
Ion Channel–Extracellular Matrix Interplay in Colorectal Cancer: A Network-Based Approach to Tumor Microenvironment Remodeling

 ,
,  , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. Results
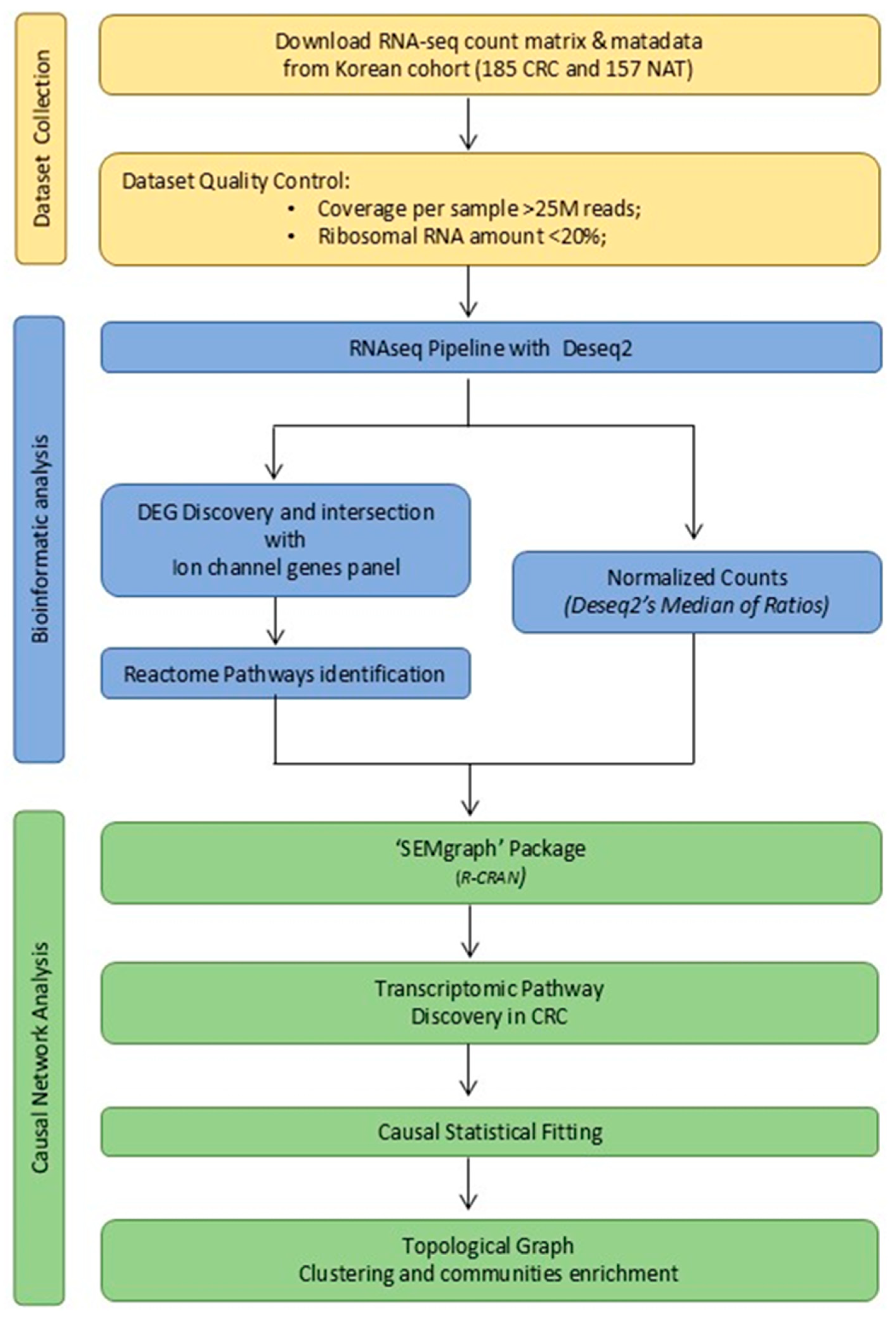
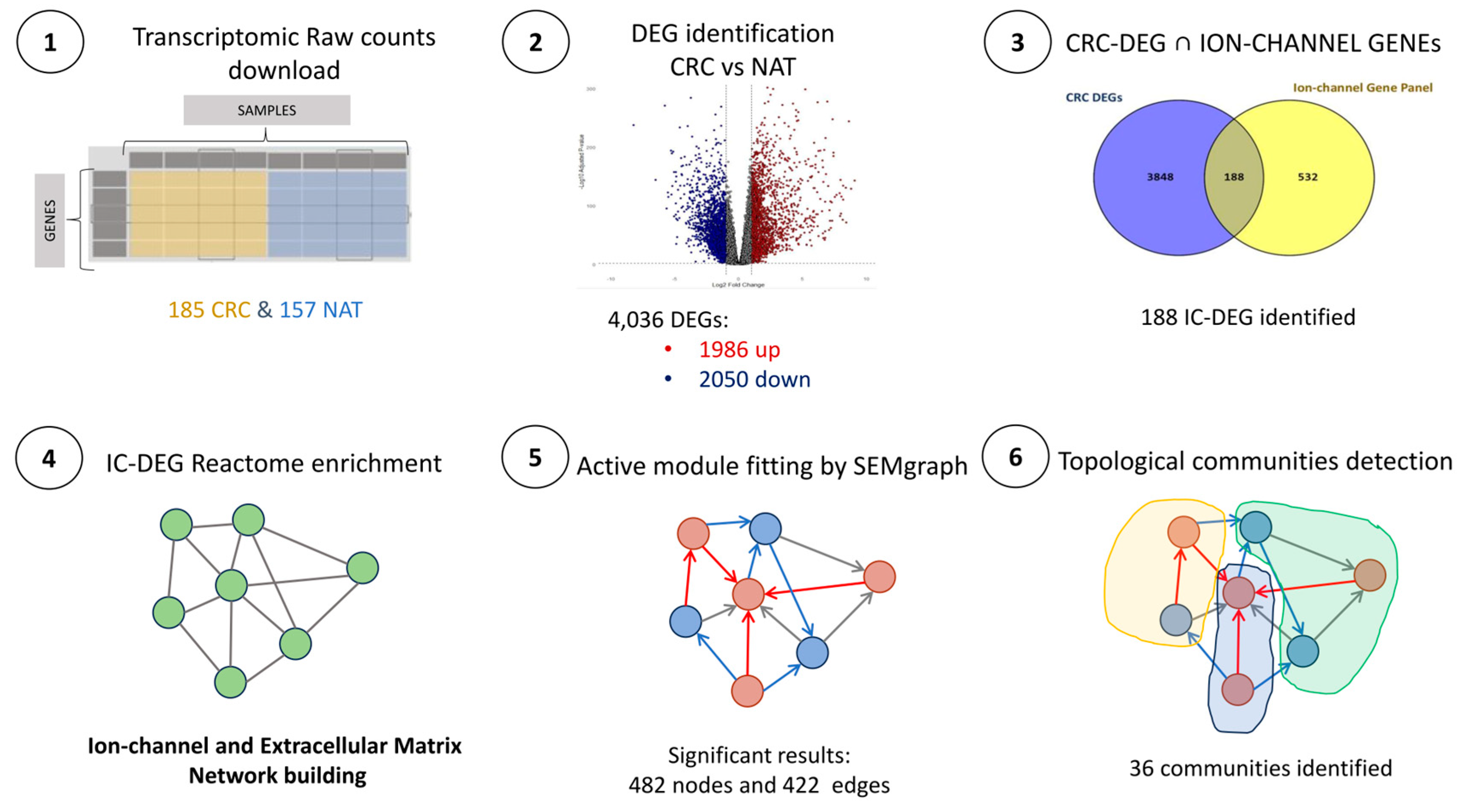
2.1. Dataset Description and Quality Control (QC)
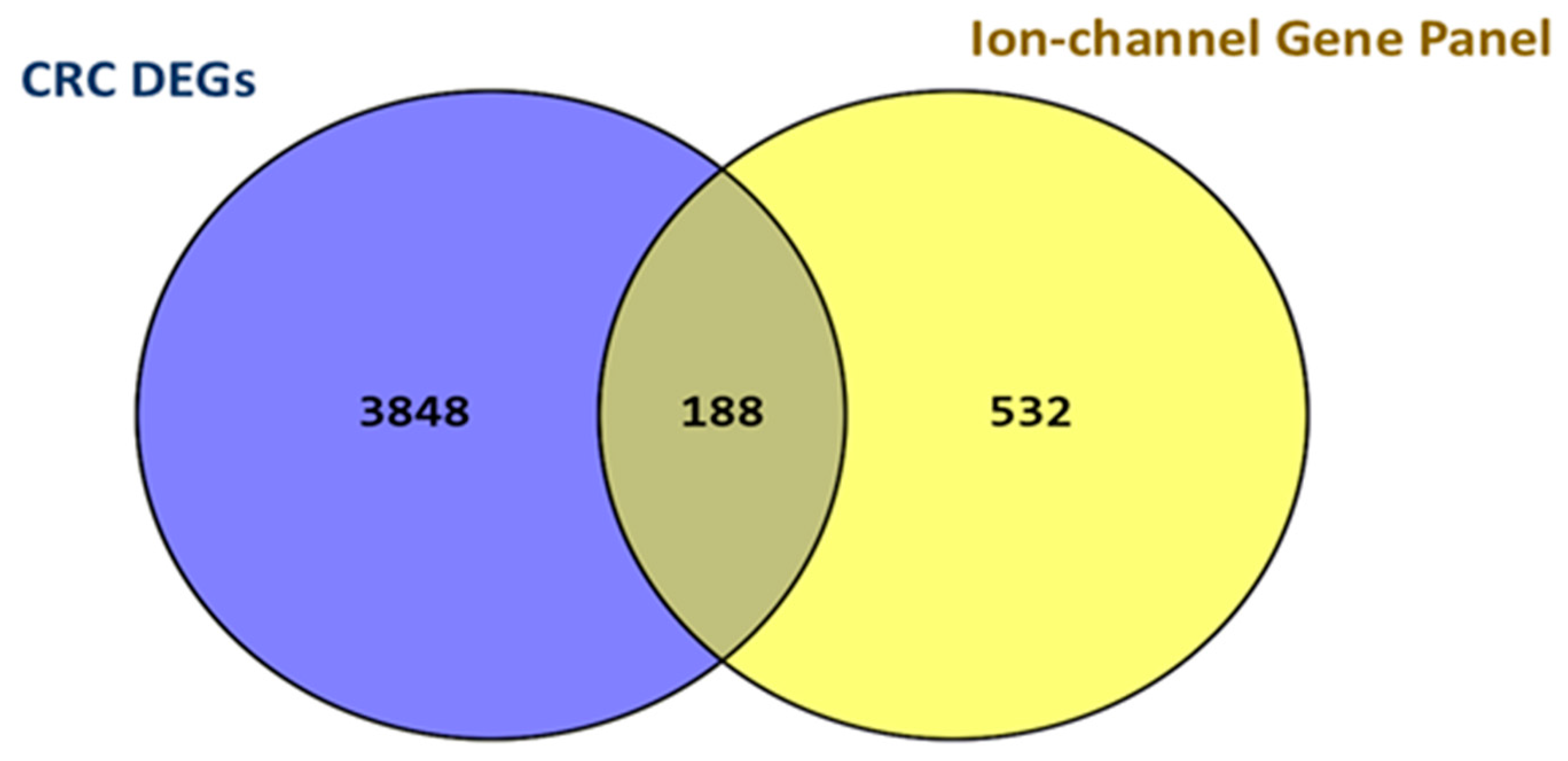
2.2. CRC DEGs Determination and Ion Channel Subset
2.3. Reactome Enrichment Analysis Results
2.4. IC-DEGs Validation in “The Cancer Genome Atlas” Database
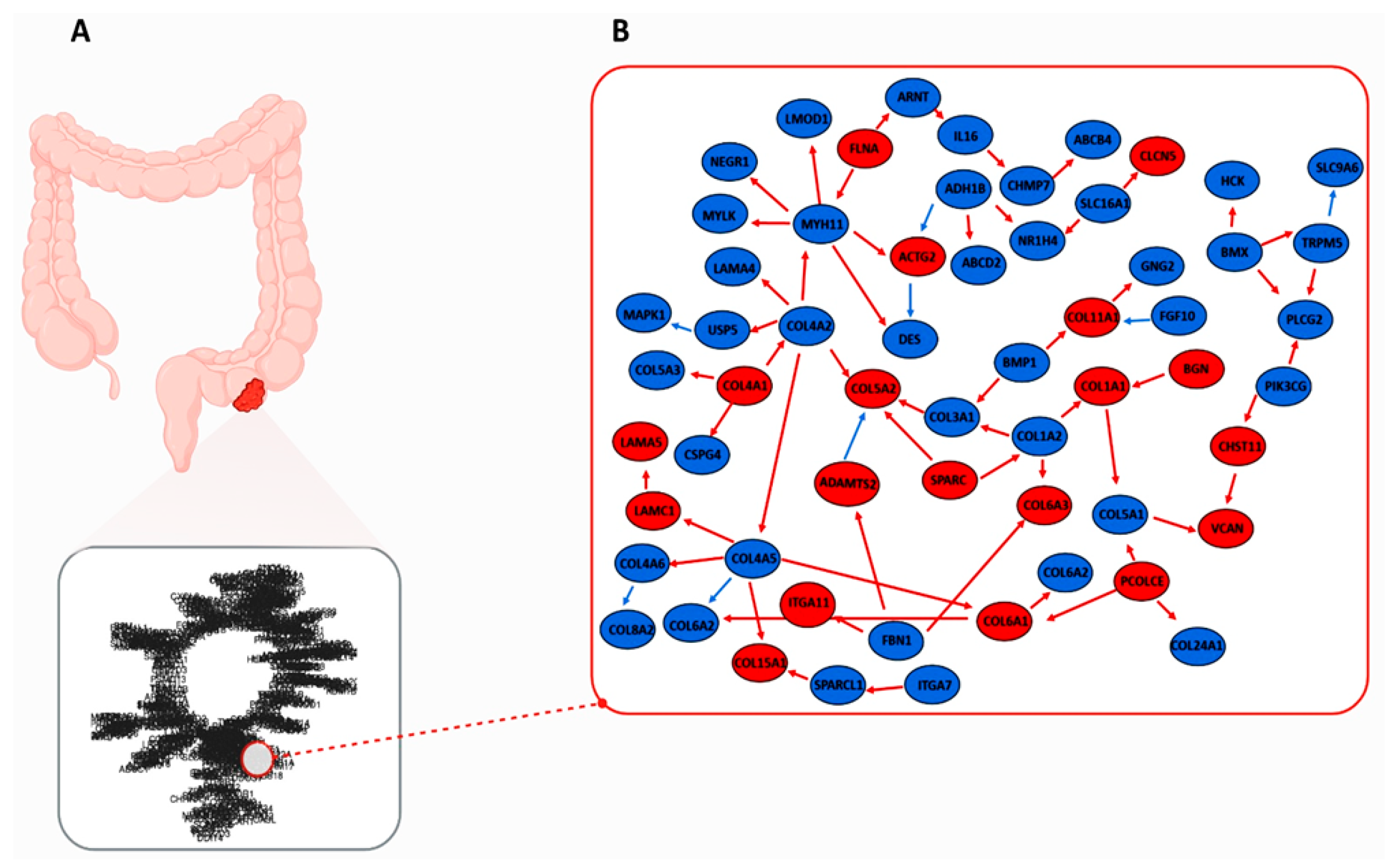
2.5. CRC-IC Module and Statistical Analysis
2.5.1. CRC-IC Module Analysis
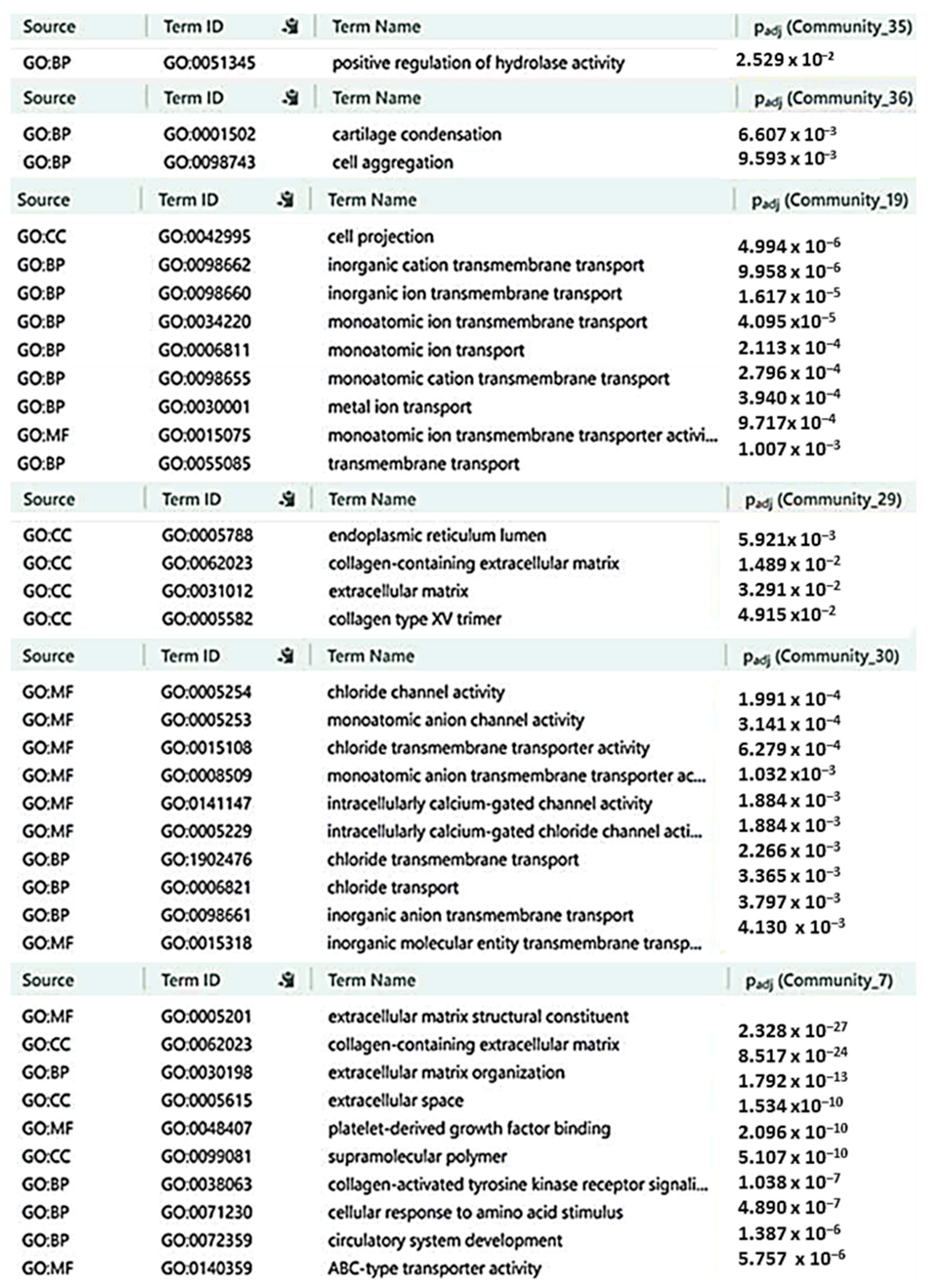
2.5.2. Gene Communities Detection Results
2.6. Characterization of CRC-IC Module Communities
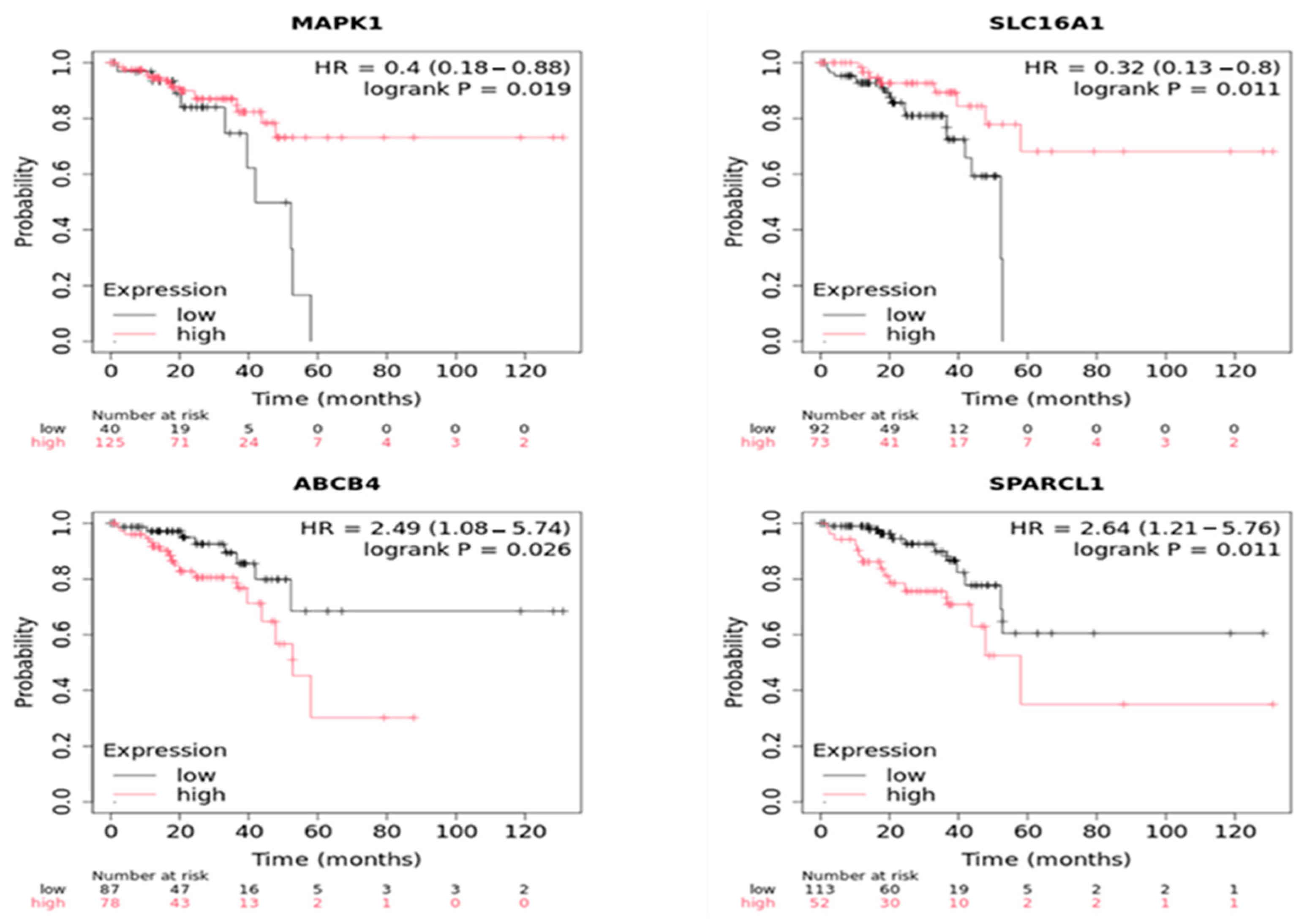
2.7. Statistical External Validation of the Active CRC-IC Module Results
3. Discussion
4. Materials and Methods
4.1. Data Collection and Study Design
4.2. Dataset Quality Control and Ion-Channel Gene Panel Selection
4.3. IC-DEGs Identification and Normalized Counts Production
4.4. IC-DEGs External Validation
4.5. Reactome Enrichment Analysis
4.6. Statistical Analysis
4.6.1. Analysis of the Active CRC-IC Module
4.6.2. Gene Communities Detection
4.7. Communities’ Enrichment Analysis
4.8. Statistical External Validation of the Active CRC-IC Module
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, L.; Xiao, H.; Jiang, J.; Li, B.; Liu, W.; Huang, W. The KMeansGraphMIL Model. Am. J. Pathol. 2025, 195, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Geng, S.; Luo, H.; Wang, W.; Mo, Y.-Q.; Luo, Q.; Wang, L.; Song, G.-B.; Sheng, J.-P.; Xu, B. Signaling Pathways Involved in Colorectal Cancer: Pathogenesis and Targeted Therapy. Signal Transduct. Target. Ther. 2024, 9, 266. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, X.; Chakravarti, D.; Shalapour, S.; DePinho, R.A. Genetic and Biological Hallmarks of Colorectal Cancer. Genes. Dev. 2021, 35, 787–820. [Google Scholar] [CrossRef]
- Gentile, R.; Feudi, D.; Sallicandro, L.; Biagini, A. Can the Tumor Microenvironment Alter Ion Channels? Unraveling Their Role in Cancer. Cancers 2025, 17, 1244. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, L.; Wang, M.; Zhou, S.; Lu, Y.; Cui, H.; Racanelli, A.C.; Zhang, L.; Ye, T.; Ding, B.; et al. Targeting Fibrosis: Mechanisms and Clinical Trials. Signal Transduct. Target. Ther. 2022, 7, 206. [Google Scholar] [CrossRef]
- Hu, M.; Feng, X.; Liu, Q.; Liu, S.; Huang, F.; Xu, H. The Ion Channels of Endomembranes. Physiol. Rev. 2024, 104, 1335–1385. [Google Scholar] [CrossRef]
- Karska, J.; Kowalski, S.; Saczko, J.; Moisescu, M.G.; Kulbacka, J. Mechanosensitive Ion Channels and Their Role in Cancer Cells. Membranes 2023, 13, 167. [Google Scholar] [CrossRef]
- Rao, V.; Perez-Neut, M.; Kaja, S.; Gentile, S. Voltage-Gated Ion Channels in Cancer Cell Proliferation. Cancers 2015, 7, 849–875. [Google Scholar] [CrossRef]
- Zhu, T.; Zhao, J.; Liu, J.; Tian, S.; Li, S.; Yuan, H. Advances in the Role of Ion Channels in Leukemia. Heliyon 2024, 10, e33452. [Google Scholar] [CrossRef]
- Mierke, C.T. Extracellular Matrix Cues Regulate Mechanosensing and Mechanotransduction of Cancer Cells. Cells 2024, 13, 96. [Google Scholar] [CrossRef]
- Anderson, K.J.; Cormier, R.T.; Scott, P.M. Role of Ion Channels in Gastrointestinal Cancer. World J. Gastroenterol. 2019, 25, 5732–5772. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Deng, Y.; Ye, J.; Luo, Y.; Weng, J.; He, Q.; Liu, F.; Li, M.; Liang, R.; Lin, Y.; et al. Store-Operated Ca2+ Entry as a Key Oncogenic Ca2+ Signaling Driving Tumor Invasion-Metastasis Cascade and Its Translational Potential. Cancer Lett. 2021, 516, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the Extracellular Matrix in Development and Disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Zhang, X.; Al-Danakh, A.; Zhu, X.; Feng, D.; Yang, L.; Wu, H.; Li, Y.; Wang, S.; Chen, Q.; Yang, D. Insights into the Mechanisms, Regulation, and Therapeutic Implications of Extracellular Matrix Stiffness in Cancer. Bioeng. Transl. Med. 2025, 10, e10698. [Google Scholar] [CrossRef]
- Lee, S.-A.; Cho, G.-J.; Kim, D.; Kim, D.-H. Biophysical Interplay between Extracellular Matrix Remodeling and Hypoxia Signaling in Regulating Cancer Metastasis. Front. Cell Dev. Biol. 2024, 12, 1335636. [Google Scholar] [CrossRef]
- Ji, C.; McCulloch, C.A. TRPV4 Integrates Matrix Mechanosensing with Ca 2+ Signaling to Regulate Extracellular Matrix Remodeling. FEBS J. 2021, 288, 5867–5887. [Google Scholar] [CrossRef]
- Dzobo, K.; Dandara, C. The Extracellular Matrix: Its Composition, Function, Remodeling, and Role in Tumorigenesis. Biomimetics 2023, 8, 146. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of Extracellular Matrix Remodelling in Tumour Progression and Metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Toss, M.S.; Miligy, I.M.; Gorringe, K.L.; AlKawaz, A.; Mittal, K.; Aneja, R.; Ellis, I.O.; Green, A.R.; Roxanis, I.; Rakha, E.A. Geometric Characteristics of Collagen Have Independent Prognostic Significance in Breast Ductal Carcinoma in Situ: An Image Analysis Study. Mod. Pathol. 2019, 32, 1473–1485. [Google Scholar] [CrossRef]
- Maqoud, F.; Zizzo, N.; Attimonelli, M.; Tinelli, A.; Passantino, G.; Antonacci, M.; Ranieri, G.; Tricarico, D. Immunohistochemical, Pharmacovigilance, and Omics Analyses Reveal the Involvement of ATP-Sensitive K+ Channel Subunits in Cancers: Role in Drug–Disease Interactions. Front. Pharmacol. 2023, 14, 1115543. [Google Scholar] [CrossRef]
- Fiscon, G.; Conte, F.; Farina, L.; Paci, P. Network-Based Approaches to Explore Complex Biological Systems towards Network Medicine. Genes 2018, 9, 437. [Google Scholar] [CrossRef] [PubMed]
- Beran, T.N.; Violato, C. Structural Equation Modeling in Medical Research: A Primer. BMC Res. Notes 2010, 3, 267. [Google Scholar] [CrossRef] [PubMed]
- Guido, D.; Maqoud, F.; Aloisio, M.; Mallardi, D.; Ura, B.; Gualandi, N.; Cocca, M.; Russo, F. Transcriptomic Module Discovery of Diarrhea-Predominant Irritable Bowel Syndrome: A Causal Network Inference Approach. Int. J. Mol. Sci. 2024, 25, 9322. [Google Scholar] [CrossRef]
- Kohestani, H.; Giuliani, A. Organization Principles of Biological Networks: An Explorative Study. Biosystems 2016, 141, 31–39. [Google Scholar] [CrossRef]
- Chartrand, G.; Zhang, P. Chromatic Graph Theory; Chapman and Hall/CRC: New York, NY, USA, 2019; ISBN 9780429438868. [Google Scholar]
- van Kesteren, E.-J. Structural Equations with Latent Variables: Computational Solutions for Modern Data Problems. Ph.D. Thesis, Utrecht University, Utrecht, The Netherlands, 2021. [Google Scholar]
- Lee, J.; Kim, J.-H.; Chu, H.B.K.; Oh, S.-T.; Kang, S.-B.; Lee, S.; Kim, D.-W.; Oh, H.-K.; Park, J.-H.; Kim, J.; et al. Comprehensive RNA-Sequencing Analysis of Colorectal Cancer in a Korean Cohort. Mol. Cells 2024, 47, 100033. [Google Scholar] [CrossRef]
- Grassi, M.; Palluzzi, F.; Tarantino, B. SEMgraph: An R Package for Causal Network Inference of High-Throughput Data with Structural Equation Models. Bioinformatics 2022, 38, 4829–4830. [Google Scholar] [CrossRef]
- Felmlee, M.A.; Jones, R.S.; Rodriguez-Cruz, V.; Follman, K.E.; Morris, M.E. Monocarboxylate Transporters (SLC16): Function, Regulation, and Role in Health and Disease. Pharmacol. Rev. 2020, 72, 466–485. [Google Scholar] [CrossRef]
- Sticova, E.; Jirsa, M. ABCB4 Disease: Many Faces of One Gene Deficiency. Ann. Hepatol. 2020, 19, 126–133. [Google Scholar] [CrossRef]
- Hu, H.; Wang, M.; Guan, X.; Yuan, Z.; Liu, Z.; Zou, C.; Wang, G.; Gao, X.; Wang, X. Loss of ABCB4 Attenuates the Caspase-Dependent Apoptosis Regulating Resistance to 5-Fu in Colorectal Cancer. Biosci. Rep. 2018, 38, BSR20171428. [Google Scholar] [CrossRef]
- Franchi, M.; Piperigkou, Z.; Mastronikolis, N.S.; Karamanos, N. Extracellular Matrix Biomechanical Roles and Adaptation in Health and Disease. FEBS J. 2024, 291, 430–440. [Google Scholar] [CrossRef]
- Haworth, A.S.; Brackenbury, W.J. Emerging Roles for Multifunctional Ion Channel Auxiliary Subunits in Cancer. Cell Calcium 2019, 80, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.J.; Huang, X. Ion Channels in Cancer: Orchestrators of Electrical Signaling and Cellular Crosstalk. Rev. Physiol. Biochem. Pharmacol. 2022, 183, 103–133. [Google Scholar] [PubMed]
- Altamura, C.; Gavazzo, P.; Pusch, M.; Desaphy, J.-F. Ion Channel Involvement in Tumor Drug Resistance. J. Pers. Med. 2022, 12, 210. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, X.; Zhao, W.; Xu, J.; Zhang, H.; Huang, T.; Wu, C.; Yang, J.; Tang, C.; Ye, Q.; et al. A Comprehensive Analysis of TRP-Related Gene Signature, and Immune Infiltration in Patients with Colorectal Cancer. Discov. Oncol. 2024, 15, 357. [Google Scholar] [CrossRef]
- Li, X.; Wu, D.; Li, Q.; Gu, J.; Gao, W.; Zhu, X.; Yin, W.; Zhu, R.; Zhu, L.; Jiao, N. Host-Microbiota Interactions Contributing to the Heterogeneous Tumor Microenvironment in Colorectal Cancer. Physiol. Genom. 2024, 56, 221–234. [Google Scholar] [CrossRef]
- Potier-Cartereau, M.; Raoul, W.; Weber, G.; Mahéo, K.; Rapetti-Mauss, R.; Gueguinou, M.; Buscaglia, P.; Goupille, C.; Le Goux, N.; Abdoul-Azize, S.; et al. Potassium and Calcium Channel Complexes as Novel Targets for Cancer Research. Rev. Physiol. Biochem. Pharmacol. 2022, 183, 157–176. [Google Scholar]
- Maeda, T.; Suzuki, A.; Koga, K.; Miyamoto, C.; Maehata, Y.; Ozawa, S.; Hata, R.-I.; Nagashima, Y.; Nabeshima, K.; Miyazaki, K.; et al. TRPM5 Mediates Acidic Extracellular PH Signaling and TRPM5 Inhibition Reduces Spontaneous Metastasis in Mouse B16-BL6 Melanoma Cells. Oncotarget 2017, 8, 78312–78326. [Google Scholar] [CrossRef]
- Li, L.; Kanemitsu, K.; Ohnishi, K.; Yamada, R.; Yano, H.; Fujiwara, Y.; Miyamoto, Y.; Mikami, Y.; Hibi, T.; Baba, H.; et al. CXCL10 Expression in Human Colorectal Cancer Tissue and Its Correlation With Serum Levels of CXCL10. Cancer Genom.-Proteom. 2024, 21, 54–64. [Google Scholar] [CrossRef]
- Waldner, M.J.; Foersch, S.; Neurath, M.F. Interleukin-6—A Key Regulator of Colorectal Cancer Development. Int. J. Biol. Sci. 2012, 8, 1248–1253. [Google Scholar] [CrossRef]
- Atreya, R.; Mudter, J.; Finotto, S.; Müllberg, J.; Jostock, T.; Wirtz, S.; Schütz, M.; Bartsch, B.; Holtmann, M.; Becker, C.; et al. Blockade of Interleukin 6 Trans Signaling Suppresses T-Cell Resistance against Apoptosis in Chronic Intestinal Inflammation: Evidence in Crohn Disease and Experimental Colitis In Vivo. Nat. Med. 2000, 6, 583–588. [Google Scholar] [CrossRef]
- Ji, Y.; Wang, Y.; Zou, J.; Liu, G.; Xia, M.; Ren, J.; Wang, D. Methyltransferase DNMT3B Promotes Colorectal Cancer Cell Proliferation by Inhibiting PLCG2. Acta Biochim. Biophys. Sin. 2024, 56, 1848–1859. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Chen, Y.; Zhang, L.; Guo, X.; Wei, X.; Shao, Y.; Wang, D.; Wu, B. Effect of CHST11, a Novel Biomarker, on the Biological Functionalities of Clear Cell Renal Cell Carcinoma. Sci. Rep. 2024, 14, 7704. [Google Scholar] [CrossRef] [PubMed]
- Hope, C.; Emmerich, P.B.; Papadas, A.; Pagenkopf, A.; Matkowskyj, K.A.; Van De Hey, D.R.; Payne, S.N.; Clipson, L.; Callander, N.S.; Hematti, P.; et al. Versican-Derived Matrikines Regulate Batf3–Dendritic Cell Differentiation and Promote T Cell Infiltration in Colorectal Cancer. J. Immunol. 2017, 199, 1933–1941. [Google Scholar] [CrossRef] [PubMed]
- Papadas, A.; Asimakopoulos, F. Versican in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1272, 55–72. [Google Scholar]
- Wang, J.; Jiang, Y.-H.; Yang, P.-Y.; Liu, F. Increased Collagen Type V A2 (COL5A2) in Colorectal Cancer Is Associated with Poor Prognosis and Tumor Progression. Onco. Targets Ther. 2021, 14, 2991–3002. [Google Scholar] [CrossRef]
- Albrethsen, J.; Knol, J.C.; Piersma, S.R.; Pham, T.V.; de Wit, M.; Mongera, S.; Carvalho, B.; Verheul, H.M.W.; Fijneman, R.J.A.; Meijer, G.A.; et al. Subnuclear Proteomics in Colorectal Cancer. Mol. Cell. Proteom. 2010, 9, 988–1005. [Google Scholar] [CrossRef]
- Chuang, J.; Wang, C.; Guo, Y.; Valenzuela, V.; Wu, J.; Fakih, M. MAP2K1 Mutations in Advanced Colorectal Cancer Predict Poor Response to Anti-EGFR Therapy and to Vertical Targeting of MAPK Pathway. Clin. Color. Cancer 2021, 20, 72–78. [Google Scholar] [CrossRef]
- Mauri, G.; Patelli, G.; Gori, V.; Lauricella, C.; Mussolin, B.; Amatu, A.; Bencardino, K.; Tosi, F.; Bonazzina, E.; Bonoldi, E.; et al. Corrigendum: Case Report: MAP2K1 K57N Mutation Is Associated with Primary Resistance to Anti-EGFR Monoclonal Antibodies in Metastatic Colorectal Cancer. Front. Oncol. 2023, 13, 1147497. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Oliveros, J.C. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 5 March 2020).
- Edgar, R. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.gtexportal.org/home (accessed on 6 March 2025).
- Available online: https://www.cancer.gov/ccg/research/genome-sequencing/tcga/using-tcga-data/citing (accessed on 6 March 2025).
- Available online: https://www.cancer.gov/ccg/research/genome-sequencing/target/using-target-data/citing (accessed on 6 March 2025).
- Győrffy, B. Integrated Analysis of Public Datasets for the Discovery and Validation of Survival-Associated Genes in Solid Tumors. Innovation 2024, 5, 100625. [Google Scholar] [CrossRef] [PubMed]
- Milacic, M.; Beavers, D.; Conley, P.; Gong, C.; Gillespie, M.; Griss, J.; Haw, R.; Jassal, B.; Matthews, L.; May, B.; et al. The Reactome Pathway Knowledgebase 2024. Nucleic Acids Res. 2024, 52, D672–D678. [Google Scholar] [CrossRef]
- Zhao, T.; Liu, H.; Roeder, K.; Lafferty, J.; Wasserman, L. The Huge Package for High-Dimensional Undirected Graph Estimation in R. J. Mach. Learn. Res. 2012, 13, 1059–1062. [Google Scholar]
- Fisher, R.A. Frequency Distribution of the Values of the Correlation Coefficient in Samples from an Indefinitely Large Population. Biometrika 1915, 10, 507. [Google Scholar] [CrossRef]
- Kou, L.; Markowsky, G.; Berman, L. A Fast Algorithm for Steiner Trees. Acta Inform. 1981, 15, 141–145. [Google Scholar] [CrossRef]
- Pepe, D.; Grassi, M. Investigating Perturbed Pathway Modules from Gene Expression Data via Structural Equation Models. BMC Bioinform. 2014, 15, 132. [Google Scholar] [CrossRef]
- Jankova, J.; van de Geer, S. Confidence Intervals for High-Dimensional Inverse Covariance Estimation. Electron. J. Statist. 2015, 9, 1205–1229. [Google Scholar] [CrossRef]
- Hu, L.; Bentler, P.M. Cutoff Criteria for Fit Indexes in Covariance Structure Analysis: Conventional Criteria versus New Alternatives. Struct. Equ. Model. 1999, 6, 1–55. [Google Scholar] [CrossRef]
- Pons, P.; Latapy, M. Computing Communities in Large Networks Using Random Walks; Springer: Berlin/Heidelberg, Germany, 2005; pp. 284–293. [Google Scholar]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. G:Profiler—Interoperable Web Service for Functional Enrichment Analysis and Gene Identifier Mapping (2023 Update). Nucleic Acids Res. 2023, 51, W207–W212. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Value |
|---|---|
| CRC Sample Number | 185 |
| NAT Sample Number | 157 |
| Age (mean ± SD) | 62.3 ± 10.5 years |
| Sex | Male: 112 (60.5%), Female: 73 (39.5%) |
| Disease Grade | Grade I: 22 (11.9%), Grade II: 58 (31.4%), Grade III: 75 (40.5%), Grade IV: 30 (16.2%) |
| Community_ID | Community Gene Number | Intersection with Reduced Graph | Intersection Percentage |
|---|---|---|---|
| 35 | 5 | 5 | 100 |
| 36 | 5 | 4 | 80 |
| 19 | 13 | 8 | 62 |
| 29 | 5 | 3 | 60 |
| 30 | 6 | 3 | 50 |
| 7 | 66 | 32 | 48 |
| 20 | 10 | 2 | 20 |
| 4 | 48 | 1 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terzi, A.; Maqoud, F.; Guido, D.; Mallardi, D.; Aloisio, M.; Ura, B.; Gualandi, N.; Russo, F.; Giannelli, G. Ion Channel–Extracellular Matrix Interplay in Colorectal Cancer: A Network-Based Approach to Tumor Microenvironment Remodeling. Int. J. Mol. Sci. 2025, 26, 5147. https://doi.org/10.3390/ijms26115147
Terzi A, Maqoud F, Guido D, Mallardi D, Aloisio M, Ura B, Gualandi N, Russo F, Giannelli G. Ion Channel–Extracellular Matrix Interplay in Colorectal Cancer: A Network-Based Approach to Tumor Microenvironment Remodeling. International Journal of Molecular Sciences. 2025; 26(11):5147. https://doi.org/10.3390/ijms26115147
Chicago/Turabian StyleTerzi, Alberta, Fatima Maqoud, Davide Guido, Domenica Mallardi, Michelangelo Aloisio, Blendi Ura, Nicolò Gualandi, Francesco Russo, and Gianluigi Giannelli. 2025. "Ion Channel–Extracellular Matrix Interplay in Colorectal Cancer: A Network-Based Approach to Tumor Microenvironment Remodeling" International Journal of Molecular Sciences 26, no. 11: 5147. https://doi.org/10.3390/ijms26115147
APA StyleTerzi, A., Maqoud, F., Guido, D., Mallardi, D., Aloisio, M., Ura, B., Gualandi, N., Russo, F., & Giannelli, G. (2025). Ion Channel–Extracellular Matrix Interplay in Colorectal Cancer: A Network-Based Approach to Tumor Microenvironment Remodeling. International Journal of Molecular Sciences, 26(11), 5147. https://doi.org/10.3390/ijms26115147