

Acquired Resistance to Decitabine Associated with the Deoxycytidine Kinase A180P Mutation: Implications for the Order of Hypomethylating Agents in Myeloid Malignancies Treatment
 , , , and
, , , and
Abstract
1. Introduction
2. Results
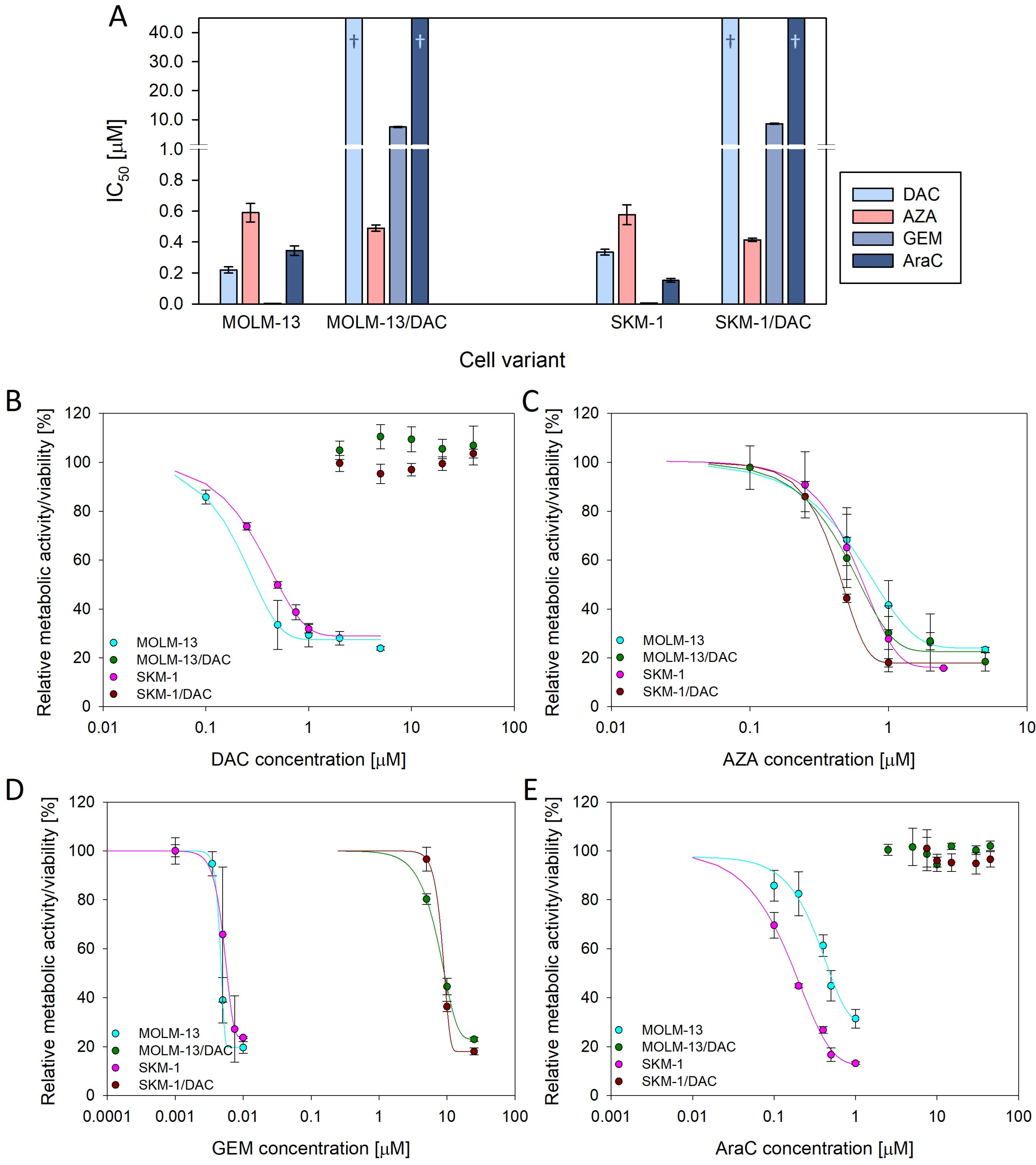
2.1. Resistance to Deoxycytidine Analogs
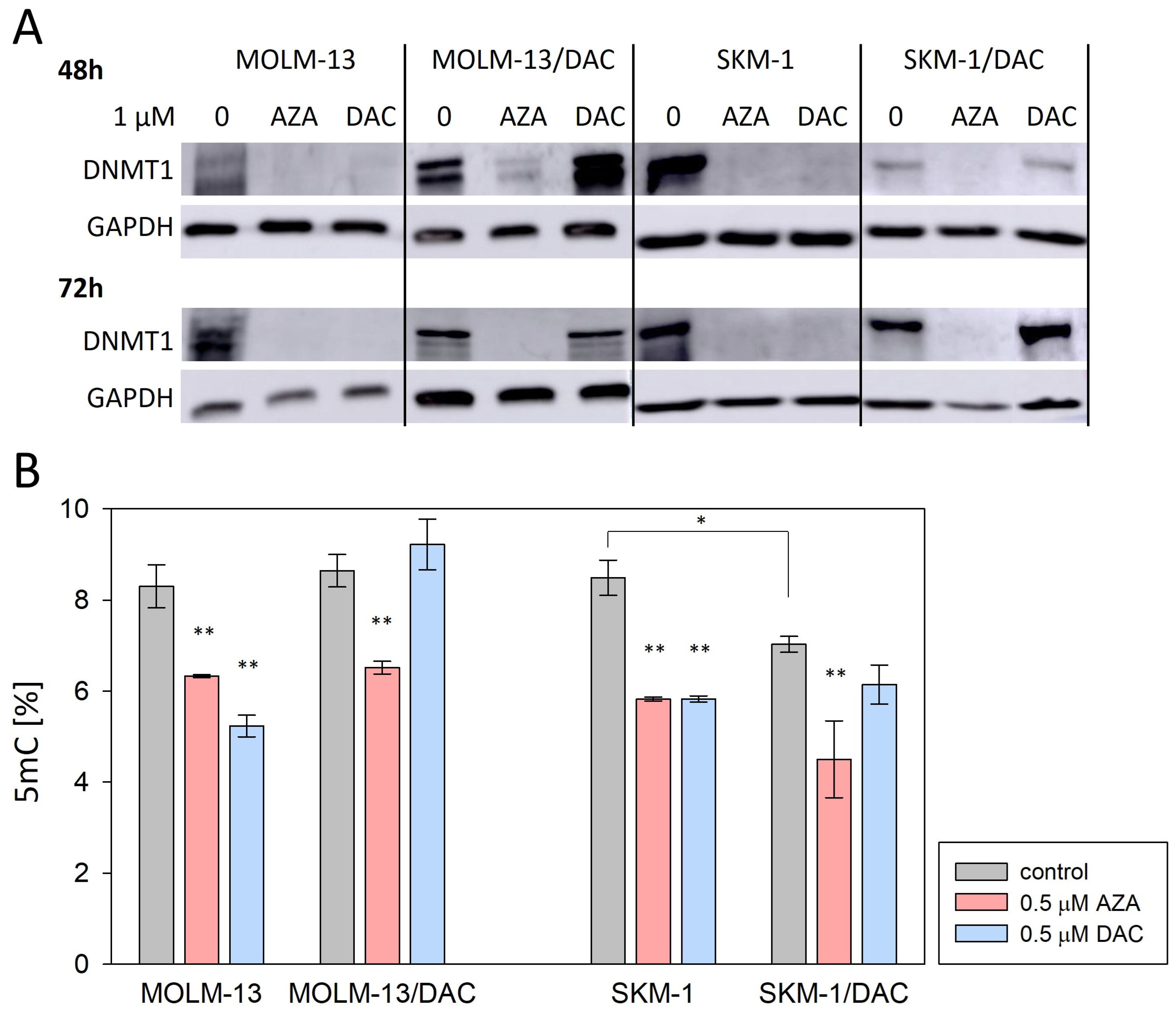
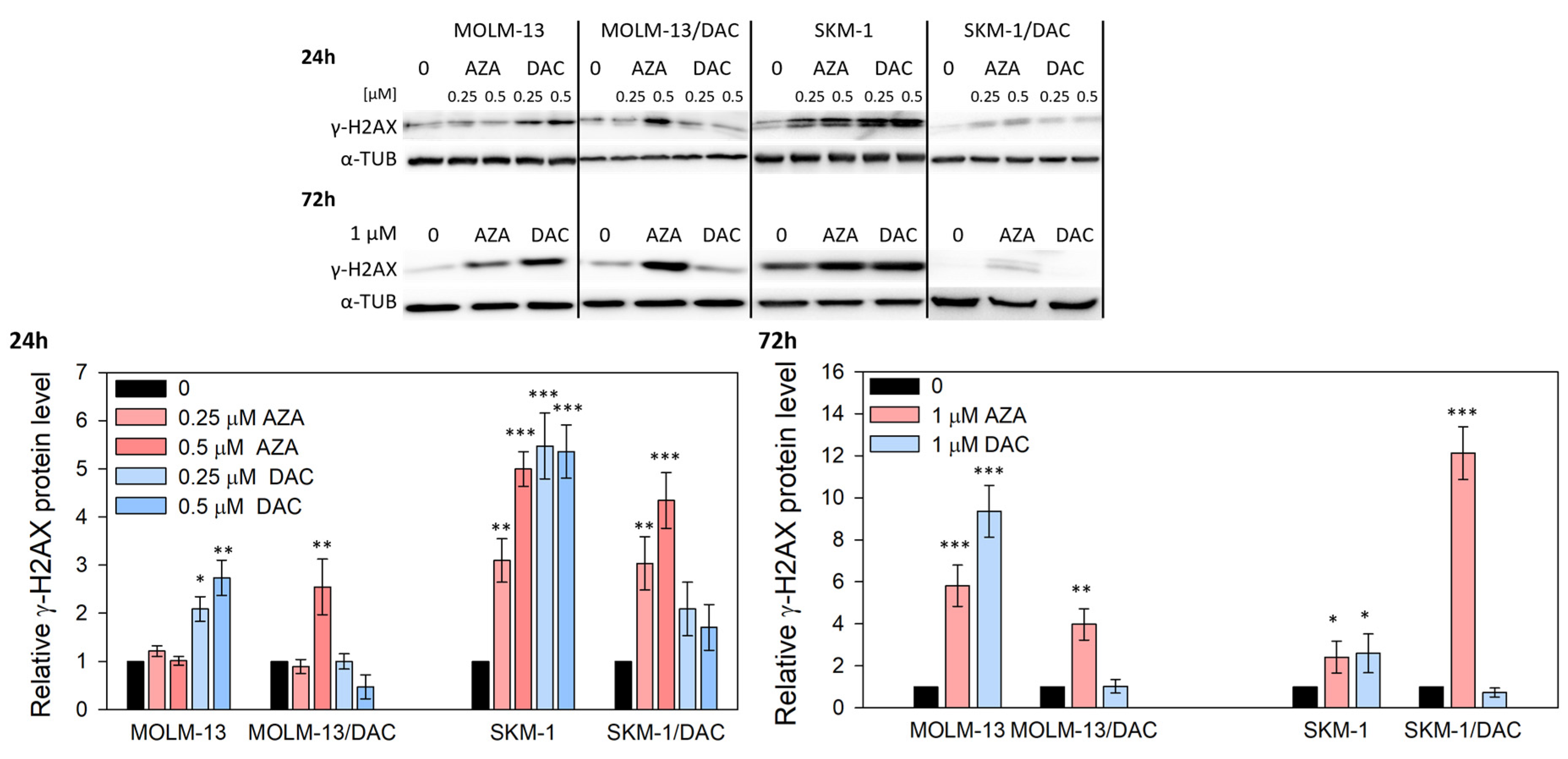
2.2. Alterations in DNA Methylation and DNA Damage Induced by HMAs
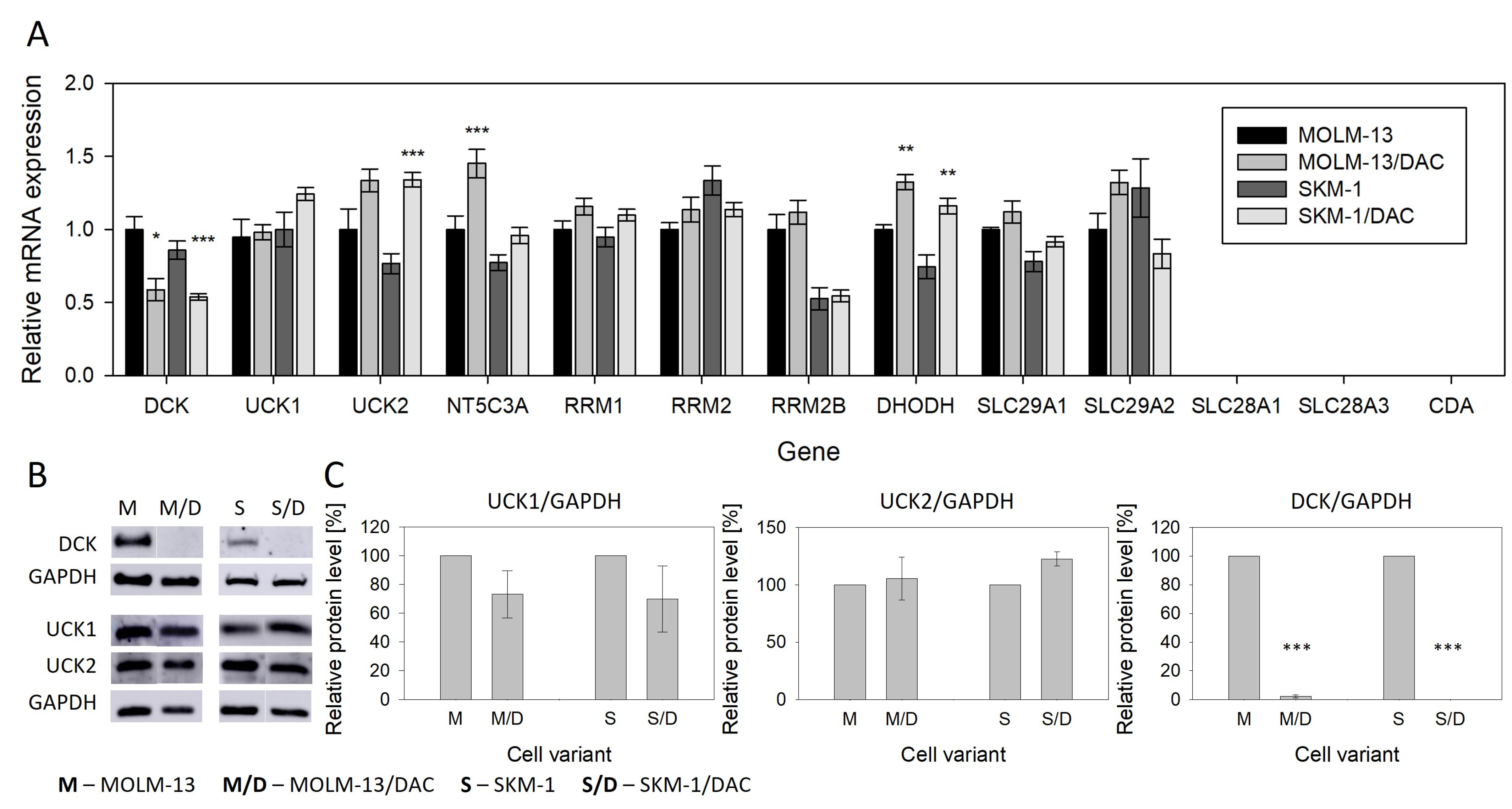
2.3. Expression of Genes Related to Metabolism and Transport of the HMAs
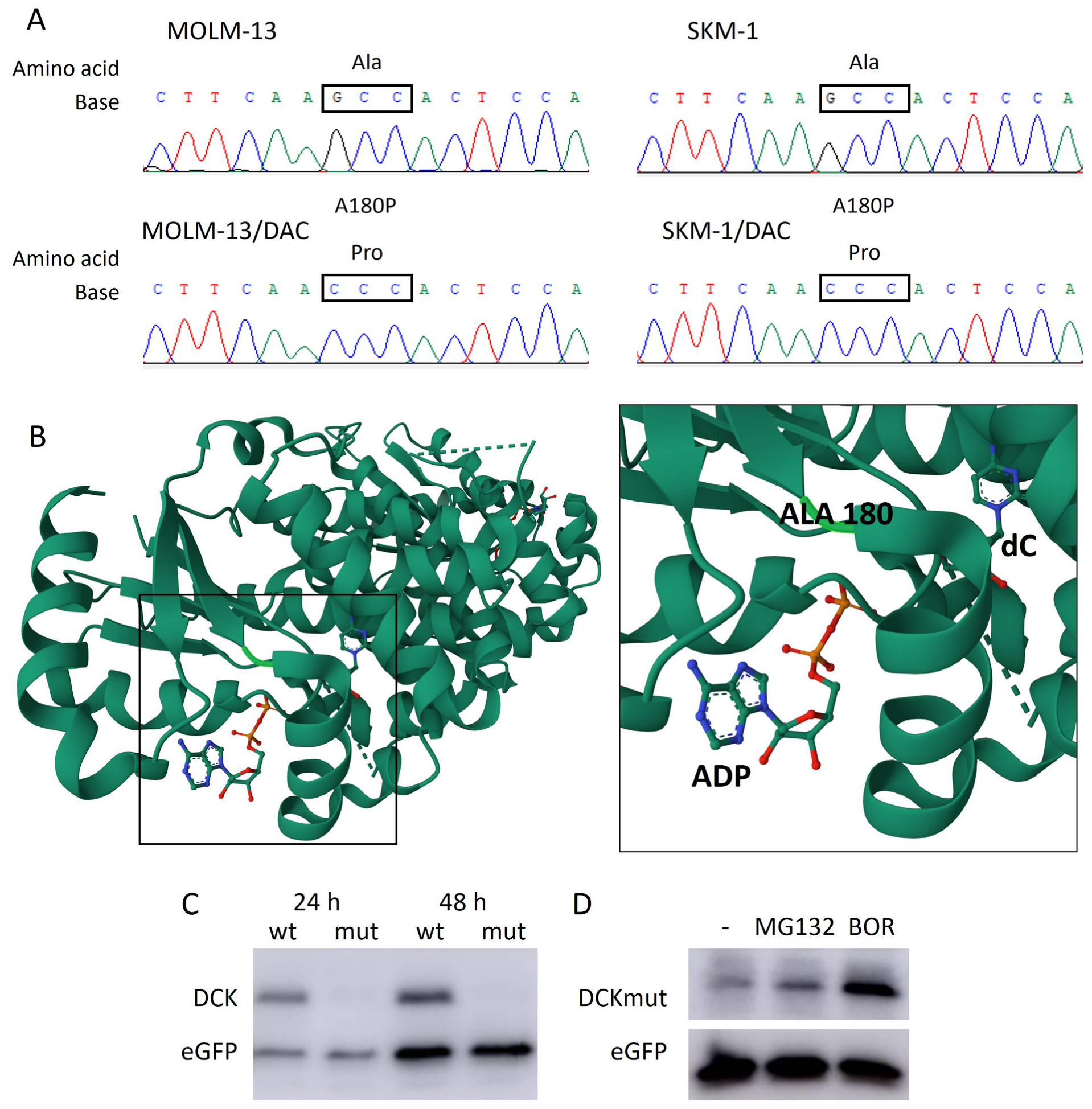
2.4. New A180P Mutation of DCK Associated with DAC Resistance
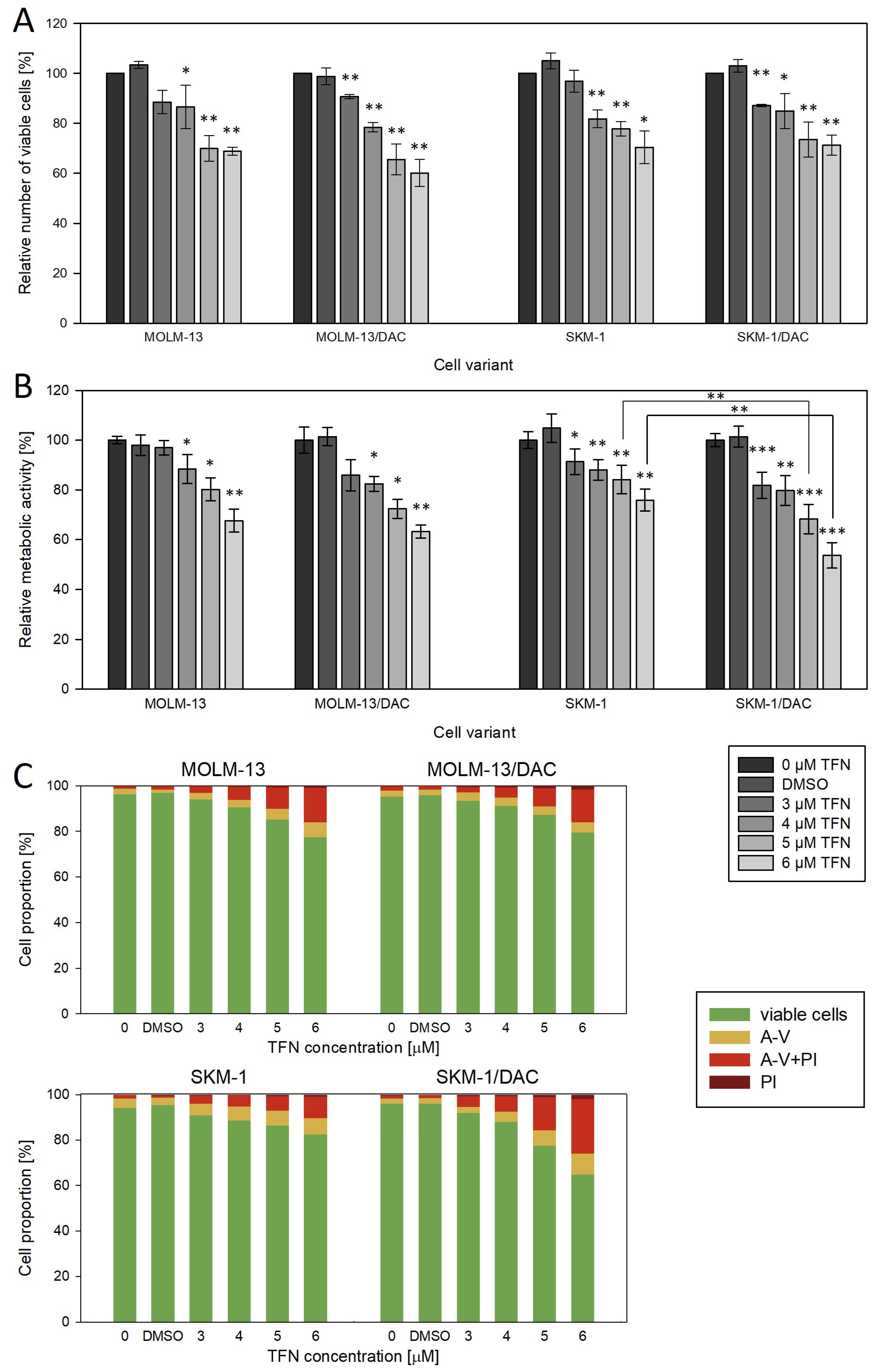
2.5. Effect of Teriflunomide, a Dihydroorotate Dehydrogenase Inhibitor
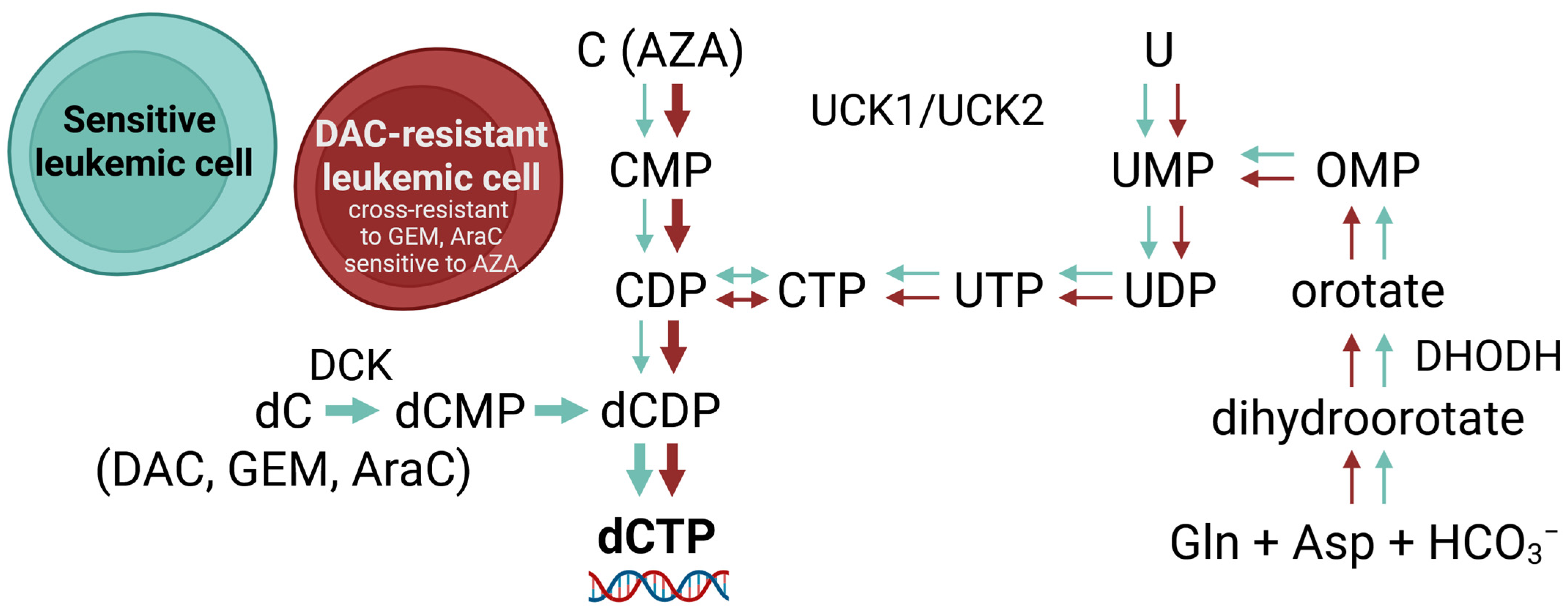
3. Discussion
4. Materials and Methods
4.1. Cell Culture Conditions
4.2. Determination of the Number and Viability of the Cells
4.3. MTS Assay and Determination of IC50 Values
4.4. RNA Isolation and Reverse Transcription
4.5. Determination of mRNA Gene Expression
4.6. Sequence Analysis
4.7. Detection of Gene Expression at the Protein Level
4.8. Preparation of Expression Vectors, Transfection, and Determination of DCKwt and DCKmut Expression
4.9. Detection of Cell Death Mode
4.10. Global DNA Methylation Status Determination
4.11. Statistical Analysis and Data Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, M.; Schnetzke, U.; Hochhaus, A.; Scholl, S. Management of Acute Myeloid Leukemia: Current Treatment Options and Future Perspectives. Cancers 2021, 13, 5722. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.K.; Sabino, A.d.P. Myelodysplastic Neoplasms: An Overview on Diagnosis, Risk-Stratification, Molecular Pathogenesis, and Treatment. Biomed. Pharmacother. 2022, 156, 113905. [Google Scholar] [CrossRef] [PubMed]
- Pleyer, L.; Greil, R. Digging Deep into “Dirty” Drugs-Modulation of the Methylation Machinery. Drug Metab. Rev. 2015, 47, 252–279. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 Recommendations from an International Expert Panel on Behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Fenaux, P.; Haase, D.; Santini, V.; Sanz, G.F.; Platzbecker, U.; Mey, U. Myelodysplastic Syndromes: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up†☆. Ann. Oncol. 2021, 32, 142–156. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Salimi, T.; Epstein, R.S. Real-World Use and Outcomes of Hypomethylating Agent Therapy in Higher-Risk Myelodysplastic Syndromes: Why Are We Not Achieving the Promise of Clinical Trials? Futur. Oncol. 2021, 17, 5163–5175. [Google Scholar] [CrossRef]
- Šimoničová, K.; Janotka, Ľ.; Kavcová, H.; Sulová, Z.; Breier, A.; Messingerova, L. Different Mechanisms of Drug Resistance to Hypomethylating Agents in the Treatment of Myelodysplastic Syndromes and Acute Myeloid Leukemia. Drug Resist. Updat. 2022, 61, 100805. [Google Scholar] [CrossRef]
- Garcia-Manero, G. Myelodysplastic Syndromes: 2023 Update on Diagnosis, Risk-Stratification, and Management. Am. J. Hematol. 2023, 98, 1307–1325. [Google Scholar] [CrossRef]
- Šimoničová, K.; Janotka, L.; Kavcova, H.; Sulova, Z.; Messingerova, L.; Breier, A. Resistance of Leukemia Cells to 5-Azacytidine: Different Responses to the Same Induction Protocol. Cancers 2023, 15, 3063. [Google Scholar] [CrossRef]
- Janotka, Ľ.; Messingerová, L.; Šimoničová, K.; Kavcová, H.; Elefantová, K.; Sulová, Z.; Breier, A. Changes in Apoptotic Pathways in MOLM-13 Cell Lines after Induction of Resistance to Hypomethylating Agents. Int. J. Mol. Sci. 2021, 22, 2076. [Google Scholar] [CrossRef] [PubMed]
- Gnyszka, A.; Jastrzebski, Z.; Flis, S. DNA Methyltransferase Inhibitors and Their Emerging Role in Epigenetic Therapy of Cancer. Anticancer Res. 2013, 33, 2989–2996. [Google Scholar] [PubMed]
- Laranjeira, A.B.A.; Hollingshead, M.G.; Nguyen, D.; Kinders, R.J.; Doroshow, J.H.; Yang, S.X. DNA Damage, Demethylation and Anticancer Activity of DNA Methyltransferase (DNMT) Inhibitors. Sci. Rep. 2023, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Juliusson, G.; Höglund, M.; Lehmann, S. Hypo, Hyper, or Combo: New Paradigm for Treatment of Acute Myeloid Leukemia in Older People. Haematologica 2020, 105, 249–251. [Google Scholar] [CrossRef]
- Sabini, E.; Ort, S.; Monnerjahn, C.; Konrad, M.; Lavie, A. Structure of Human DCK Complexed with 2’-Deoxycytidine and ADP, P 43 21 2 Space Group; 2003. Available online: https://www.rcsb.org/pages/policies (accessed on 14 March 2025). [CrossRef]
- Sabini, E.; Ort, S.; Monnerjahn, C.; Konrad, M.; Lavie, A. Structure of Human DCK Suggests Strategies to Improve Anticancer and Antiviral Therapy. Nat. Struct. Biol. 2003, 10, 513–519. [Google Scholar] [CrossRef]
- Steinhaus, R.; Proft, S.; Schuelke, M.; Cooper, D.N.; Schwarz, J.M.; Seelow, D. MutationTaster2021. Nucleic Acids Res. 2021, 49, W446–W451. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Calabrese, R.; Capriotti, E.; Fariselli, P.; Martelli, P.L.; Casadio, R. Functional Annotations Improve the Predictive Score of Human Disease-Related Mutations in Proteins. Hum. Mutat. 2009, 30, 1237–1244. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Imanishi, S.; Umezu, T.; Ohtsuki, K.; Kobayashi, C.; Ohyashiki, K.; Ohyashiki, J.H. Constitutive Activation of the ATM/BRCA1 Pathway Prevents DNA Damage-Induced Apoptosis in 5-Azacytidine-Resistant Cell Lines. Biochem. Pharmacol. 2014, 89, 361–369. [Google Scholar] [CrossRef]
- Murase, M.; Iwamura, H.; Komatsu, K.; Saito, M.; Maekawa, T.; Nakamura, T.; Yokokawa, T.; Shimada, Y. Lack of Cross-Resistance to FF-10501, an Inhibitor of Inosine-5′-Monophosphate Dehydrogenase, in Azacitidine-Resistant Cell Lines Selected from SKM-1 and MOLM-13 Leukemia Cell Lines. Pharmacol. Res. Perspect. 2016, 4, e00206. [Google Scholar] [CrossRef] [PubMed]
- Hur, E.H.; Jung, S.-H.; Goo, B.-K.; Moon, J.; Choi, Y.; Choi, D.R.; Chung, Y.-J.; Lee, J.-H. Establishment and Characterization of Hypomethylating Agent Resistant Cell Lines, MOLM/AZA-1 and MOLM/DEC-5. Oncotarget 2017, 8, 11748–11762. [Google Scholar] [CrossRef] [PubMed]
- Oellerich, T.; Schneider, C.; Thomas, D.; Knecht, K.M.; Buzovetsky, O.; Kaderali, L.; Schliemann, C.; Bohnenberger, H.; Angenendt, L.; Hartmann, W.; et al. Selective Inactivation of Hypomethylating Agents by SAMHD1 Provides a Rationale for Therapeutic Stratification in AML. Nat. Commun. 2019, 10, 3475. [Google Scholar] [CrossRef] [PubMed]
- Yoshida-Sakai, N.; Watanabe, T.; Yamamoto, Y.; Ureshino, H.; Kamachi, K.; Kurahashi, Y.; Fukuda-Kurahashi, Y.; Kimura, S. Adult T-cell Leukemia-lymphoma Acquires Resistance to DNA Demethylating Agents through Dysregulation of Enzymes Involved in Pyrimidine Metabolism. Int. J. Cancer 2021, 150, 1184–1197. [Google Scholar] [CrossRef]
- Qin, T.; Jelinek, J.; Si, J.; Shu, J.; Issa, J.P.J. Mechanisms of Resistance to 5-Aza-2’-Deoxycytidine in Human Cancer Cell Lines. Blood 2009, 113, 659–667. [Google Scholar] [CrossRef]
- Hosokawa, M.; Saito, M.; Nakano, A.; Iwashita, S.; Ishizaka, A.; Ueda, K.; Iwakawa, S. Acquired Resistance to Decitabine and Cross-Resistance to Gemcitabine during the Long-Term Treatment of Human HCT116 Colorectal Cancer Cells with Decitabine. Oncol. Lett. 2015, 10, 761–767. [Google Scholar] [CrossRef]
- Prébet, T.; Gore, S.D.; Esterni, B.; Gardin, C.; Itzykson, R.; Thepot, S.; Dreyfus, F.; Rauzy, O.B.; Recher, C.; Adès, L.; et al. Outcome of High-Risk Myelodysplastic Syndrome after Azacitidine Treatment Failure. J. Clin. Oncol. 2011, 29, 3322–3327. [Google Scholar] [CrossRef]
- Apuri, S.; Al Ali, N.; Padron, E.; Lancet, J.E.; List, A.F.; Komrokji, R.S. Evidence for Selective Benefit of Sequential Treatment With Hypomethylating Agents in Patients With Myelodysplastic Syndrome. Clin. Lymphoma Myeloma Leuk. 2017, 17, 211–214. [Google Scholar] [CrossRef]
- Harel, S.; Cherait, A.; Berthon, C.; Willekens, C.; Park, S.; Rigal, M.; Brechignac, S.; Thépot, S.; Quesnel, B.; Gardin, C.; et al. Outcome of Patients with High Risk Myelodysplastic Syndrome (MDS) and Advanced Chronic Myelomonocytic Leukemia (CMML) Treated with Decitabine after Azacitidine Failure. Leuk. Res. 2015, 39, 501–504. [Google Scholar] [CrossRef]
- Borthakur, G.; El Ahdab, S.; Garcia-Manero, G.; Ravandi, F.; Ferrajoli, A.; Newman, B.A.; Issa, J.P.; Kantarjian, H. Activity of Decitabine in Patients with Myelodysplastic Syndrome Previously Treated with Azacitidine. Leuk. Lymphoma 2008, 49, 690–695. [Google Scholar] [CrossRef]
- Duong, V.H.; Bhatnagar, B.; Zandberg, D.P.; Vannorsdall, E.J.; Tidwell, M.L.; Chen, Q.; Baer, M.R. Lack of Objective Response of Myelodysplastic Syndromes and Acute Myeloid Leukemia to Decitabine after Failure of Azacitidine. Leuk. Lymphoma 2014, 56, 1718–1722. [Google Scholar] [CrossRef] [PubMed]
- Pandit, B.; Royzen, M. Recent Development of Prodrugs of Gemcitabine. Genes 2022, 13, 466. [Google Scholar] [CrossRef] [PubMed]
- Buocikova, V.; Tyciakova, S.; Pilalis, E.; Mastrokalou, C.; Urbanova, M.; Matuskova, M.; Demkova, L.; Medova, V.; Longhin, E.M.; Rundén-Pran, E.; et al. Decitabine-Induced DNA Methylation-Mediated Transcriptomic Reprogramming in Human Breast Cancer Cell Lines; the Impact of DCK Overexpression. Front. Pharmacol. 2022, 13, 991751. [Google Scholar] [CrossRef] [PubMed]
- Derissen, E.J.B.; Beijnen, J.H. Intracellular Pharmacokinetics of Pyrimidine Analogues Used in Oncology and the Correlation with Drug Action. Clin. Pharmacokinet. 2020, 59, 1521–1550. [Google Scholar] [CrossRef]
- Vadeikienė, R.; Jakštys, B.; Ugenskienė, R.; Šatkauskas, S.; Juozaitytė, E. Systemic Optimization of Gene Electrotransfer Protocol Using Hard-to-Transfect UT-7 Cell Line as a Model. Biomedicines 2022, 10, 2687. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, Z.; Wang, Y.; Du, W.; Song, X.; Lai, W.; Wang, H.; Zhu, B.; Xiong, J. A CRISPR-Cas9 screen reveals genetic determinants of the cellular response to decitabine. Embo Rep. 2025, 26, 1528–1565. [Google Scholar] [CrossRef]
- Dahn, M.L.; Cruickshank, B.M.; Jackson, A.J.; Dean, C.; Holloway, R.W.; Hall, S.R.; Coyle, K.M.; Maillet, H.; Waisman, D.M.; Goralski, K.B.; et al. Decitabine Response in Breast Cancer Requires Efficient Drug Processing and Is Not Limited by Multidrug Resistance. Mol. Cancer Ther. 2020, 19, 1110–1122. [Google Scholar] [CrossRef]
- Gruber, E.; Franich, R.L.; Shortt, J.; Johnstone, R.W.; Kats, L.M. Distinct and overlapping mechanisms of resistance to azacytidine and guadecitabine in acute myeloid leukemia. Leukemia 2020, 34, 3388–3392. [Google Scholar] [CrossRef]
- Stegmann, A.; Honders, W.; Willemze, R.; van Haperen, V.R.; Landegent, J. Transfection of wild-type deoxycytidine kinase (dck) cDNA into an AraC- and DAC-resistant rat leukemic cell line of clonal origin fully restores drug sensitivity. Blood 1995, 85, 1188–1194. [Google Scholar] [CrossRef]
- Nakano, T.; Saiki, Y.; Kudo, C.; Hirayama, A.; Mizuguchi, Y.; Fujiwara, S.; Soga, T.; Sunamura, M.; Matsumura, N.; Motoi, F.; et al. Acquisition of chemoresistance to gemcitabine is induced by a loss-of-function missense mutation of DCK. Biochem. Biophys. Res. Commun. 2015, 464, 1084–1089. [Google Scholar] [CrossRef]
- Qin, T.; Castoro, R.; El Ahdab, S.; Jelinek, J.; Wang, X.; Si, J.; Shu, J.; He, R.; Zhang, N.; Chung, W.; et al. Mechanisms of Resistance to Decitabine in the Myelodysplastic Syndrome. PLoS ONE 2011, 6, e23372. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Geng, S.; Weng, J.; Deng, C.; Lu, Z.; Luo, C.; Du, X. The HENT1 and DCK Genes Underlie the Decitabine Response in Patients with Myelodysplastic Syndrome. Leuk. Res. 2015, 39, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Shi, W.; Li, X.; Chang, C.; Xu, F.; He, Q.; Wu, D.; Su, J.; Zhou, L.; Song, L.; et al. High Expression of the Human Equilibrative Nucleoside Transporter 1 Gene Predicts a Good Response to Decitabine in Patients with Myelodysplastic Syndrome. J. Transl. Med. 2016, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Slusarczyk, M.; Lopez, M.H.; Balzarini, J.; Mason, M.; Jiang, W.G.; Blagden, S.; Thompson, E.; Ghazaly, E.; McGuigan, C. Application of ProTide Technology to Gemcitabine: A Successful Approach to Overcome the Key Cancer Resistance Mechanisms Leads to a New Agent (NUC-1031) in Clinical Development. J. Med. Chem. 2014, 57, 1531–1542. [Google Scholar] [CrossRef]
- Daifuku, R.; Koratich, M.; Stackhouse, M. Vitamin E Phosphate Nucleoside Prodrugs: A Platform for Intracellular Delivery of Monophosphorylated Nucleosides. Pharmaceuticals 2018, 11, 16. [Google Scholar] [CrossRef]
- Blagden, S.P.; Rizzuto, I.; Suppiah, P.; O’Shea, D.; Patel, M.; Spiers, L.; Sukumaran, A.; Bharwani, N.; Rockall, A.; Gabra, H.; et al. Anti-Tumour Activity of a First-in-Class Agent NUC-1031 in Patients with Advanced Cancer: Results of a Phase I Study. Br. J. Cancer 2018, 119, 815–822. [Google Scholar] [CrossRef]
- Momparler, R.L.; Momparler, L.F. Chemotherapy of L1210 and L1210/ARA-C Leukemia with 5-Aza-2′-Deoxycytidine and 3-Deazauridine. Cancer Chemother. Pharmacol. 1989, 25, 51–54. [Google Scholar] [CrossRef]
- Meinsma, R.; van Kuilenburg, A.B.P. Purification, Activity, and Expression Levels of Two Uridine-Cytidine Kinase Isoforms in Neuroblastoma Cell Lines. Nucleosides Nucleotides Nucleic Acids 2016, 35, 613–618. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| In Silico Tool | Predicted Mutation Effect | Score a |
|---|---|---|
| MutationTaster [17] | Disease causing (probably deleterious) | Probability value 0.999 |
| PolyPhen-2 [18] | Probably damaging | Score 0.998; sensitivity 0.27; specificity 0.99 |
| SNPs&GO [19] | Disease | Reliability index 4 |
| SIFT [20] | Tolerated | Probability value 0.09 |
| Gene | Primer Sequences (5′-3′) | TA (°C) | PCR Product Size (bp) |
|---|---|---|---|
| ACTB | GACGACATGGAGAAAATCTG ATGATCTGGGTCATCTTCTC | 60 | 131 |
| CDA | GCAACATAGAAAATGCCTGCT TAGCAATTGCCCTGAAATCC | 60 | 102 |
| DCK | GTCTCAGAAAAATGGTGGGAATG ACAGGTTTCTCTGCATCTTTGAG | 60 | 150 |
| UCK1 | CGTGTGTGAGAAGATCATGG TGGTCAAAATTGTACTGTCCTTT | 60 | 150 |
| UCK2 | GACATCAGCGAGAGAGGCAG TCTTGCGTGAAGGGGTGTAG | 60 | 187 |
| NT5C3A | GTCAAGCCTGCCTTTGAGGA TTCCTCAAGGCACCATCATGT | 60 | 198 |
| RRM1 | TGGAATTGGGGTACAAGGTC GAGAGCCCTCATAGGTTTCG | 60 | 176 |
| RRM2 | TTTACACTGTGATTTTGCTTGC TGTTCTATCCGAACAGCATTG | 60 | 102 |
| RRM2B | ATAAACAGGCACAGGCTTCC GAACCTGCACCTCCTGACTAA | 60 | 186 |
| SLC29A1 | CATGCTGCCCCTGCTGTTAT CTGCACCTTCACCAGGATGG | 60 | 136 |
| SLC29A2 | TCCTTCCTGTACCAGTGCGT ATGTCCACCTTGACCAGCG | 60 | 104 |
| SLC28A1 | AGGTTCTGCCCATCATTGTC CAAGTAGGGCCGGATCAGTA | 60 | 197 |
| SLC28A3 | GACTCACATCCATGGCTCCT TTCCAGGGAAAGTGGAGTTG | 60 | 183 |
| DHODH | CTGAACACCTGATGCCGACT AGTCTTGAAATCTGGCCCGT | 60 | 111 |
| DCK a | CAGGATCTGGCTTAGCGG CATTTGGCTGCCTGTAGTCT | 63 | 914 |
| UCK1 a | AGATGGCTTCGGCGGGA AGTCCCTGAACACACATGCC | 65 | 890 |
| UCK2 a | AACCATGGCCGGGGACAG GATGAGCAGTGCCTCCTGAC | 65 | 858 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simonicova, K.; Janotka, L.; Kavcova, H.; Borovska, I.; Sulova, Z.; Breier, A.; Messingerova, L. Acquired Resistance to Decitabine Associated with the Deoxycytidine Kinase A180P Mutation: Implications for the Order of Hypomethylating Agents in Myeloid Malignancies Treatment. Int. J. Mol. Sci. 2025, 26, 5083. https://doi.org/10.3390/ijms26115083
Simonicova K, Janotka L, Kavcova H, Borovska I, Sulova Z, Breier A, Messingerova L. Acquired Resistance to Decitabine Associated with the Deoxycytidine Kinase A180P Mutation: Implications for the Order of Hypomethylating Agents in Myeloid Malignancies Treatment. International Journal of Molecular Sciences. 2025; 26(11):5083. https://doi.org/10.3390/ijms26115083
Chicago/Turabian StyleSimonicova, Kristina, Lubos Janotka, Helena Kavcova, Ivana Borovska, Zdena Sulova, Albert Breier, and Lucia Messingerova. 2025. "Acquired Resistance to Decitabine Associated with the Deoxycytidine Kinase A180P Mutation: Implications for the Order of Hypomethylating Agents in Myeloid Malignancies Treatment" International Journal of Molecular Sciences 26, no. 11: 5083. https://doi.org/10.3390/ijms26115083
APA StyleSimonicova, K., Janotka, L., Kavcova, H., Borovska, I., Sulova, Z., Breier, A., & Messingerova, L. (2025). Acquired Resistance to Decitabine Associated with the Deoxycytidine Kinase A180P Mutation: Implications for the Order of Hypomethylating Agents in Myeloid Malignancies Treatment. International Journal of Molecular Sciences, 26(11), 5083. https://doi.org/10.3390/ijms26115083