
Phosphazenyl Phosphine Proton Sponges: Does the Proton-Chelating Effect Enhance Their Basicity?




Abstract
1. Introduction
2. Results and Discussion
2.1. Hydrogen Bond in Phosphine Dimers
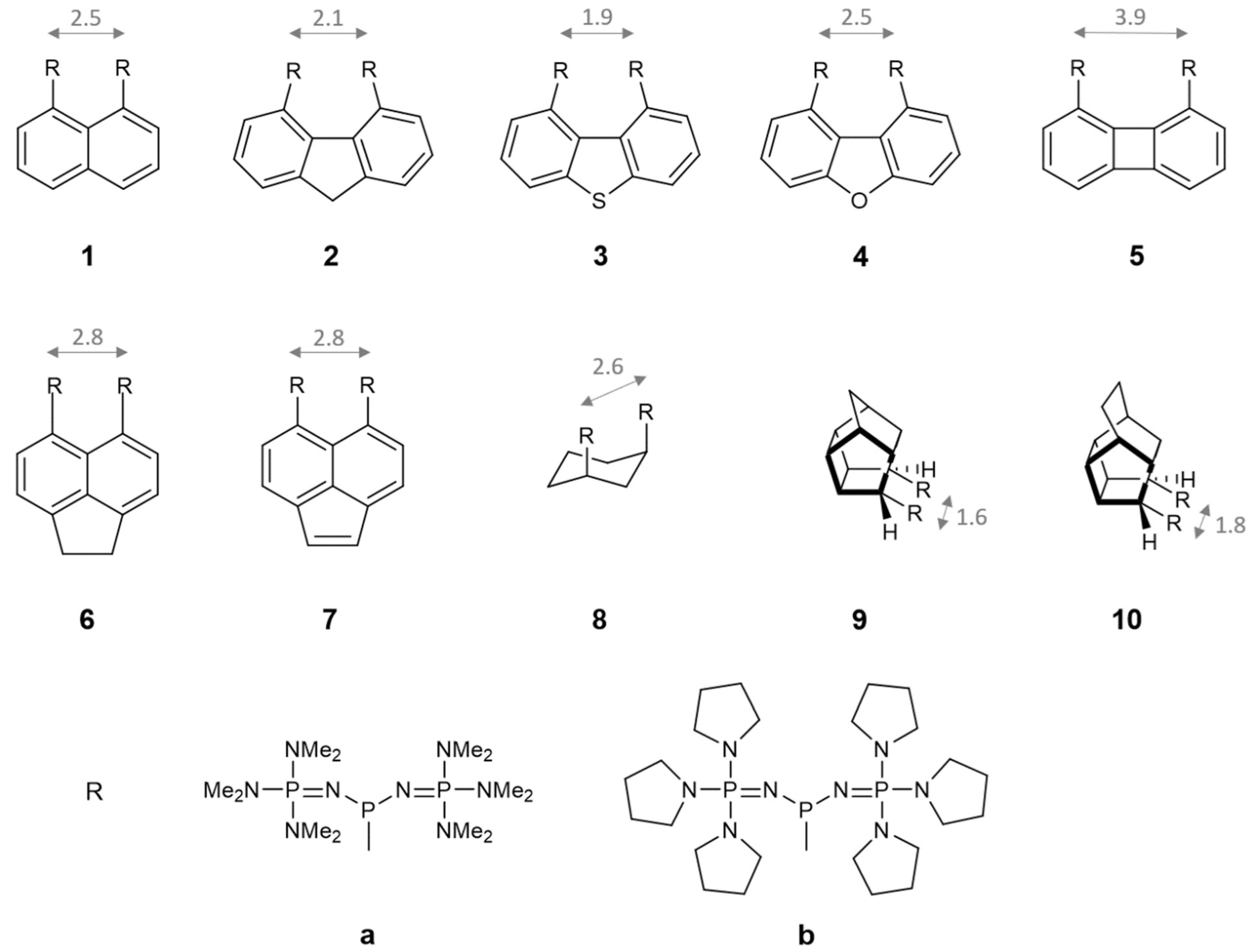
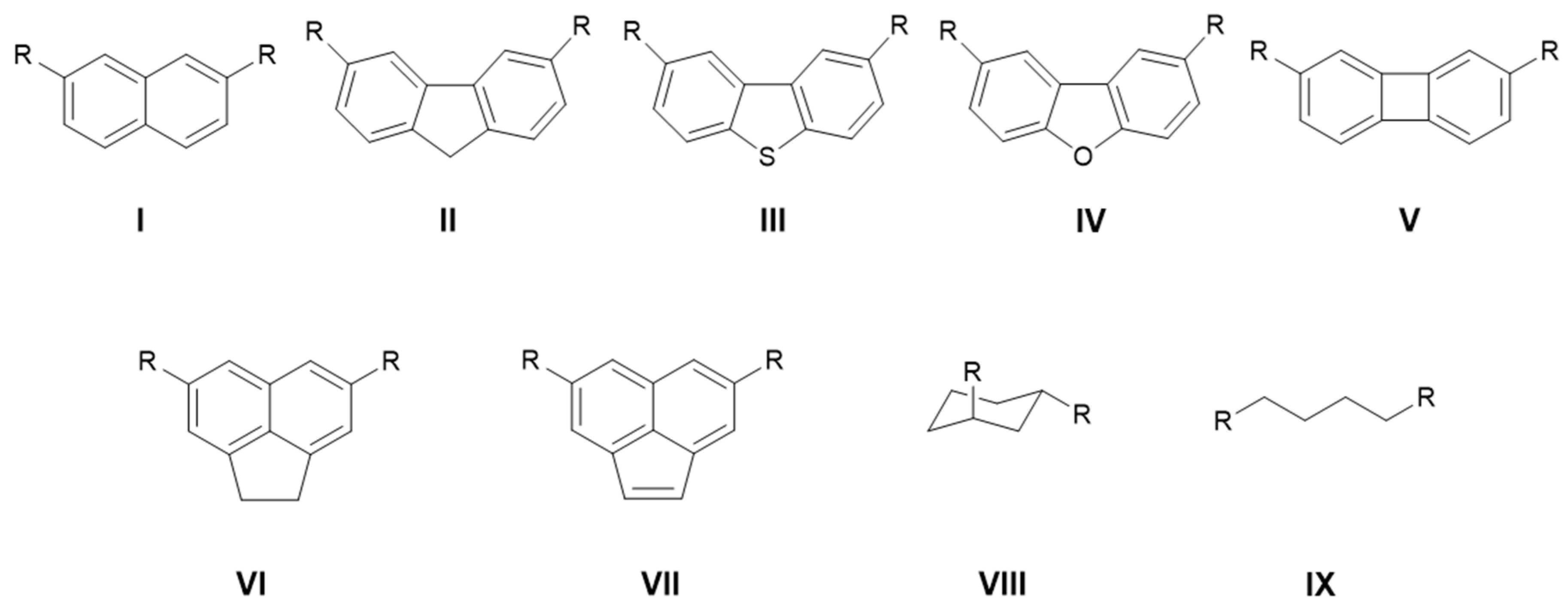
2.2. Proton-Chelating Effect in Bisphosphazenyl Phosphines
2.3. Basicity in Acetonitrile
2.4. AIM and NBO Analysis
3. Computational Details
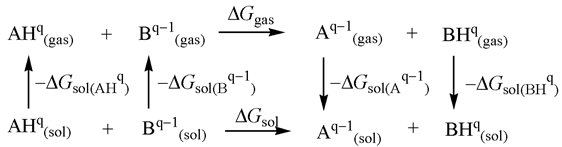
ΔGsol(A) − ΔGsol(BH+) + ΔGsol(AH+)}/2.303RT
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maksić, Z.B.; Kovačević, B.; Vianello, R. Advances in determining the absolute proton affinities of neutral organic molecules in the gas phase and their interpretation: A theoretical account. Chem. Rev. 2012, 112, 5240–5270. [Google Scholar] [CrossRef]
- Raczynska, E.D.; Gal, J.-F.; Maria, P.-C. Enhanced Basicity of Push–Pull Nitrogen Bases in the Gas Phase. Chem. Rev. 2016, 116, 13454–13511. [Google Scholar] [CrossRef]
- Raczynska, E.D.; Gal, J.-F.; Maria, P.-C. Strong Bases and beyond: The Prominent Contribution of Neutral Push–Pull Organic Molecules towards Superbases in the Gas Phase. Int. J. Mol. Sci. 2024, 25, 5591. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Margetić, D. Superbases for organic synthesis: Guanidines, amidines, phosphaznes and related organocatalysts. In Perspectives; Ishikawa, T., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2009; pp. 315–320. [Google Scholar]
- Puleo, T.R.; Sujansky, S.J.; Wright, S.E.; Bandar, J.S. Organic Superbases in Recent Synthetic Methodology Research. Chem. A Eur. J. 2021, 27, 4216–4229. [Google Scholar] [CrossRef]
- Weitkamp, R.F.; Neumann, B.; Stammler, H.-G.; Hoge, B. Phosphorus-Containing Superbases: Recent Progress in the Chemistry of Electron-Abundant Phosphines and Phosphazenes. Chem. Eur. J. 2021, 27, 10807–10825. [Google Scholar] [CrossRef]
- Vazdar, K.; Margetić, D.; Kovačević, B.; Sundermeyer, J.; Leito, I.; Jahn, U. Design of Novel Uncharged Organic Superbases: Merging Basicity and Functionality. Acc. Chem. Res. 2021, 54, 3108–3123. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Dopke, J.; Verkade, J.G. Synthesis of New Exceedingly Strong Non-ionic Bases: RN:P(MeNCH2CH2)3N. J. Am. Chem. Soc. 1993, 115, 5015–5020. [Google Scholar] [CrossRef]
- Buß, F.; Röthel, M.B.; Werra, J.A.; Rotering, P.; Wilm, L.F.B.; Daniliuc, C.G.; Löwe, P.; Dielmann, F. Tris(tetramethylguanidinyl)phosphine: The Simplest Non-ionic Phosphorus Superbase and Strongly Donating Phosphine Ligand. Chem. Eur. J. 2022, 28, e202104021. [Google Scholar] [CrossRef]
- Mehlmann, P.; Mück-Lichtenfeld, C.; Tan, T.T.Y.; Dielmann, F. Tris(imidazolin-2-ylidenamino)phosphine: A Crystalline Phosphorus(III) Superbase That Splits Carbon Dioxide. Chem. Eur. J. 2017, 23, 5929–5933. [Google Scholar] [CrossRef]
- Ullrich, S.; Kovačević, B.; Xie, X.; Sundermeyer, J. Phosphazenyl Phosphines: The Most Electron-Rich Uncharged Phosphorus Brønsted and Lewis Bases. Angew. Chem. Int. Ed. 2019, 58, 10335–10339. [Google Scholar] [CrossRef]
- Schwesinger, R.; Schlemper, H. Peralkylated Polyaminophosphazenes—Extremely Strong, Neutral Nitrogen Bases. Angew. Chem. Int. Ed. Engl. 1987, 26, 1167–1169. [Google Scholar] [CrossRef]
- Kögel, J.F.; Oelkers, B.; Kovačević, B.; Sundermeyer, J. A New Synthetic Pathway to the Second and Third Generation of Superbasic Bisphosphazene Proton Sponges: The Run for the Best Chelating Ligand for a Proton. J. Am. Chem. Soc. 2013, 35, 17768–17774. [Google Scholar] [CrossRef] [PubMed]
- Alder, R.W.; Bowman, P.S.; Steele, W.R.S.; Winterman, D.R. The remarkable basicity of 1,8-bis(dimethylamino)naphthalene. Chem. Commun. 1968, 13, 723–724. [Google Scholar]
- Kovačević, B.; Maksić, Z.B. The Proton Affinity of the Superbase 1,8-Bis(tetramethylguanidino)naphthalene (TMGN) and Some Related Compounds: A Theoretical Study. Chem. Eur. J. 2002, 8, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Rabb, V.; Kipke, J.; Gschwind, R.M.; Sundermeyer, J. 1,8-Bis(tetramethylguanidino)naphthalene (TMGN): A New, Superbasic and Kinetically Active “Proton Sponge”. Chem. Eur. J. 2002, 8, 1682–1693. [Google Scholar] [CrossRef]
- Belding, L.; Dudding, T. Synthesis and theoretical investigation of a 1,8-bis(bis(diisopropylamino)cyclopropeniminyl)naphthalene proton sponge derivative. Chem. Eur. J. 2014, 20, 1032–1037. [Google Scholar] [CrossRef]
- Belding, L.; Stoyanov, P.; Dudding, T. Synthesis, Theoretical Analysis, and Experimental pKa Determination of a Fluorescent, Nonsymmetric, In−Out Proton Sponge. J. Org. Chem. 2016, 81, 6–13. [Google Scholar] [CrossRef]
- Raab, V.; Gauchenova, E.; Merkoulov, A.; Harms, K.; Sundermeyer, J.; Kovačević, B.; Maksić, Z.B. 1,8-Bis(hexamethyltriaminophosphazenyl)naphthalene, HMPN: A Superbasic Bisphosphazene “Proton Sponge”. J. Am. Chem. Soc. 2005, 127, 15738–15743. [Google Scholar] [CrossRef]
- Kögel, J.F.; Xie, X.; Baal, E.; Gesevičius, D.; Oelkers, B.; Kovačević, B.; Sundermeyer, J. Superbasic Alkyl-Substituted Bisphosphazene Proton Sponges: Synthesis, Structural Features, Thermodynamic and Kinetic Basicity, Nucleophilicity and Coordination Chemistry. Chem. Eur. J. 2014, 20, 7670–7685. [Google Scholar] [CrossRef]
- Saha, A.; Ganguly, B. Exploiting the (–C–H···C–) Interaction to Design Cage-Functionalized Organic Superbases and Hyperbases: A Computational Study. ACS Omega 2023, 8, 38546–38556. [Google Scholar] [CrossRef]
- Kögel, J.F.; Ullrich, S.; Xie, X.; Finger, L.H.; Kovačević, B.; Saame, J.; Haljasorg, T.; Leito, I.; Sundermeyer, J. The Next Generation of Phosphorus Bisylide Superbases—Synthesis, Structures, Basicity and Proton Self-Exchange. Chem. Eur. J. 2025, 31, e202404692. [Google Scholar] [CrossRef] [PubMed]
- Reiter, S.A.; Nogai, S.D.; Karaghiosoff, K.; Schmidbaur, H. Insignificance of P-H⋯P hydrogen bonding: Structural chemistry of neutral and protonated 1,8-di(phosphinyl)naphthalene. J. Am. Chem. Soc. 2004, 126, 15833–15843. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Elguero, J. Systematic ab initio study of 15N-15N and 15N-1H spin-spin coupling constants across N-H+-N hydrogen bonds: Predicting N-N and N-H coupling constants and relating them to hydrogen bond type. J. Phys. Chem. A 2006, 110, 7496–7502. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Elguero, J.; Alkorta, I. Probing P-H+-P hydrogen bonds: Structures, binding energies, and spin-spin coupling constants. J. Phys. Chem. A 2007, 111, 3416–3422. [Google Scholar] [CrossRef]
- Haav, K.; Saame, J.; Kutt, A.; Leito, I. Basicity of Phosphanes and Diphosphanes in Acetonitrile. Eur. J. Org. Chem. 2012, 2012, 2167–2172. [Google Scholar] [CrossRef]
- Yamagdni, R.; Kebarle, P. Gas-phase basicities of amines: Hydrogen bonding in proton-bound amine dimers and proton-induced cyclization of .alpha.,.omega.-diamines. J. Am. Chem. Soc. 1973, 95, 3504–3510. [Google Scholar] [CrossRef]
- Grabowski, S.J. Theoretical studies of strong hydrogen bonds. Annu. Rep. Prog. Chem., Sect. C 2006, 102, 131–165. [Google Scholar] [CrossRef]
- Meot-Ner, M. Update 1 of: Strong Ionic Hydrogen Bonds. Chem. Rev. 2012, 112, PR22–PR103. [Google Scholar] [CrossRef]
- Despotović, I.; Kovačević, B.; Maksić, Z.B. Hyperstrong Neutral Organic Bases: Phosphazeno Azacalix[3](2,6)pyridines. Org. Lett. 2007, 23, 4709–4712. [Google Scholar] [CrossRef]
- Ahmet, A.; Neese, F.; Bistoni, G. HFLD: A Nonempirical London Dispersion-Corrected Hartree–Fock Method for the Quantification and Analysis of Noncovalent Interaction Energies of Large Molecular Systems. J. Chem. Theory Comput. 2019, 15, 5894–5907. [Google Scholar] [CrossRef]
- Glasovac, Z.; Kovačević, B. Modeling pKa of the Brønsted Bases as an Approach to the Gibbs Energy of the Proton in Acetonitrile. Int. J. Mol. Sci. 2022, 23, 10576. [Google Scholar] [CrossRef] [PubMed]
- Afonin, A.V.; Vashchenkoa, A.V.; Sigalov, M.V. Estimating the energy of intramolecular hydrogen bonds from 1H NMR and QTAIM calculations. Org. Biomol. Chem. 2016, 14, 11199–11211. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Foresman, J.B.; Keith, T.A.; Wiberg, K.B.; Snoonian, J.; Frisch, M.J. Solvent Effects. 5. Influence of Cavity Shape, Truncation of Electrostatics, and Electron Correlation on ab Initio Reaction Field Calculations. J. Phys. Chem. 1996, 100, 16098–16104. [Google Scholar] [CrossRef]
- Schaftenaar, G.; Nordik, J.H. Molden: A Pre- and Post-processing Program for Molecular and Electronic Structures. J. Comput. Aided Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software update: The ORCA program system, version 5.0 WIRES. Comput. Molec. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Neese, F.; Hansen, A.; Liakos, D.G. Efficient and accurate approximations to the local coupled cluster singles doubles method using a truncated pair natural orbital basis. J. Chem. Phys. 2009, 131, 064103. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A. Efficient and accurate local approximations to coupled-electron pair approaches: An attempt to revive the pair natural orbital method. J. Chem. Phys. 2009, 130, 114108. [Google Scholar] [CrossRef]
- Riplinger, C.; Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013, 138, 034106. [Google Scholar] [CrossRef]
- Datta, D.; Kossmann, S.; Neese, F. Analytic energy derivatives for the calculation of the first-order molecular properties using the domain-based local pair-natural orbital coupled-cluster theory. J. Chem. Phys. 2016, 145, 114101. [Google Scholar] [CrossRef]
- Riplinger, C.; Pinski, P.; Becker, U.; Valeev, E.F.; Neese, F. Sparse maps-A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory. J. Chem. Phys. 2016, 144, 024109. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.B.; Bistoni, G.; Sparta, M.; Saitow, M.; Riplinger, C.; Auer, A.A.; Neese, F. Decomposition of Intermolecular Interaction Energies within the Local Pair Natural Orbital Coupled Cluster Framework. J. Theo. Comp. Chem. 2016, 12, 4778–4792. [Google Scholar] [CrossRef] [PubMed]
- Bistoni, G.; Riplinger, C.; Minenkov, Y.; Cavallo, L.; Auer, A.A.; Neese, F. Treating Subvalence Correlation Effects in Domain Based Pair Natural Orbital Coupled Cluster Calculations: An Out-of-the-Box Approach. J. Theo. Comp. Chem. 2017, 13, 3220–3227. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The SHARK Integral Generation and Digestion System. J. Comp. Chem. 2022, 44, 381–396. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PH3 | P(CH3)3 | P(Ph)3 | P(tmg)3 | P(imme)3 | (dmaP)3P | (pyrrP)3P | N(CH3)3 | |
|---|---|---|---|---|---|---|---|---|
| HF | 4.4 | 9.1 | 9.0 | 15.2 (18.3) | 17.9 (23.8) | 18.4 (18.8) | pta (23.8) | 14.9 |
| PH4+ | PH(CH3)3+ | PH(Ph)3+ | PH(tmg)3+ | PH(imme)3+ | (dma)P3PH+ | (pyrr)P3PH+ | NH(CH3)3+ | |
| NH3 | pt b | 10.3 | 8.3 | 3.6 | 6.0 | 3.3 | 4.9 | 20.1 |
| Molecule | PA | GB | Molecule | PA | GB | ΔPA | En | Ep |
|---|---|---|---|---|---|---|---|---|
| 1aa | 292.9 | 285.1 | 1a | 281.5 | 273.5 | 11.4 | 3.6 | −7.8 |
| 1bb | 301.0 | 292.4 | 1b | 284.5 | 276.7 | 16.5 | 2.9 | −13.7 |
| 2aa | 292.7 | 284.8 | 2a | 280.9 | 274.5 | 11.8 | 2.1 | −9.7 |
| 2bb | 297.7 | 290.8 | 2b | 288.1 | 279.3 | 9.6 | −1.5 | −11.1 |
| 3aa | 291.2 | 283.9 | 3a | 278.7 | 272.3 | 12.6 | 3.2 | −9.4 |
| 3bb | 296.8 | 291.1 | 3b | 283.8 | 276.5 | 13.0 | −1.2 | −14.2 |
| 4aa | 291.1 | 283.7 | 4a | 280.1 | 272.5 | 10.9 | 1.3 | −9.6 |
| 4bb | 295.8 | 289.0 | 4b | 284.3 | 276.9 | 11.5 | −3.9 | −15.4 |
| 5aa | 294.0 | 286.4 | 5a | 280.8 | 273.9 | 13.2 | −2.0 | −15.2 |
| 5bb | 295.5 | 288.4 | 5b | 286.7 | 278.6 | 8.8 | −9.0 | −17.9 |
| 6aa | 292.0 | 285.4 | 6a | 283.0 | 275.6 | 9.1 | 0.7 | −8.4 |
| 6bb | 299.1 | 292.8 | 6b | 288.6 | 280.8 | 10.5 | 0.5 | −10.1 |
| 7aa | 290.7 | 283.4 | 7a | 279.9 | 273.6 | 10.8 | −2.9 | −13.7 |
| 7bb | 296.8 | 289.4 | 7b | 285.7 | 277.9 | 11.1 | 0.5 | −10.6 |
| 8aa | 296.8 | 288.6 | 8a | 282.6 | 276.0 | 14.2 | −4.0 | −18.2 |
| 8bb | 307.0 | 296.3 | 8b | 288.8 | 282.2 | 18.1 | 1.1 | −17.0 |
| 9aa | 292.8 | 285.4 | 9a | 282.1 | 275.6 | 10.7 | 0.0 | −10.7 |
| 9bb | 300.6 | 294.2 | 9b | 289.7 | 281.3 | 10.8 | −5.7 | −16.6 |
| 10aa | 296.6 | 288.4 | 10a | 282.0 | 275.6 | 14.6 | 5.7 | −8.9 |
| 10bb | 305.3 | 297.8 | 10b | 288.8 | 282.1 | 16.5 | −2.7 | −19.3 |
| (dmaP)3P | 297.4 | 291.3 | (pyrrP)3P | 307.5 | 300.2 |
| Molecule | PA | GB | Molecule | PA | GB | ΔPA | En | Ep |
|---|---|---|---|---|---|---|---|---|
| Iaa | 286.2 | 277.5 | Ia | 281.5 | 274.1 | 4.7 | −0.2 | −4.9 |
| Ibb | 293.1 | 285.4 | Ib | 287.3 | 279.5 | 5.9 | −3.0 | −8.9 |
| IIaa | 288.6 | 281.5 | IIa | 282.4 | 275.0 | 6.1 | −0.3 | −6.4 |
| IIbb | 293.5 | 285.4 | IIb | 289.2 | 282.2 | 4.3 | −9.4 | −13.7 |
| IIIaa | 284.1 | 275.3 | IIIa | 281.1 | 273.2 | 3.0 | −3.3 | −6.3 |
| IIIbb | 289.7 | 281.6 | IIIb | 287.9 | 281.2 | 1.8 | −10.0 | −11.8 |
| IVaa | 287.2 | 280.2 | Iva | 281.2 | 273.2 | 6.0 | 0.6 | −5.4 |
| IVbb | 294.7 | 284.5 | IVb | 288.4 | 281.8 | 6.4 | −8.9 | −15.3 |
| Vaa | 285.3 | 277.5 | Vbb | 280.5 | 273.3 | 4.8 | 0.2 | −4.6 |
| Vbb | 290.6 | 285.1 | Vb | 287.6 | 279.9 | 3.0 | −1.6 | −4.5 |
| VIaa | 287.5 | 279.8 | Via | 282.8 | 275.9 | 4.7 | −0.3 | −5.0 |
| VIbb | 291.3 | 283.7 | VIb | 286.9 | 280.3 | 4.4 | −1.7 | −6.1 |
| VIIaa | 286.3 | 279.6 | VIIa | 281.1 | 273.9 | 5.3 | 0.1 | −5.2 |
| VIIbb | 292.8 | 284.7 | VIIb | 287.0 | 279.9 | 5.8 | −1.9 | −7.7 |
| VIIIaa | 288.6 | 279.6 | VIIIa | 282.7 | 276.3 | 5.9 | −3.9 | −9.8 |
| VIIIbb | 293.2 | 286.2 | VIIIb | 289.3 | 281.1 | 4.1 | −8.8 | −11.7 |
| IXaa | 287.7 | 280.5 | IXa | 282.0 | 274.7 | 5.6 | −2.0 | −7.6 |
| IXbb | 289.5 | 282.3 | IXb | 288.0 | 280.2 | 1.6 | −5.7 | −7.3 |
| Molecule | M1 | M2 | Molecule | M1 | M2 | ||||
|---|---|---|---|---|---|---|---|---|---|
| pKa1 | pKa2 | pKa1 | pKa2 | pKa1 | pKa2 | pKa1 | pKa2 | ||
| 1aa | 34.0 | 29.9 | 36.2 | 29.7 | 6aa | 35.0 | 29.4 | 38.2 | 27.4 |
| 1bb | 36.0 | 30.0 | 36.5 | 28.4 | 6bb | 37.0 | 30.7 | 37.2 | 29.4 |
| 2aa | 34.2 | 28.5 | 36.2 | 27.9 | 7aa | 33.4 | 28.7 | 33.2 | 31.2 |
| 2bb | 38.9 | 33.1 | 37.5 | 31.0 | 7bb | 34.9 | 30.2 | 35.9 | 28.9 |
| 3aa | 34.1 | 26.2 | 35.2 | 25.6 | 8aa | 36.3 | 33.5 | 36.4 | 31.8 |
| 3bb | 39.5 | 30.7 | 33.7 | 32.1 | 8bb | 37.5 | 35.8 | 38.5 | 29.8 |
| 4aa | 33.5 | 28.3 | 34.7 | 27.8 | 9aa | 35.3 | 31.2 | 37.0 | 29.2 |
| 4bb | 37.2 | 31.4 | 36.4 | 31.6 | 9bb | 40.2 | 36.6 | 39.6 | 34.8 |
| 5aa | 30.5 | 31.3 | 34.0 | 28.3 | 10aa | 37.8 | 30.3 | 36.9 | 29.4 |
| 5bb | 32.8 | 32.8 | 31.9 | 33.5 | 10bb | 42.0 | 34.6 | 39.2 | 35.2 |
| Compound | BCP | ρ(rc) a | V(rc) b | E c |
|---|---|---|---|---|
| 1aa, H+ | P···H–+P | 0.0220 | −0.0147 | −2.96 |
| 1bb, H+ | P···H–+P | 0.0225 | −0.0151 | −3.02 |
| 2aa, H+ | P···H–+P | 0.0262 | −0.0171 | −3.40 |
| 2bb, H+ | P···H–+P | 0.0226 | −0.0164 | −3.27 |
| Nimine···H–+P | 0.0236 | −0.0175 | −3.46 | |
| 3aa, H+ | P···H–+P | 0.0293 | −0.0195 | −3.84 |
| 3bb, H+ | P···H–+P | 0.0229 | −0.0167 | −3.34 |
| 4aa, H+ | P···H–+P | 0.0208 | −0.0132 | −2.71 |
| 4bb, H+ | P···H–+P | 0.0350 | −0.0238 | −4.53 |
| 5aa, H+ | Nimine···H–+P | 0.0147 | −0.0093 | −2.02 |
| 5bb, H+ | Nimine···H–+P | 0.0185 | −0.0124 | −2.58 |
| 6aa, H+ | P···H–+P | 0.0173 | −0.0093 | −2.02 |
| 6bb, H+ | P···H–+P | 0.0220 | −0.0136 | −2.77 |
| 7aa, H+ | P···H–+P | 0.0170 | −0.0091 | −2.02 |
| 7bb, H+ | P···H–+P | 0.0213 | −0.0131 | −2.71 |
| 8aa, H+ | P···H–+P | 0.0191 | −0.0113 | −2.40 |
| 8bb, H+ | P···H–+P | 0.0171 | −0.0104 | −2.21 |
| 9aa, H+ | P···H–+P | 0.0197 | −0.0116 | −2.46 |
| 9bb, H+ | P···H–+P | 0.0188 | −0.0111 | −2.33 |
| 10aa, H+ | P···H–+P | 0.0205 | −0.0124 | −2.58 |
| 10bb, H+ | P···H–+P | 0.0210 | −0.0135 | −2.77 |
| TPPN, H+ | N···H–+N | 0.0545 | −0.0460 | −8.42 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glasovac, Z.; Barić, D.; Despotović, I.; Kovačević, B. Phosphazenyl Phosphine Proton Sponges: Does the Proton-Chelating Effect Enhance Their Basicity? Int. J. Mol. Sci. 2025, 26, 5058. https://doi.org/10.3390/ijms26115058
Glasovac Z, Barić D, Despotović I, Kovačević B. Phosphazenyl Phosphine Proton Sponges: Does the Proton-Chelating Effect Enhance Their Basicity? International Journal of Molecular Sciences. 2025; 26(11):5058. https://doi.org/10.3390/ijms26115058
Chicago/Turabian StyleGlasovac, Zoran, Danijela Barić, Ines Despotović, and Borislav Kovačević. 2025. "Phosphazenyl Phosphine Proton Sponges: Does the Proton-Chelating Effect Enhance Their Basicity?" International Journal of Molecular Sciences 26, no. 11: 5058. https://doi.org/10.3390/ijms26115058
APA StyleGlasovac, Z., Barić, D., Despotović, I., & Kovačević, B. (2025). Phosphazenyl Phosphine Proton Sponges: Does the Proton-Chelating Effect Enhance Their Basicity? International Journal of Molecular Sciences, 26(11), 5058. https://doi.org/10.3390/ijms26115058