
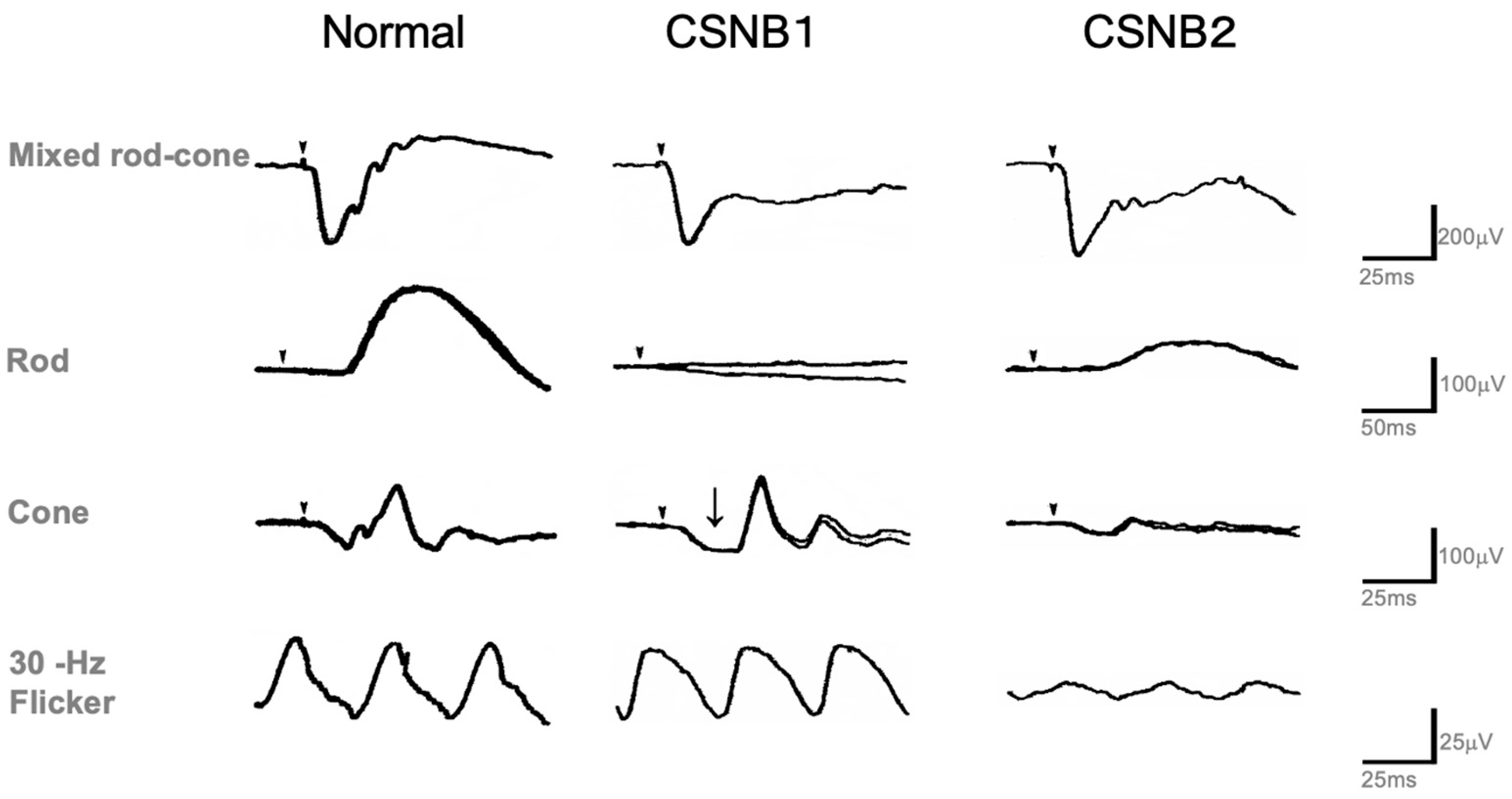
2.1. Congenital Stationary Night Blindness (CSNB) with Normal Fundus: A New Classification
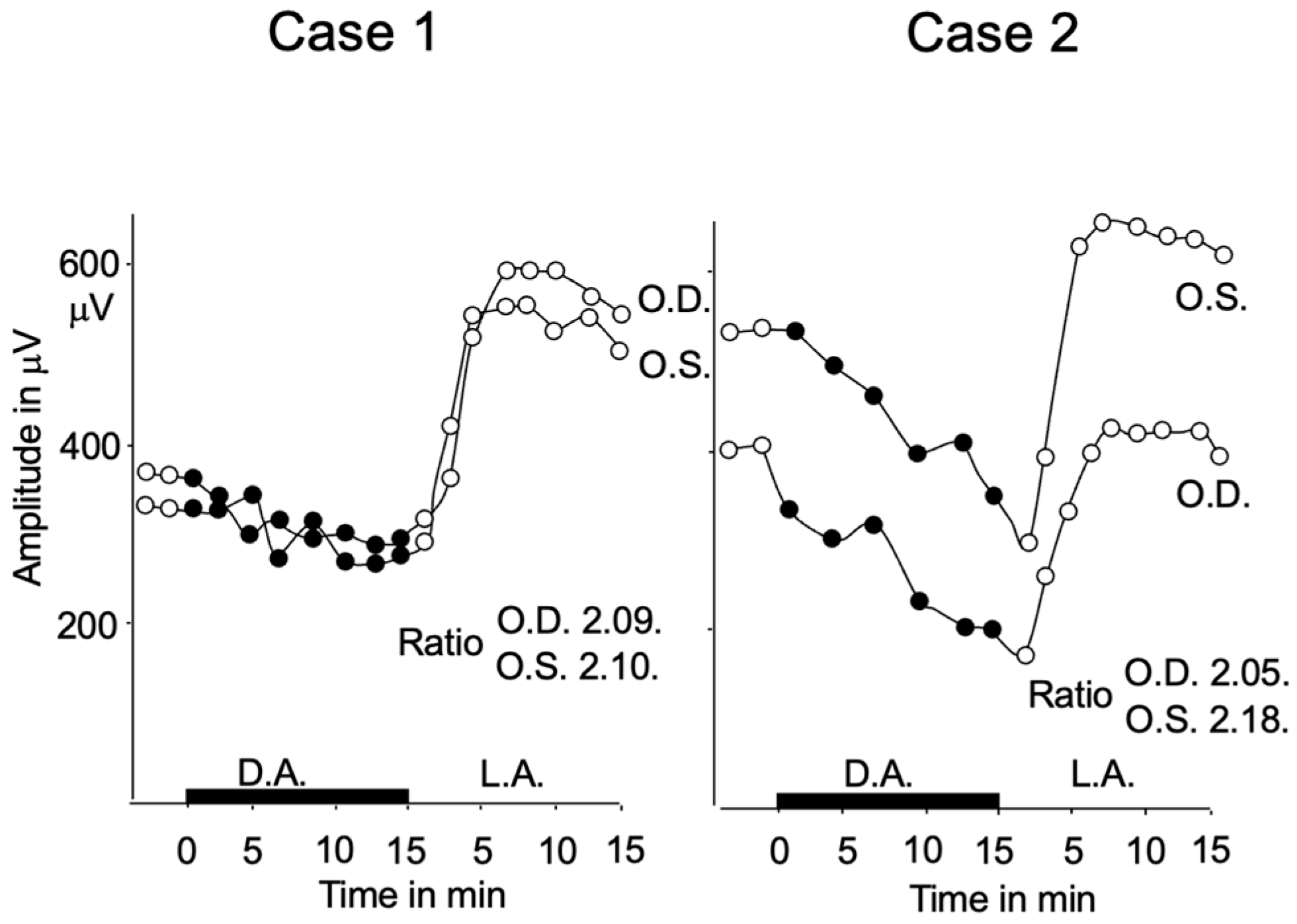
2.2. Subjective Dark Adaptation
2.3. Visual Acuity and Refractive Error
2.4. Standard Full-Field ERG (ISCEV Protocol)
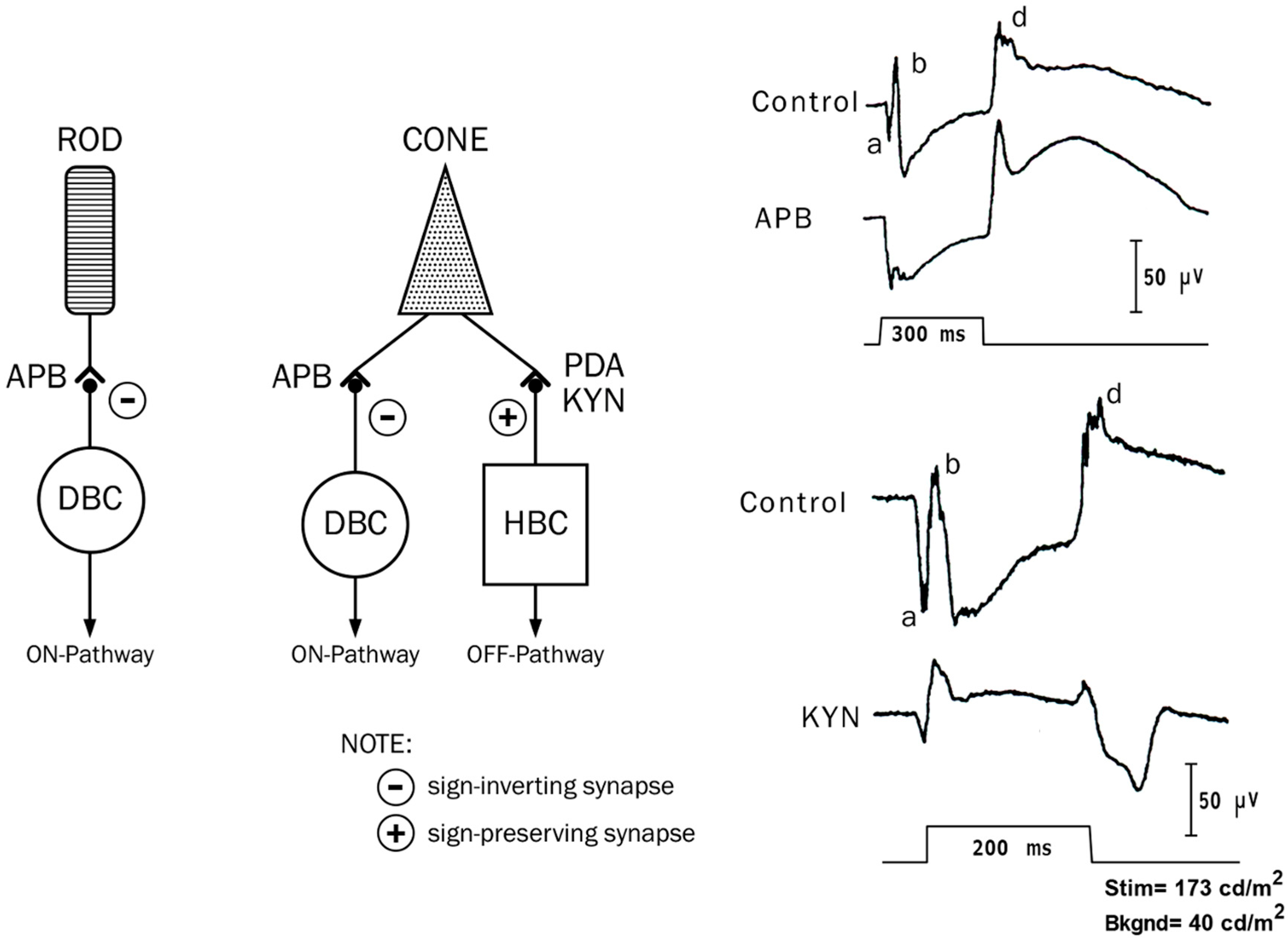
2.5. ON and OFF Responses in Photopic ERG
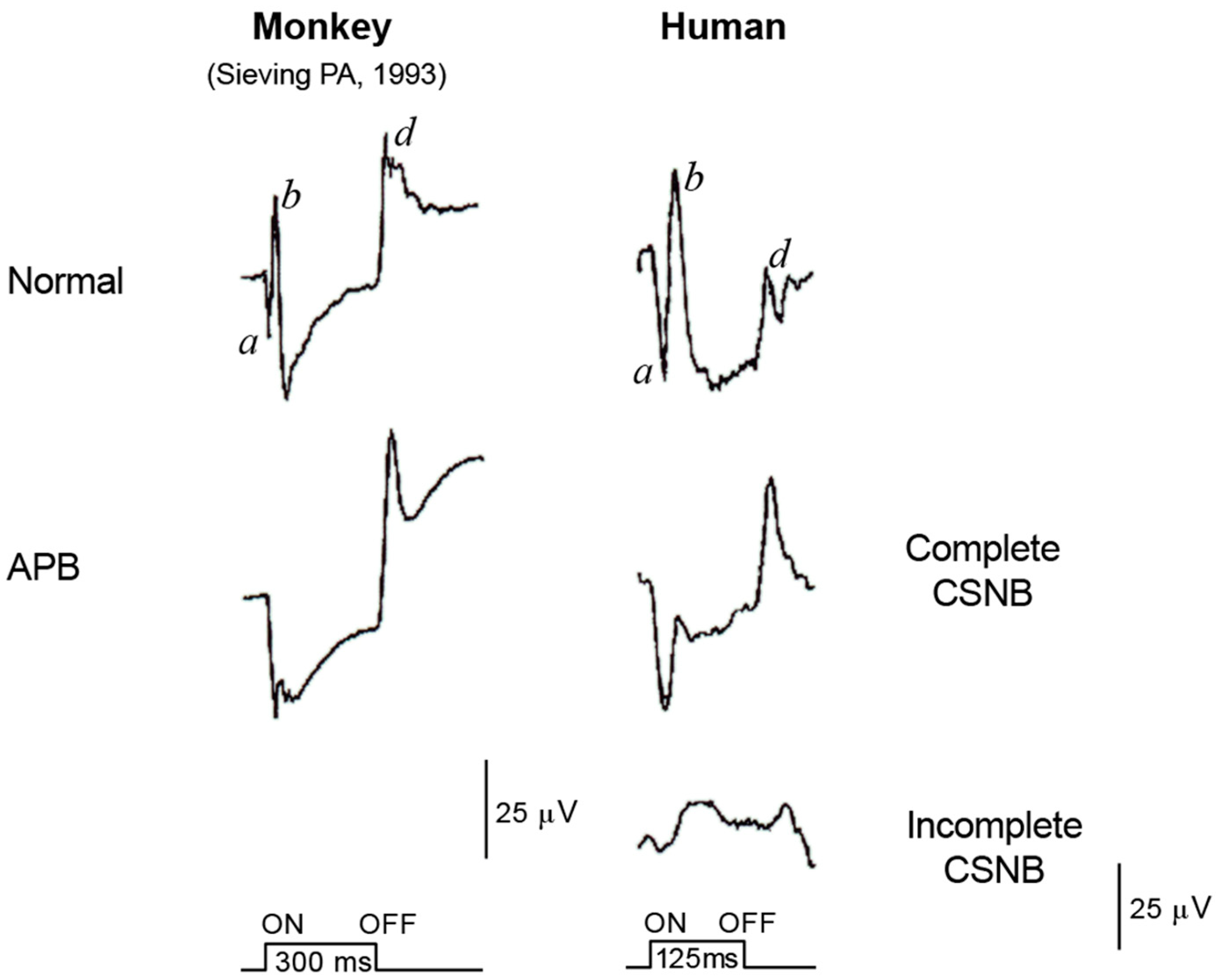
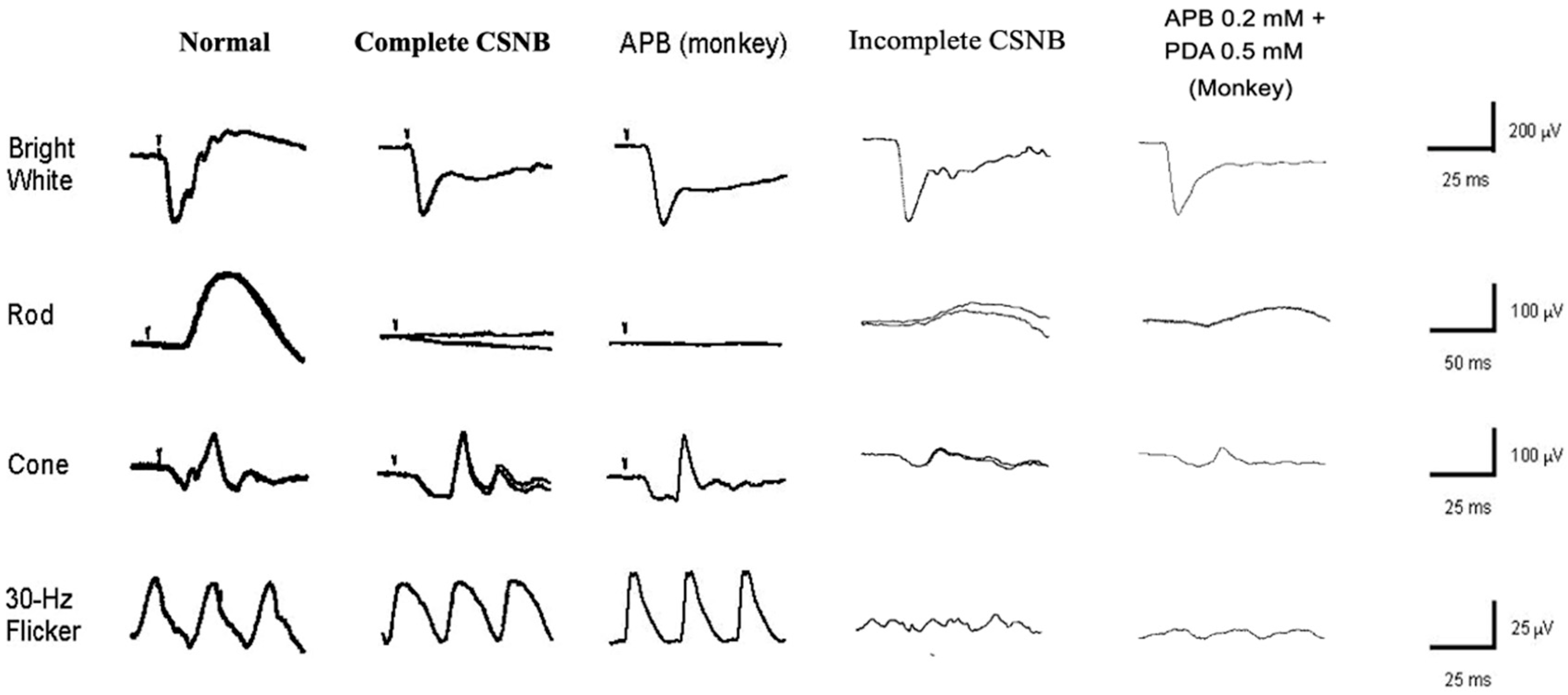
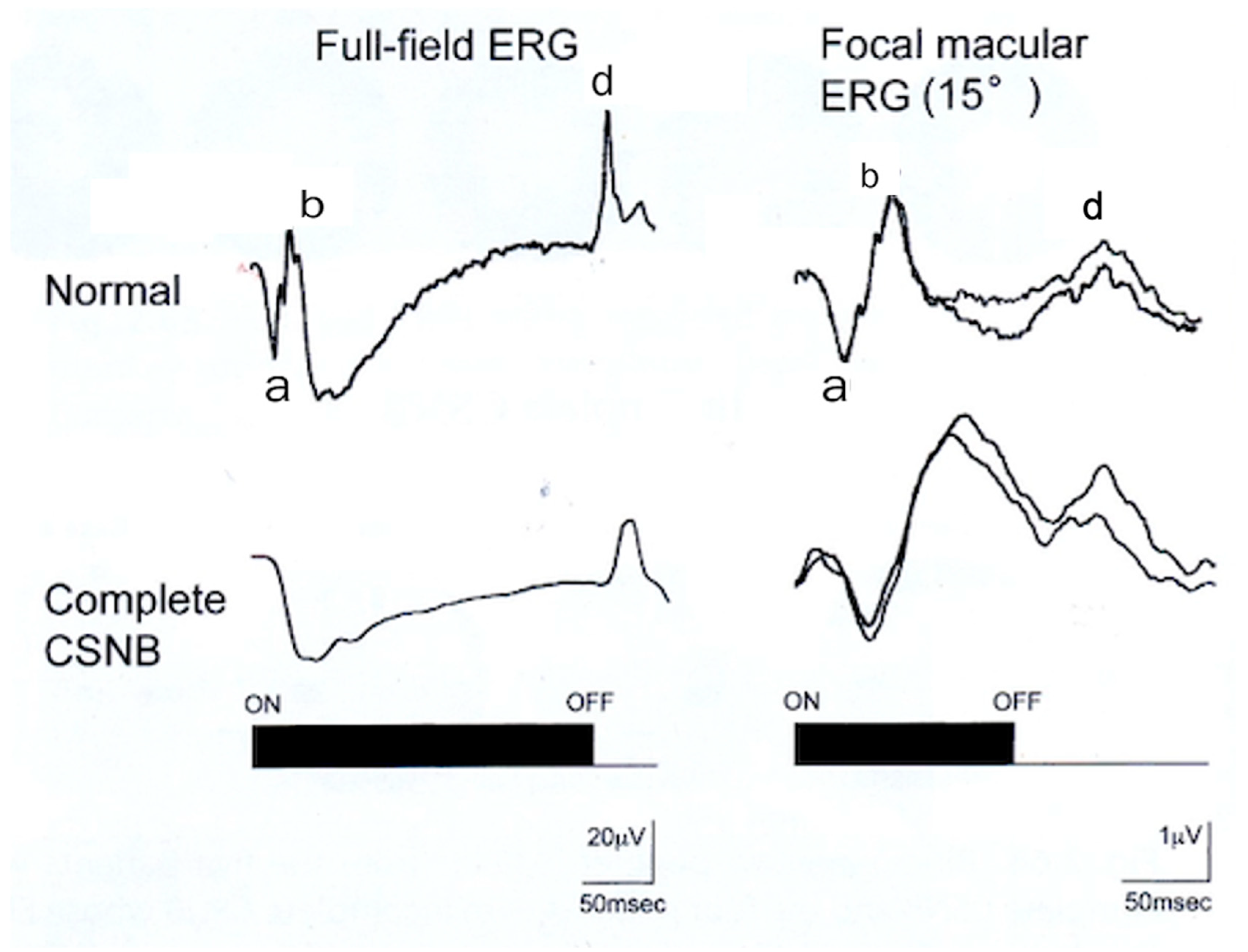
2.6. ON and OFF Responses in CSNB
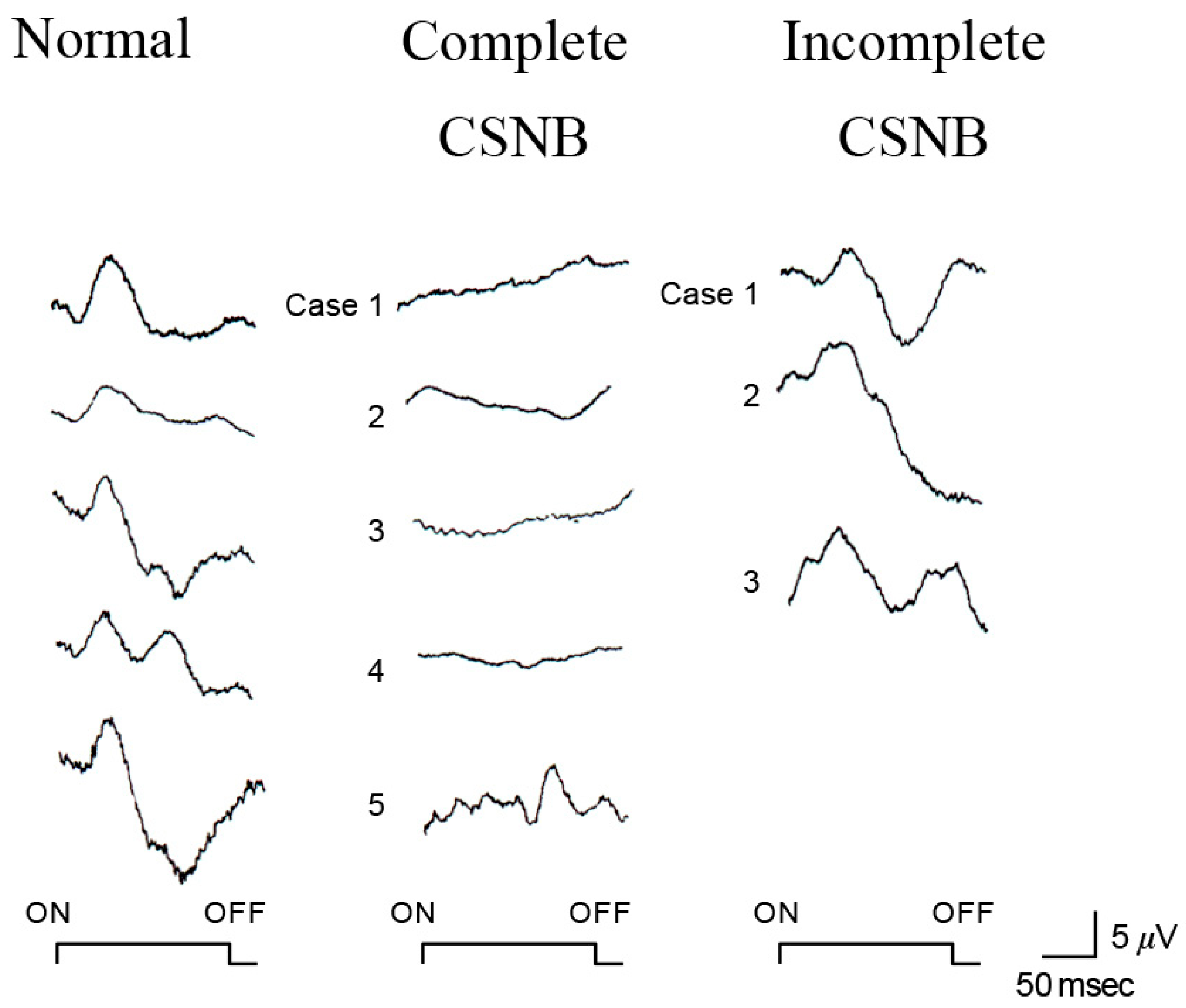
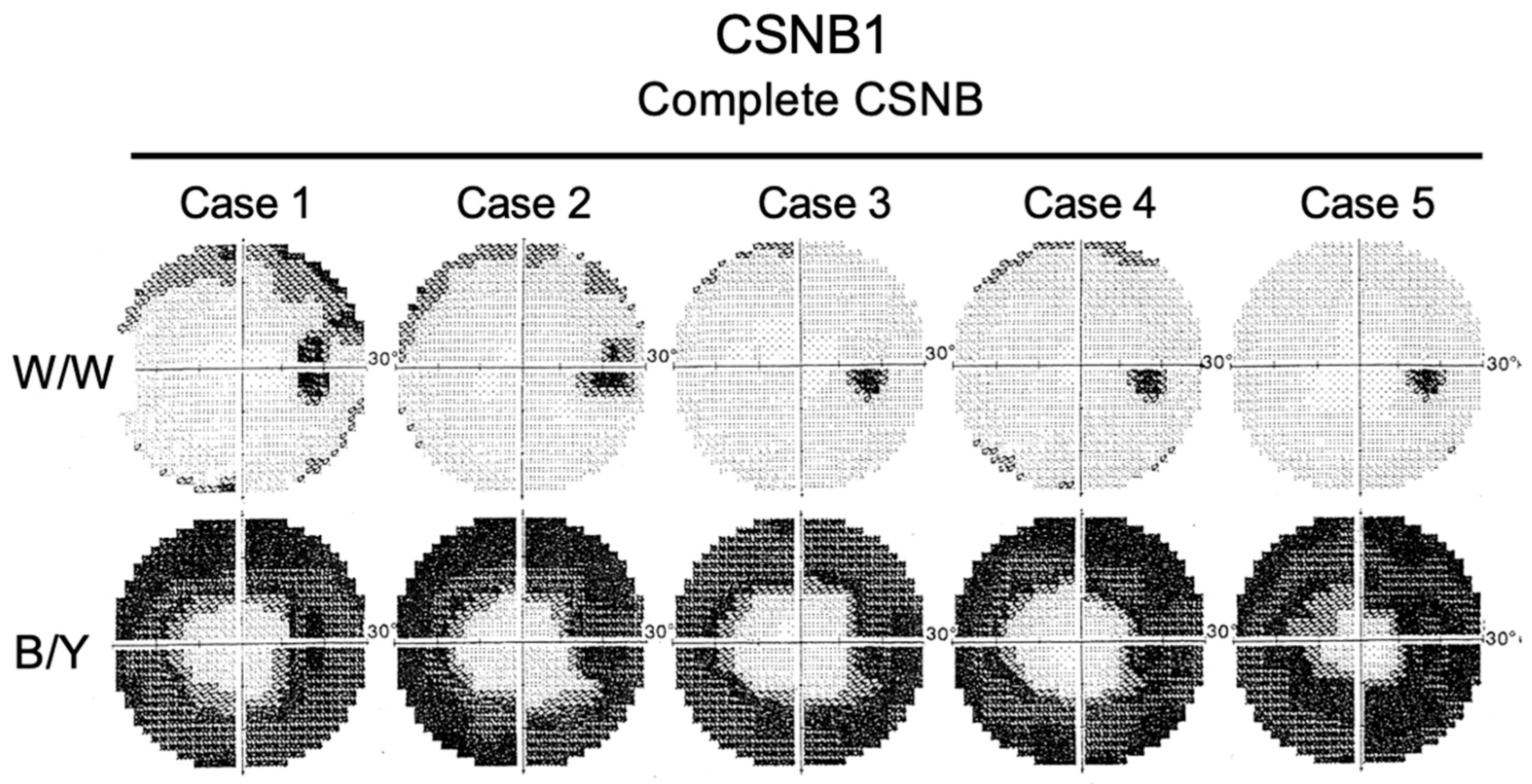
2.7. Short-Wavelength Cone (S-Cone) ERG and Subjective Blue Sensitivity
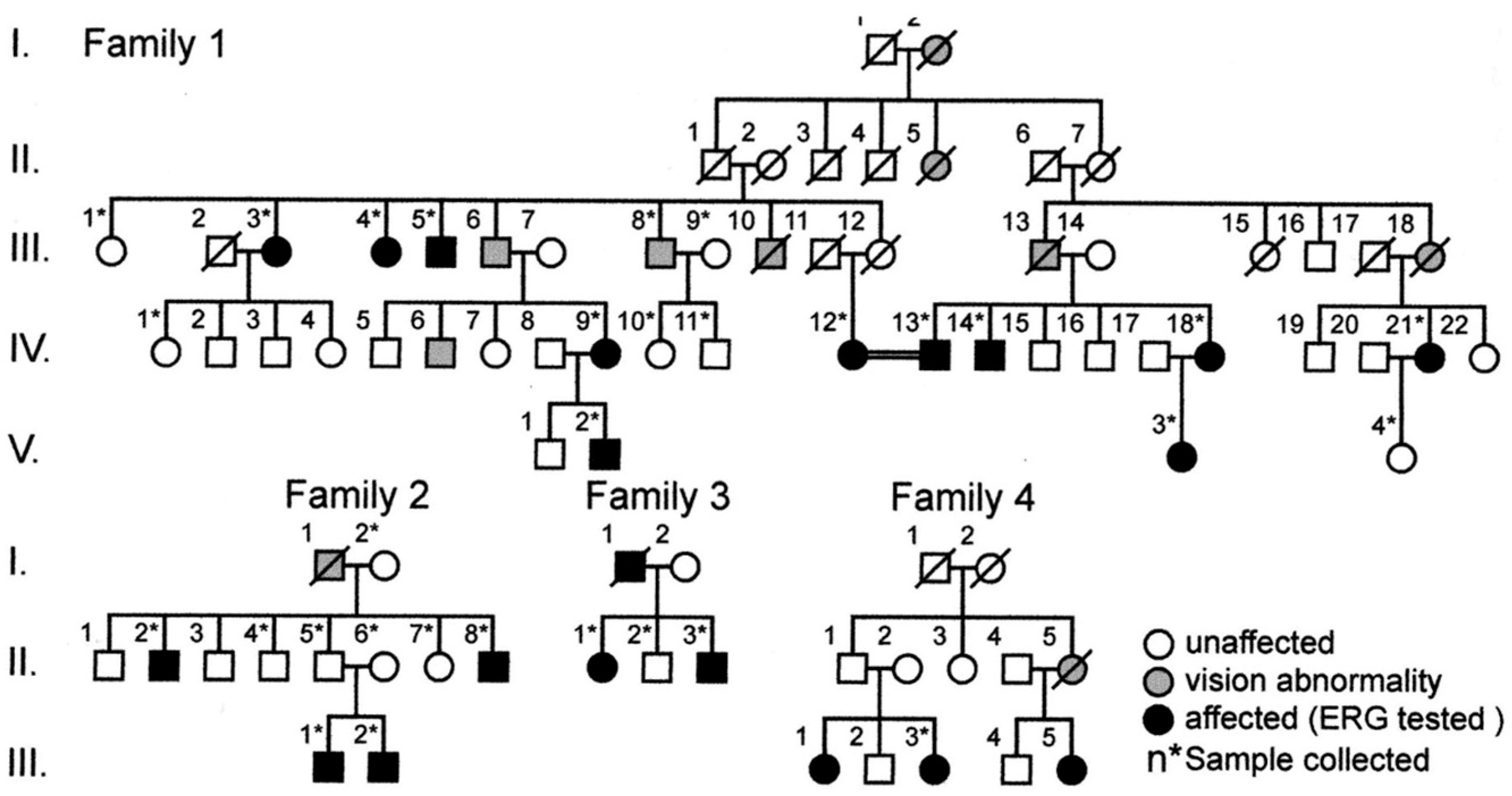
2.8. Molecular Genetics
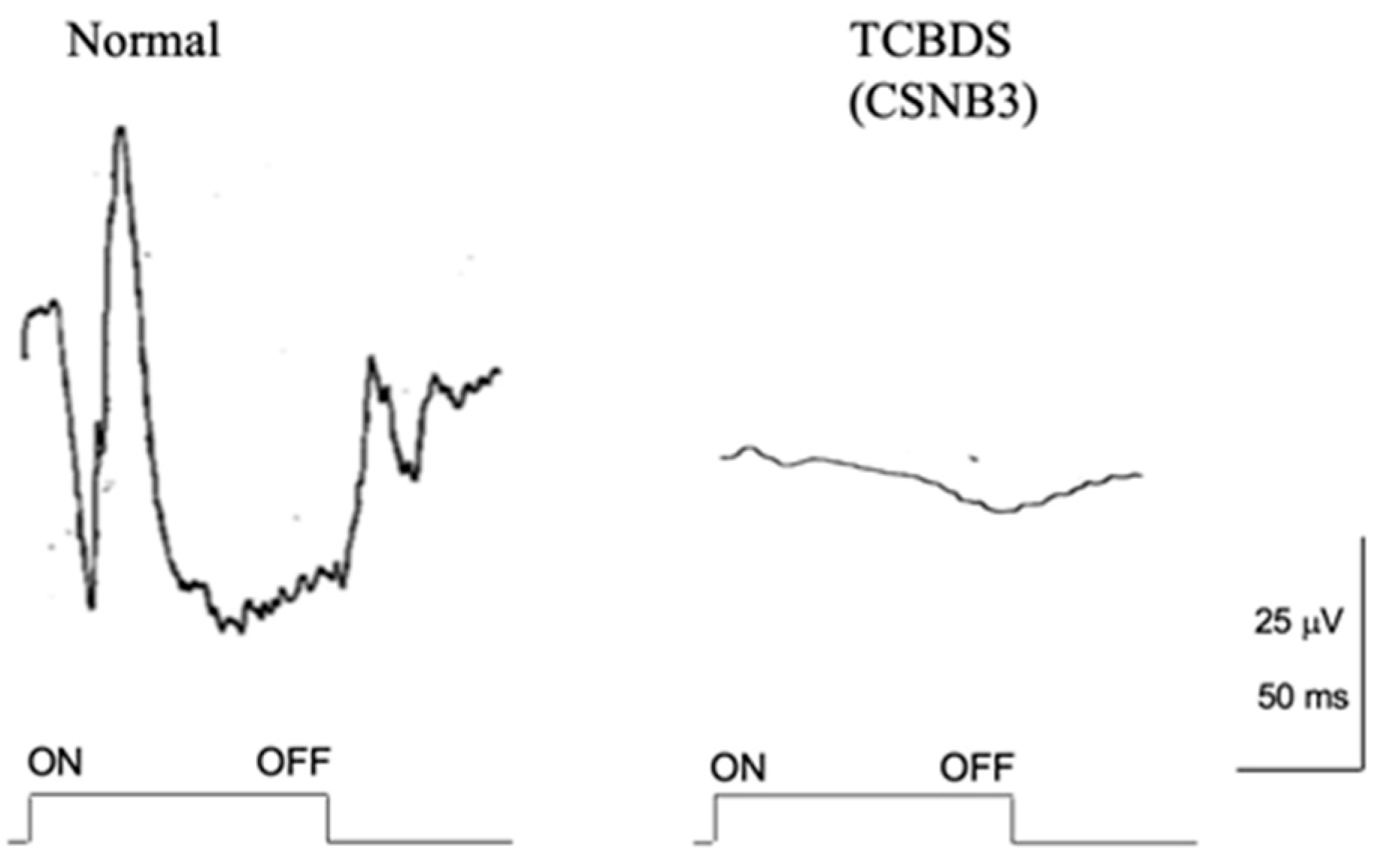
2.9. Total Complete Bipolar Cell Dysfunction Syndrome (TCBDS)
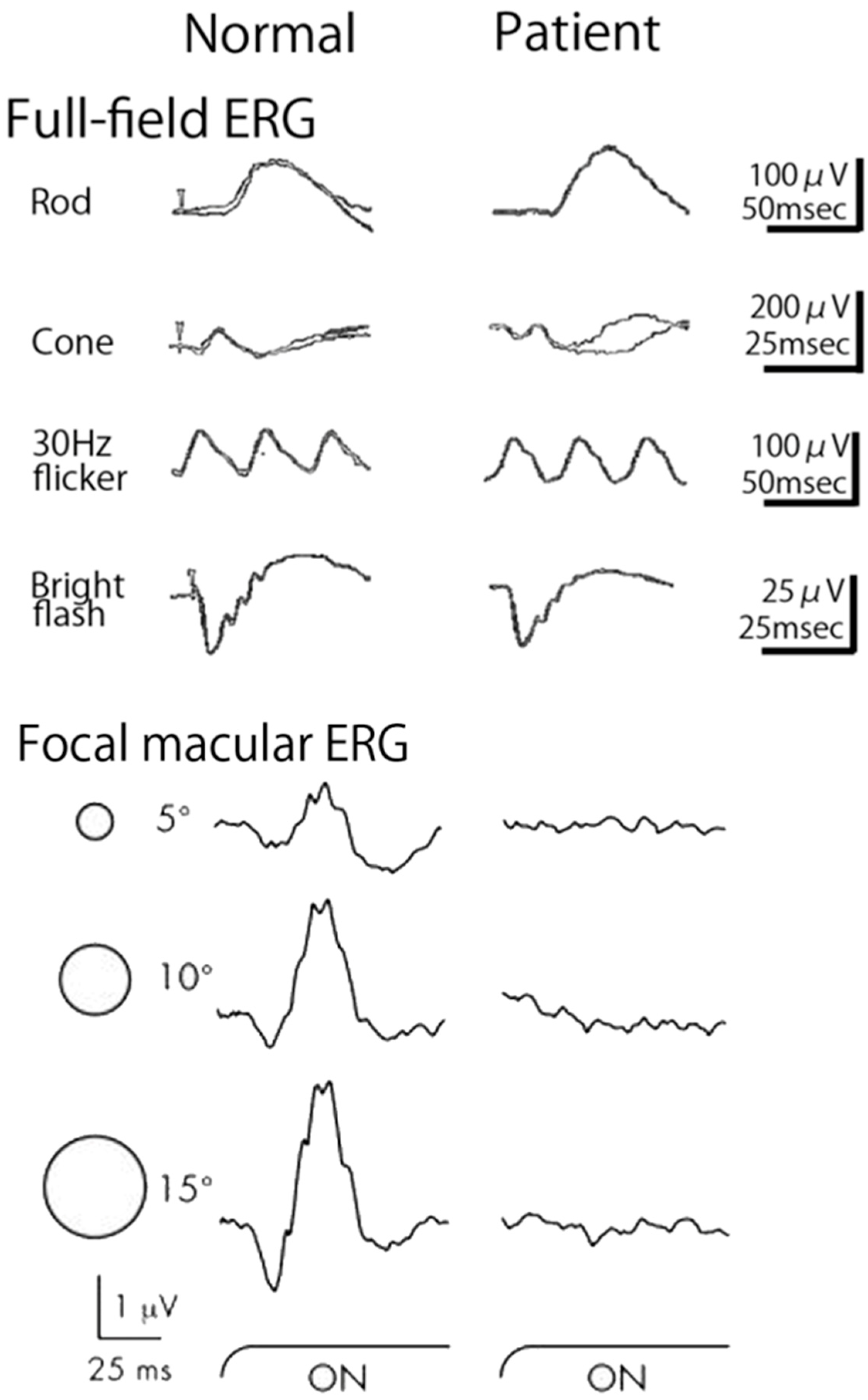
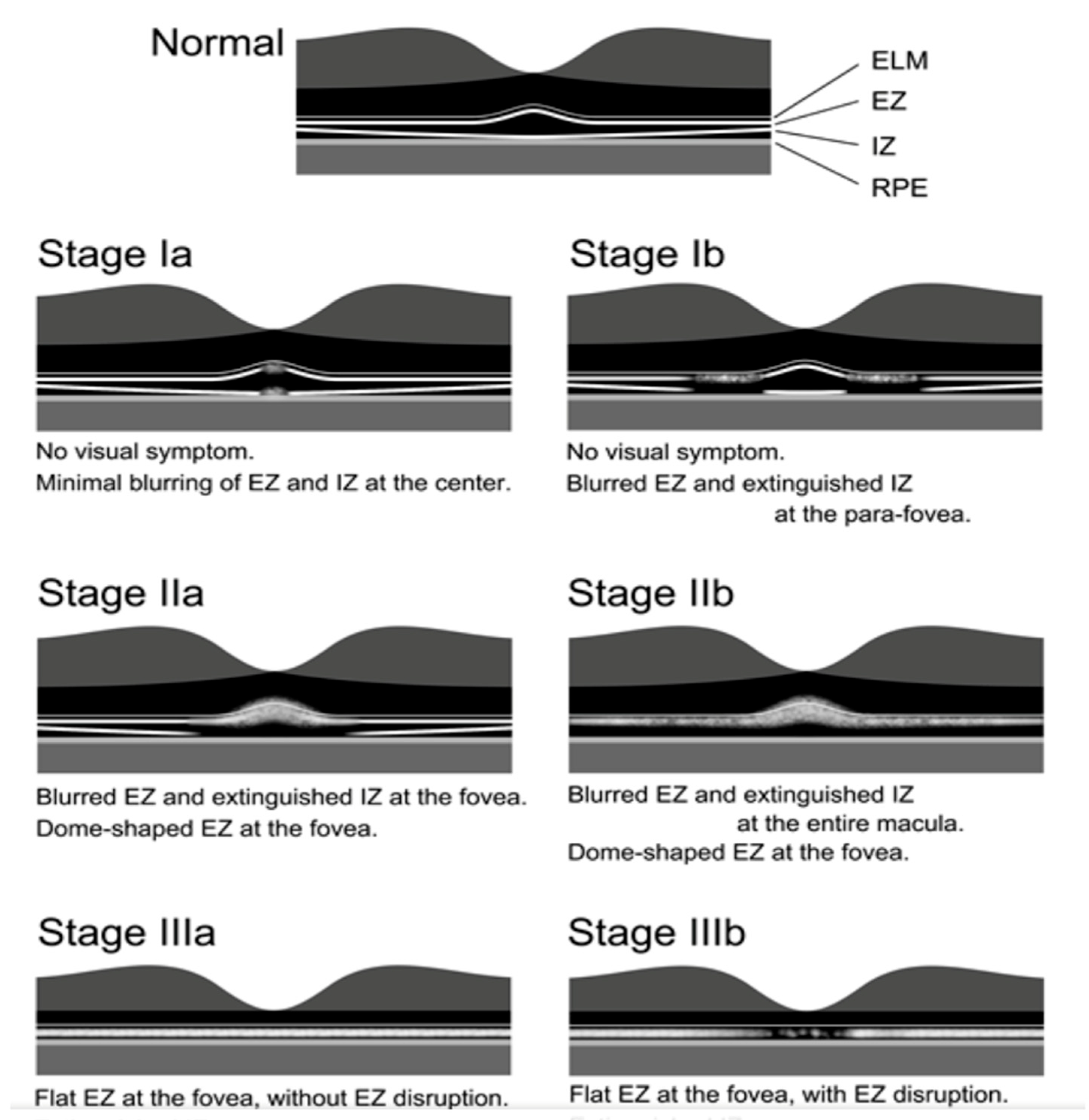
2.10. Occult Macular Dystrophy
Funding
Conflicts of Interest
References
- Oguchi, C. Uber eine Abart von Hemeralopie. Acte Soc. Ophthalmol. Jpn. 1907, 11, 123–134. [Google Scholar]
- Fuchs, S.; Nakazawa, M.; Maw, M.; Tamaio, M.; Ogucjhi, Y.; Gal, A. A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat. Genet. 1995, 10, 360–362. [Google Scholar] [CrossRef]
- Nakamura, M.; Yamamoto, S.; Okada, M.; Ito, S.; Tano, Y.; Miyake, Y. Novel mutations in arrestin gene and associated clinical features in Japanese patients with Oguchi disease. Ophthalmology 2004, 111, 1410–1414. [Google Scholar] [CrossRef]
- Yamamoto, S.; Sippel, K.C.; Berson, E.L.; Dryja, T.P. Defects in the rhodopsin kinase gene in patients with the Oguchi form of stationary night blindness. Nat. Genet. 1997, 15, 175–178. [Google Scholar] [CrossRef]
- Miyake, Y.; Horiguchi, M.; Suzuki, S.; Kondo, M.; Tanikawa, A. Electrophysiological findings in patients with Oguchi’s disease. Jpn. J. Ophthalmol. 1996, 40, 511–519. [Google Scholar]
- Miyake, Y.; Yagasaki, K.; Horiguchi, M.; Kawase, Y.; Kanda, T. Congenital stationary night blindness with negative electroretinogram. A new classification. Arch. Ophthalmol. 1986, 104, 1013–1020. [Google Scholar] [CrossRef]
- Miyake, Y.; Yagasaki, K.; Horiguchi, M.; Kawase, Y. On- and off-responses in photopic electroretinogram in complete and incomplete types of congenital stationary night blindness. Jpn. J. Ophthalmol. 1987, 31, 81–87. [Google Scholar]
- Miyake, Y. Establishment of the concept of new clinical entities. Complete and incomplete form of congenital stationary night blindness. Jpn. Ophthalmol. Soc. 2002, 106, 737–756. [Google Scholar]
- Miyake, Y. Hereditary retinal and allied diseases. Complete and incomplete types of CSNB. In Electrodiagnosis of Retinal Diseases; Springer: Tokyo, Japan; Berlin, Germany; New York, NY, USA, 2008; pp. 90–118. [Google Scholar]
- Miyake, Y.; Yasuma, T.; Awaya, S. Familial cone-rod dysfunction syndrome. Jpn. Clin. Ophthalmol. 1980, 34, 377–385. [Google Scholar]
- Miyake, Y.; Ichikawa, K.; Shiose, Y.; Kawase, Y. Hereditary macular dystrophy without visible fundus abnormality. Am. J. Ophthalmol. 1989, 108, 292–299. [Google Scholar] [CrossRef]
- Miyake, Y.; Horiguchi, M.; Tomita, N.; Kondo, M.; Tanikawa, A.; Takahashi, H.; Suzuki, S.; Terasaki, H. Occult macular dystrophy. Am. J. Ophthalmol. 1996, 122, 644–653. [Google Scholar] [CrossRef]
- Schubert, G.; Bornschein, H. Beiteitrag zur Analyse des menshlichen Electroretinogram. Ophthalmologica 1952, 123, 396–412. [Google Scholar] [CrossRef]
- Marmor, M.F.; Fulton, A.B.; Holder, G.E.; Miyake, Y.; Brigell Bach, M. International Society for Clinical Electrophysiology of Vision ISCEV Standard for full-field clinical electroretinography (2008 update). Doc. Ophthalmol. 2009, 118, 69–77. [Google Scholar] [CrossRef]
- Sieving, P.A. Photopic on- and off-pathway abnormalities in retinal dystrophies. Trans. Am. Ophthalmol. Soc. 1993, 91, 701–773. [Google Scholar]
- Slaughter, M.M.; Miller, R.F. 2 amino-4-phosphenobutric acid: A new pharmacological tool for retina research. Science 1981, 211, 182–185. [Google Scholar] [CrossRef]
- Slaughter, M.M.; Miller, R.F. An excitatory amino acid antagonist blocks cone input to signal conserving second-order retinal neuros. Science 1983, 219, 1230–1232. [Google Scholar] [CrossRef]
- Kolb, H.; Lipets, L.E. The anatomical basis for color vision in the vertebrate retina. In The Perception of Colour; Gouras, P., Ed.; Macimillan: London, UK, 1991; pp. 128–145. [Google Scholar]
- McKay, C.J.; Saeki, M.; Gouras, P.; Roy, M. Congenital and acquired nyctalopia eliminate the S-cone without disturbing color vision. Investig. Ophthalmol. Vis. Sci. (Supple) 1995, 36, 3925. [Google Scholar]
- Kamiyama, M.; Yamamoto, S.; Nitta, K.; Hayasaka, S. Undetectable S-cone electroretinogram b-wave in complete congenital stationary night blindness. Br. J. Ophthalmol. 1996, 80, 637–639. [Google Scholar] [CrossRef]
- Terasaki, H.; Miyake, Y.; Nomura, R.; Horiguchi, M.; Suzuki, K.; Kondo, M. Blue-on-yellow perimetry in the complete type of congenital stationary night blindness. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2761–2764. [Google Scholar]
- Boycott, K.M.; Pearce, W.G.; Musarella, M.A.; Weleber, R.G.; Maybaum, T.A.; Birch, D.G.; Miyake, Y.; Young, R.S.; Bech-Hansen, N.T. Evidence for genetic heterogeneity in X-linked congenital stationary night blindness. Am. J. Hum. Genet. 1998, 62, 865–875. [Google Scholar] [CrossRef]
- Strom, T.M.; Nyakatura, G.; Apfelstedt-Sylla, E.; Hellebrand, H.; Lorenz, B.; Weber, B.H.; Wutz, K.; Gutwillinger, N.; Rüther, K.; Drescher, B.; et al. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998, 19, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Bech-Hansen, N.T.; Naylor, M.J.; Maybaum, T.A.; Pearce, W.G.; Koop, B.; Fishman, G.A.; Mets, M.; Musarella, M.A.; Boycott, K.M. Loss-of-function mutations in a calcium-channel altha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998, 13, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Ito, D.; Terasaki, H.; Miyake, Y. Novel CACNA1F mutation in Japanese patients with incomplete congenital stationary night blindness. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1610–1616. [Google Scholar]
- Katta, M.; de Guimaraes, T.A.C.; Fujinami-Yokokawa, Y.; Fujinami, K.; Georgiou, M.; Mahroo, O.A.; Webster, A.R.; Michaelides, M. Congenital stationary night blindness: Structure, function and genotype-phenotype correlations in cohort of 122 patients. Ophthalmol. Retina 2024, 8, 932–941. [Google Scholar] [CrossRef]
- Bech-Hansen, N.T.; Naylor, M.J.; Maybaum, T.A.; Sparkes, R.L.; Koop, B.; Birch, D.G.; Bergen, A.A.; Prinsen, C.F.; Polomeno, R.C.; Gal, A.; et al. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat. Genet. 2000, 26, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Pusch, C.M.; Zeitz, C.; Brandau, O.; Pesch, K.; Achatz, H.; Feil, S.; Scharfe, C.; Maurer, J.; Jacobi, F.K.; Pinckers, A.; et al. The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein. Nat. Genet. 2000, 26, 324–327. [Google Scholar] [CrossRef]
- Dryja, T.P.; McGee, T.L.; Berson, E.L.; Fishman, G.A.; Sandberg, M.A.; Alexander, K.R.; Derlacki, D.J.; Rajagopalan, A.S. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc. Natl. Acad. Sci. USA 2005, 102, 4884–4889. [Google Scholar] [CrossRef]
- Audo, I.; Kohl, S.; Leroy, B.P.; Munier, F.L.; Guillonneau, X.; Mohand-Said, S. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2009, 85, 720–7229. [Google Scholar] [CrossRef] [PubMed]
- van Genderen, M.M.; Bijveld, M.M.; Claassen, Y.B.; Florijn, R.J.; Pearring, J.N.; Meire, F.M.; McCall, M.A.; Riemslag, F.C.; Gregg, R.G.; Bergen, A.A.; et al. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am. J. Hum. Genet. 2009, 85, 730–736. [Google Scholar] [CrossRef]
- Nakamura, M.; Sanuki, R.; Yasuma, T.R.; Onishi, A.; Nishiguchi, K.M.; Koike, C. TRPM1 mutations are associated with the complete form congenital stationary night blindness. Mol. Vis. 2010, 16, 425–437. [Google Scholar]
- Audo, I.; Bujakowska, K.; Orhan, E.; Poloschek, C.M.; Defoort-Dhellemmes, S.; Drumare, I.; Kohl, S.; Luu, T.D.; Lecompte, O.; Zrenner, E.; et al. Whole-exome sequencing identifies mutations in GPR179 leading to autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2012, 90, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Jacobson, S.G.; Hamel, C.P.; Bujakowska, K.; Neuillé, M.; Orhan, E.; Zanlonghi, X.; Lancelot, M.E.; Michiels, C.; Schwartz, S.B.; et al. Whole-exome sequencing identifies LRIT3 mutatons as a cause of autosomal-recessive complete congenital night blindness. Am. J. Hum. Genet. 2013, 92, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Kloeckener-Gruissem, B.; Forster, U.; Kohl, S.; Magyar, I.; Wissinger, B.; Mátyás, G.; Borruat, F.-X.; Schorderet, D.F.; Zrenner, E.; et al. Mutations in CABP4, the gene encoding the Ca2+-binding protein 4, cause autosomal recessive night blindness. Am. J. Hum. Genet. 2006, 79, 657–667. [Google Scholar] [CrossRef]
- Littink, K.W.; van Genderen, M.M.; Collin, R.W.J.; Roosing, S.; de Brouwer, A.P.M.; Riemslag, F.C.C.; Venselaar, H.; Thiadens, A.A.H.J.; Hoyng, C.B.; Rohrschneider, K.; et al. A novel homozygous nonsense mutation in CABP4 causes congenital cone-rod synaptic disorder. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2344–2359. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.O.; Alrashed, M.; Alkuraya, F.S. Clinical characterization of the CABP4 related retinal phenotype. Br. J. Ophthalmol. 2013, 97, 262–265. [Google Scholar] [CrossRef]
- Khan, A.O. CABP4 mutations do not cause congenital stationary night blindness. Ophthalmology 2013, 121, e15. [Google Scholar] [CrossRef]
- Bijveld, M.M.; Florijn, R.J.; Bergen, A.A.; van den Born, L.I.; Kamermans, M.; Prick, L.; Riemslag, F.C.; van Schooneveld, M.J.; Kappers, A.M.; van Genderen, M.M. Genotype and phenotype of 101 dutch patients with congenital stationary night blindness. Ophthalmology 2013, 120, 2072–2081. [Google Scholar] [CrossRef]
- Schatz, P.; Abdalla Elsayed, M.E.A.; Khan, A.O. Mutation modal imaging in CABP-4 related retinopathy. Ophthalmic Genet. 2017, 38, 459–464. [Google Scholar] [CrossRef]
- Smirnov, V.M.; Zeitz, C.; Soumittra, N.; Audo, I.; Defoort-Dhellemmes, S. Retinal findings in a patient of French ancestry with CABP-4 related retinal disease. Doc. Ophthalmol. 2018, 136, 135–143. [Google Scholar] [CrossRef]
- Tan, J.K.; Arno, G.; Josifova, D.; Mohamed, M.D.; Mahroo, O.A. Unusual findings in a patient with CABP4-associated cone-rod synaptic disorder. Doc. Ophthalmol. 2024, 148, 1115–1120. [Google Scholar] [CrossRef]
- Wycisk, K.A.; Zeitz, C.; Feil, S.; Wittmer, M.; Forster, U.; Neidhardt, J.; Wissinger, B.; Zrenner, E.; Wilke, R.; Kohl, S.; et al. Mutation in the auxiliary calcium-channel subunit CACNA2D4 causes autosomal recessive cone dystrophy. Am. J. Hum. Genet. 2006, 79, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Miyake, Y.; Hara, A. Simultaneous recording pf electroretinogram and visual evoked response. Focal stimulation under direct observation. Arch. Ophthalmol. 1977, 95, 1205–1208. [Google Scholar] [CrossRef]
- Miyake, Y. Studies of local macular ERG. Jpn. Ophthamol. Soc. 1988, 92, 1419–1449. [Google Scholar]
- Miyake, Y.; Shiroyama, N.; Ota, I.; Horiguchi, M. Oscillatory potentials in electroretinograms of the human macular region. Investig. Ophthalmol. Vis. Sci. 1988, 29, 1631–1635. [Google Scholar]
- Sutter, E.E.; Tran, D. The field topography of ERG components in man. 1. The photopic luminance response. Vis. Res. 1992, 32, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Tsunoda, K. Occult macular dystrophy. Jpn. J. Ophthalmol. 2015, 59, 71–80. [Google Scholar] [CrossRef]
- Nakanishi, A.; Ueno, S.; Kawano, K.; Ito, Y.; Kominami, T.; Yasuda, S.; Kondo, M.; Tsunoda, K.; Iwata, T.; Terasaki, H. Pathologic changes of cone photoreceptors in eyes with occult macular dystrophy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7243–7249. [Google Scholar] [CrossRef]
- Fujinami, K.; Kameya, S.; Kikuchi, S.; Ueno, S.; Kondo, M.; Hayashi, T.; Shinoda, K.; Machida, S.; Kuniyoshi, K.; Kawamura, Y.; et al. Novel RP1L1 variants and genotype-photoreceptor microstructural phenotype association in cohort of Japanese patients with occult macular dystrophy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4837–4846. [Google Scholar] [CrossRef]
- Nakamura, N.; Tsunoda, K.; Mizuno, Y.; Usui, T.; Hatase, T.; Ueno, S.; Kuniyoshi, K.; Hayashi, T.; Katagiri, S.; Kondo, M.; et al. Clinical stages of occult macular dystrophy based on optical coherence tomographic findings. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4691–5700. [Google Scholar] [CrossRef]
- Akahori, M.; Tsunoda, K.; Miyake, Y.; Fukuda, Y.; Ishiura, H.; Tsuji, S.; Usui, T.; Hatase, T.; Nakamura, M.; Ohde, H.; et al. Dominant mutations in RP1L1are responsible for occult macular dystrophy. Am. J. Hum. Genet. 2010, 10, 424–429. [Google Scholar] [CrossRef]
- Noel, N.C.L.; MacDonald, I.M. RP1L1 and inherited photoreceptor disease. A review. Surv. Ophthalmol. 2020, 65, 725–739. [Google Scholar] [CrossRef] [PubMed]
- Conte, I.; Lestingi, M.; den Hollander, A.; Alfano, G.; Ziviello, C.; Pugliese, M.; Circolo, D.; Caccioppoli, C.; Ciccodicola, A.; Banfi, S. Identification and characterization of the retinitis pigmentosa 1-like gene (RP1L1) a novel candidate for retinal degenerations. Eur. J. Human. Genet. EJHC 2003, 11, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Bowne, S.J.; Daiger, S.P.; Malone, K.A.; Heckenlively, J.R.; Kennan, A.; Humphries, P.; Hughbanks-Wheaton, D.; Birch, D.G.; Liu, Q.; Pierce, E.A.; et al. Characterization of RPL1L, a highly polymorphic paralog of retinitis pigmentosa 1 (RP1) gene. Mol. Vis. 2003, 9, 129–137. [Google Scholar]
- Tsunoda, K.; Usui, T.; Hatase, T.; Yamai, S.; Fujinami, K.; Hanazono, G.; Shinoda, K.; Ohde, H.; Akahori, M.; Iwata, T.; et al. Clinical characteristics of occult macular dystrophy in family with mutation of RP1L1 gene. Retina 2012, 32, 1135–1147. [Google Scholar] [CrossRef]
- Fujinami, K.; Yang, L.; Joo, K.; Tsunoda, K.; Kameya, S.; Hanazono, G.; Fujinami-Yokokawa, Y.; Arno, G.; Kondo, M.; Nakamura, N.; et al. Clinical and genetic characteristics of east Asian patients with occult macular dystrophy (Miyake Disease): East Asia Occult Macular Dystrophy Studies Report Number 3. Ophthalmology 2019, 126, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Hanazono, G.; Fujinami, K.; Hatase, T.; Kawamura, Y.; Iwata, T.; Miyake, Y.; Tsunoda, K. Parafoveal photoreceptor abnormalities in asymptomatic patients with RP1L1 mutations in families with occult macular dystrophy (Miyake Disease). Investig. Ophthalmol. Vis. Sci. 2017, 58, 6020–6029. [Google Scholar] [CrossRef]
- Fujinami-Yokokawa, Y.; Joo, K.; Liu, X.; Tsunoda, K.; Kondo, M.; Ahn, S.J.; Robson, A.G.; Naka, I.; Ohashi, J.; Li, H.; et al. Distinct clinical effects of two RP1L1 hotspots in east Asian with occult macular dystrophy (Miyake Disease): EAOMO Report 4. Investig. Ophthalmol. Vis. Sci. 2024, 65, 41–47. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyake, Y. Review of Four Refined Clinical Entities in Hereditary Retinal Disorders from Japan. Int. J. Mol. Sci. 2025, 26, 5166. https://doi.org/10.3390/ijms26115166
Miyake Y. Review of Four Refined Clinical Entities in Hereditary Retinal Disorders from Japan. International Journal of Molecular Sciences. 2025; 26(11):5166. https://doi.org/10.3390/ijms26115166
Chicago/Turabian StyleMiyake, Yozo. 2025. "Review of Four Refined Clinical Entities in Hereditary Retinal Disorders from Japan" International Journal of Molecular Sciences 26, no. 11: 5166. https://doi.org/10.3390/ijms26115166
APA StyleMiyake, Y. (2025). Review of Four Refined Clinical Entities in Hereditary Retinal Disorders from Japan. International Journal of Molecular Sciences, 26(11), 5166. https://doi.org/10.3390/ijms26115166