
Triple-Band Warm White-Light Emission from Type II Band-Aligned Aggregation-Induced Enhanced Emission Organic Cation-Incorporated Two-Dimensional Lead Iodide Perovskite

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
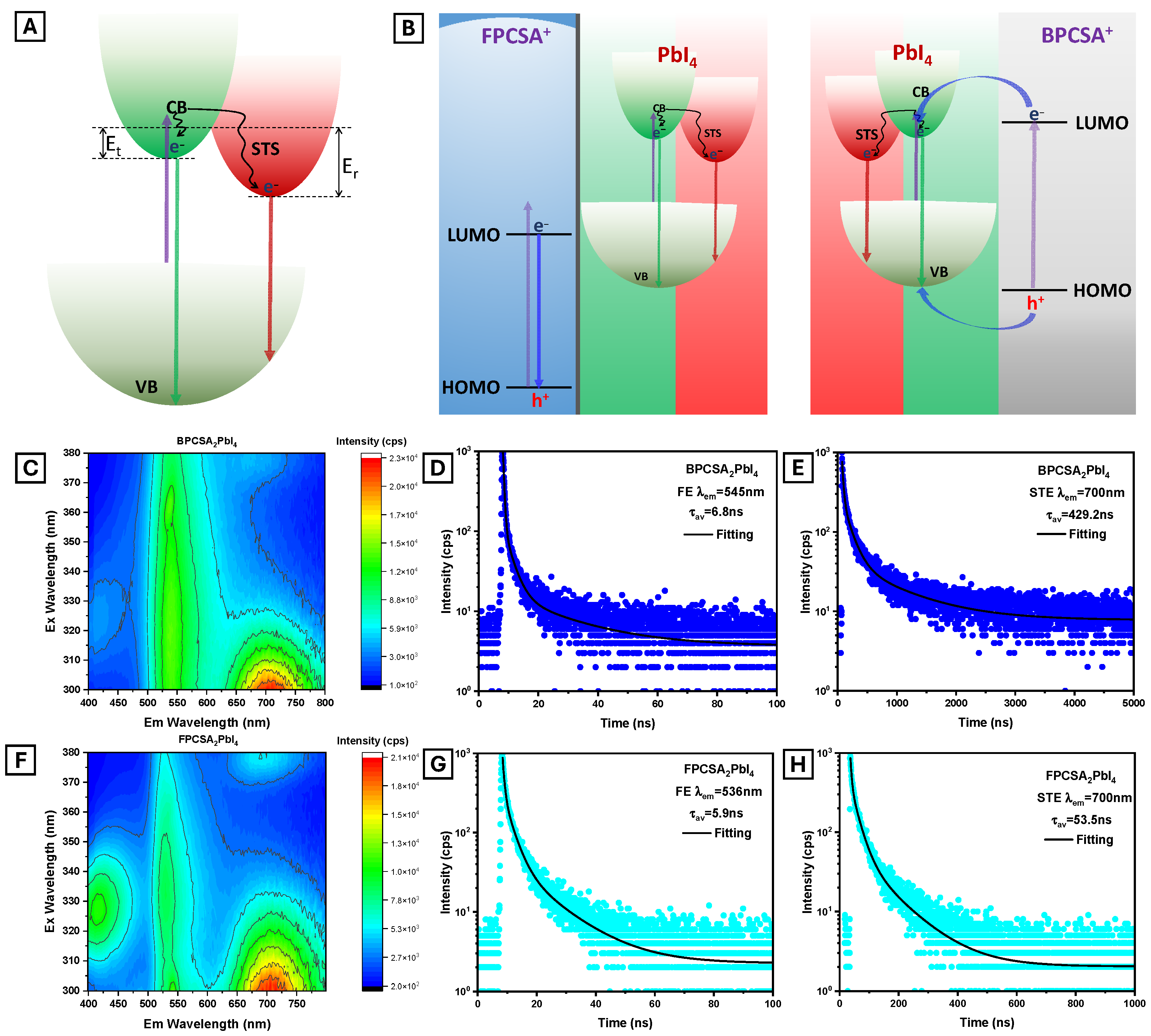
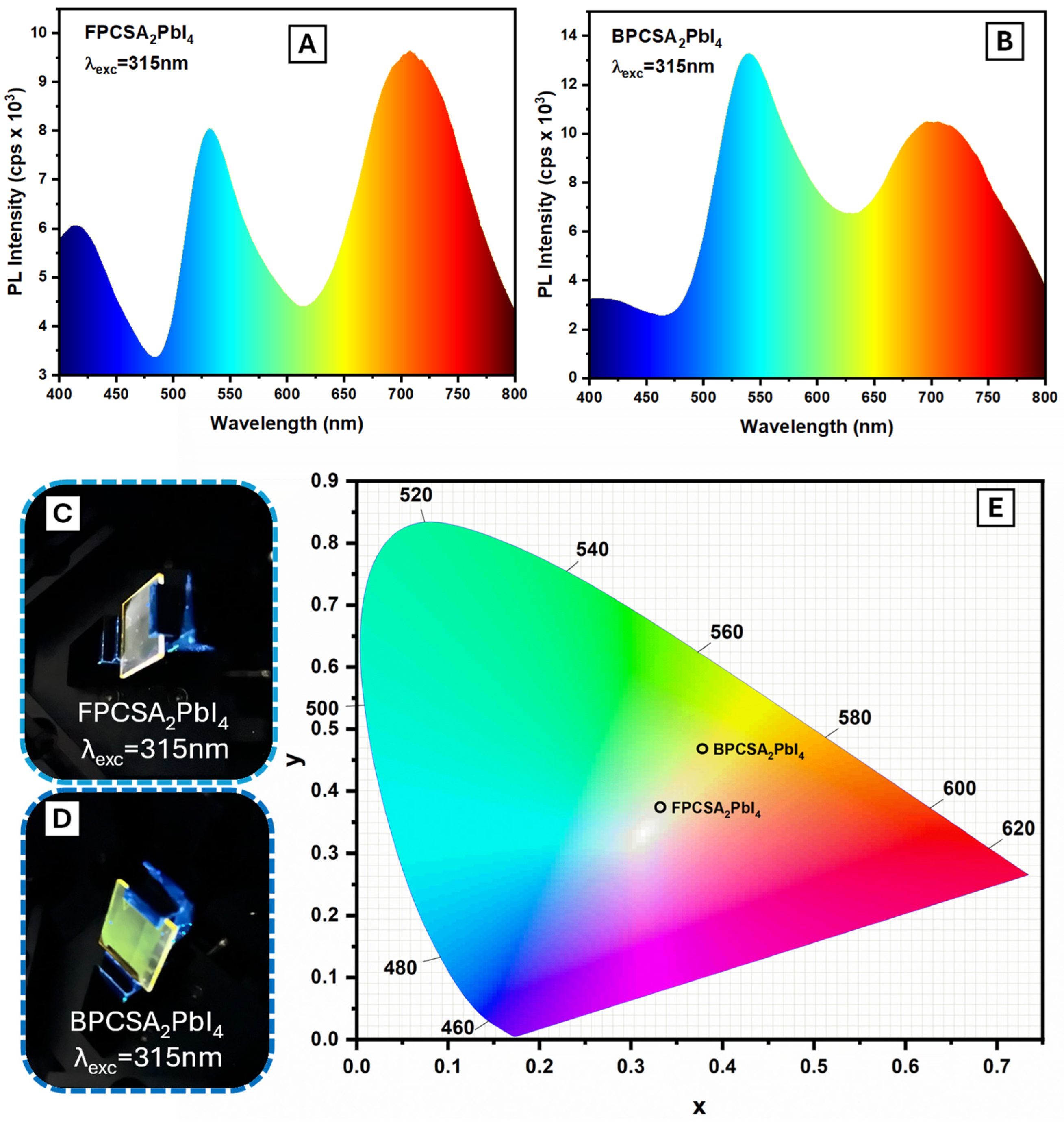
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Synthesis of BPCSA
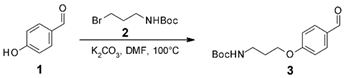
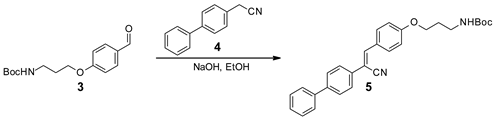

3.3. Synthesis of FPCSA

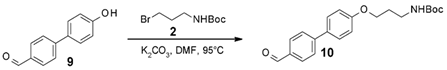
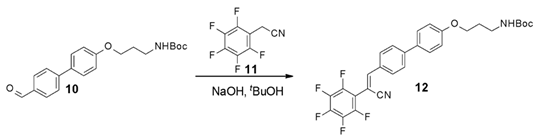
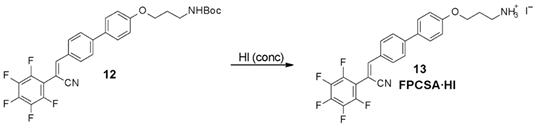
3.4. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Q.; Ma, D. Management of charges and excitons for high-performance white organic light-emitting diodes. Chem. Soc. Rev. 2010, 39, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ying, L.; Yang, W.; Cao, Y. Progress and perspective of polymer white light-emitting devices and materials. Chem. Soc. Rev. 2009, 38, 3391–3400. [Google Scholar] [CrossRef] [PubMed]
- Kamtekar, K.T.; Monkman, A.P.; Bryce, M.R. Recent advances in white organic light-emitting materials and devices (WOLEDs). Adv. Mater. 2010, 22, 572–582. [Google Scholar] [CrossRef]
- Abbel, R.; Grenier, C.; Pouderoijen, M.J.; Stouwdam, J.W.; Leclere, P.E.; Sijbesma, R.P.; Meijer, E.W.; Schenning, A.P. White-light emitting hydrogen-bonded supramolecular copolymers based on π-conjugated oligomers. J. Am. Chem. Soc. 2009, 131, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tseng, M.C.; Chen, Y.; Yeung, F.S.Y.; He, H.; Cheng, Y.; Cai, J.; Chen, E.; Kwok, H.-S. Color-conversion displays: Current status and future outlook. Light Sci. Appl. 2024, 13, 301. [Google Scholar] [CrossRef]
- Li, W.; Ma, N.; Devakumar, B.; Huang, X. High-color-quality blue-light-pumped full-spectrum white-light-emitting diodes realized by efficient green-emitting CaY2HfScAl3O12: Ce3+ phosphors. J. Lumin. 2023, 264, 120183. [Google Scholar] [CrossRef]
- Gaffuri, P.; Stolyarova, E.; Llerena, D.; Appert, E.; Consonni, M.; Robin, S.; Consonni, V. Potential substitutes for critical materials in white LEDs: Technological challenges and market opportunities. Renew. Sustain. Energy Rev. 2021, 143, 110869. [Google Scholar] [CrossRef]
- Cho, J.; Park, J.H.; Kim, J.K.; Schubert, E.F. White light-emitting diodes: History, progress, and future. Laser Photonics Rev. 2017, 11, 1600147. [Google Scholar] [CrossRef]
- Farinola, G.M.; Ragni, R. Electroluminescent materials for white organic light emitting diodes. Chem. Soc. Rev. 2011, 40, 3467–3482. [Google Scholar] [CrossRef]
- Li, F.; Nie, C.; You, L.; Jin, X.; Zhang, Q.; Qin, Y.; Zhao, F.; Song, Y.; Chen, Z.; Li, Q. White light emitting device based on single-phase CdS quantum dots. Nanotechnology 2018, 29, 205701. [Google Scholar] [CrossRef]
- Pan, M.; Liao, W.M.; Yin, S.Y.; Sun, S.S.; Su, C.Y. Single-phase white-light-emitting and photoluminescent color-tuning coordination assemblies. Chem. Rev. 2018, 118, 8889–8935. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Wang, R.; Chen, J.; Li, F.; Zeng, H. Research progress of full electroluminescent white light-emitting diodes based on a single emissive layer. Light Sci. Appl. 2021, 10, 206. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, C.; Quan, Z.; Cheng, Z.; Lin, J. Tunable luminescence properties of CaIn2O4: Eu3+ phosphors. J. Phys. Chem. C 2007, 111, 16601–16607. [Google Scholar] [CrossRef]
- Burgess, D.S. Nanoparticle white-light emitter based on up-conversion demonstrated. Photonics Spectra 2005, 39, 121. [Google Scholar]
- Shang, M.; Li, C.; Lin, J. How to produce white light in a single-phase host? Chem. Soc. Rev. 2014, 43, 1372–1386. [Google Scholar] [CrossRef]
- Luo, J.; Li, X.; Hou, Q.; Peng, J.B.; Yang, W.; Cao, Y. High-efficiency white-light emission from a single copolymer: Fluorescent blue, green, and red chromophores on a conjugated polymer backbone. Adv. Mater. 2007, 19, 1113–1117. [Google Scholar] [CrossRef]
- Xie, Z.; Huang, Q.; Yu, T.; Wang, L.; Mao, Z.; Li, W.; Yang, Z.; Zhang, Y.; Liu, S.; Xu, J.; et al. Hydrogen-bonding-assisted intermolecular charge transfer: A new strategy to design single-component white-light-emitting materials. Adv. Funct. Mater. 2017, 27, 1703918. [Google Scholar] [CrossRef]
- Dai, Q.; Duty, C.E.; Hu, M.Z. Semiconductor-nanocrystals-based white light-emitting diodes. Small 2010, 6, 1577–1588. [Google Scholar] [CrossRef]
- Sun, C.Y.; Wang, X.L.; Zhang, X.; Qin, C.; Li, P.; Su, Z.M.; Zhu, D.X.; Shan, G.G.; Shao, K.Z.; Wu, H.; et al. Efficient and tunable white-light emission of metal–organic frameworks by iridium-complex encapsulation. Nat. Commun. 2013, 4, 2717. [Google Scholar] [CrossRef]
- Xu, D.D.; Dong, W.W.; Li, M.K.; Han, H.M.; Zhao, J.; Li, D.S.; Zhang, Q. Encapsulating organic dyes in metal–organic frameworks for color-tunable and high-efficiency white-light-emitting properties. Inorg. Chem. 2022, 61, 21107–21114. [Google Scholar] [CrossRef]
- Chiu, N.C.; Smith, K.T.; Stylianou, K.C. Metal-organic frameworks for white light emission: From synthesis to device fabrication. Coord. Chem. Rev. 2022, 459, 214441. [Google Scholar] [CrossRef]
- Dohner, E.R.; Hoke, E.T.; Karunadasa, H.I. Self-assembly of broadband white-light emitters. J. Am. Chem. Soc. 2014, 136, 1718–1721. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Gu, H.; Han, Y.; Yin, J.; Xing, G.; Cui, B.B. Broadband white-light emission from a novel two-dimensional metal halide assembled by Pb–Cl hendecahedrons. J. Mater. Chem. C 2022, 10, 9465–9470. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Xu, X.; Li, J.; Song, J.; Lan, S.; Liu, S.; Cai, B.; Han, B.; Precht, J.T.; et al. Efficient and bright white light-emitting diodes based on single-layer heterophase halide perovskites. Nat. Photonics 2021, 15, 238–244. [Google Scholar] [CrossRef]
- Veldhuis, S.A.; Boix, P.P.; Yantara, N.; Li, M.; Sum, T.C.; Mathews, N.; Mhaisalkar, S.G. Perovskite materials for light-emitting diodes and lasers. Adv. Mater. 2016, 28, 6804–6834. [Google Scholar] [CrossRef]
- Luo, J.; Hu, M.; Niu, G.; Tang, J. Lead-free halide perovskites and perovskite variants as phosphors toward light-emitting applications. ACS Appl. Mater. Interfaces 2019, 11, 31575–31584. [Google Scholar] [CrossRef]
- Chen, J.; Xiang, H.; Wang, J.; Wang, R.; Li, Y.; Shan, Q.; Xu, X.; Dong, Y.; Wei, C.; Zeng, H. Perovskite white light emitting diodes: Progress, challenges, and opportunities. ACS Nano 2021, 15, 17150–17174. [Google Scholar] [CrossRef] [PubMed]
- Stoumpos, C.C.; Cao, D.H.; Clark, D.J.; Young, J.; Rondinelli, J.M.; Jang, J.I.; Hupp, J.T.; Kanatzidis, M.G. Ruddlesden–Popper hybrid lead iodide perovskite 2D homologous semiconductors. Chem. Mater. 2016, 28, 2852–2867. [Google Scholar] [CrossRef]
- Smith, M.D.; Karunadasa, H.I. White-light emission from layered halide perovskites. Acc. Chem. Res. 2018, 51, 619–627. [Google Scholar] [CrossRef]
- Cortecchia, D.; Yin, J.; Bruno, A.; Lo, S.Z.A.; Gurzadyan, G.G.; Mhaisalkar, S.; Brédas, J.-L.; Soci, C. Polaron self-localization in white-light emitting hybrid perovskites. J. Mater. Chem. C 2017, 5, 2771–2780. [Google Scholar] [CrossRef]
- Li, J.; Wang, H.; Li, D. Self-trapped excitons in two-dimensional perovskites. Front. Optoelectron. 2020, 13, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.T.; Song, K.S. The self-trapped exciton. J. Phys. Chem. Solids 1990, 51, 679–716. [Google Scholar] [CrossRef]
- Straus, D.B.; Kagan, C.R. Electrons, excitons, and phonons in two-dimensional hybrid perovskites: Connecting structural, optical, and electronic properties. J. Phys. Chem. Lett. 2018, 9, 1434–1447. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Zhou, G.; Chen, D.; Zhao, J.; Wang, Y.; Liu, Q. Broadband photoluminescence in 2D organic–inorganic hybrid perovskites:(C7H18N2) PbBr4 and (C9H22N2) PbBr4. J. Phys. Chem. Lett. 2020, 11, 2934–2940. [Google Scholar] [CrossRef]
- Gautier, R.; Paris, M.; Massuyeau, F. Exciton self-trapping in hybrid lead halides: Role of halogen. J. Am. Chem. Soc. 2019, 141, 12619–12623. [Google Scholar] [CrossRef]
- Wu, X.; Trinh, M.T.; Niesner, D.; Zhu, H.; Norman, Z.; Owen, J.S.; Yaffe, O.; Kudisch, B.J.; Zhu, X.Y. Trap states in lead iodide perovskites. J. Am. Chem. Soc. 2015, 137, 2089–2096. [Google Scholar] [CrossRef]
- Biswas, S.; Zhao, R.; Alowa, F.; Zacharias, M.; Sharifzadeh, S.; Coker, D.F.; Seferos, D.S.; Scholes, G.D. Exciton polaron formation and hot-carrier relaxation in rigid Dion–Jacobson-type two-dimensional perovskites. Nat. Mater. 2024, 23, 937–943. [Google Scholar] [CrossRef]
- Kahmann, S.; Tekelenburg, E.K.; Duim, H.; Kamminga, M.E.; Loi, M.A. Extrinsic nature of the broad photoluminescence in lead iodide-based Ruddlesden–Popper perovskites. Nat. Commun. 2020, 11, 2344. [Google Scholar] [CrossRef]
- Li, X.; Hoffman, J.M.; Kanatzidis, M.G. The 2D halide perovskite rulebook: How the spacer influences everything from the structure to optoelectronic device efficiency. Chem. Rev. 2021, 121, 2230–2291. [Google Scholar] [CrossRef]
- Jemli, K.; Audebert, P.; Galmiche, L.; Trippé-Allard, G.; Garrot, D.; Lauret, J.S.; Deleporte, E. Two-dimensional perovskite activation with an organic luminophore. ACS Appl. Mater. Interfaces 2015, 7, 21763–21769. [Google Scholar] [CrossRef]
- Passarelli, J.V.; Fairfield, D.J.; Sather, N.A.; Hendricks, M.P.; Sai, H.; Stern, C.L.; Stupp, S.I. Enhanced out-of-plane conductivity and photovoltaic performance in n = 1 layered perovskites through organic cation design. J. Am. Chem. Soc. 2018, 140, 7313–7323. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Cervantes, C.; Román-Román, P.I.; Vazquez-Chavez, J.; Hernández-Rodríguez, M.; Solis-Ibarra, D. Thousand-fold Conductivity Increase in 2D Perovskites by Polydiacetylene Incorporation and Doping. Angew. Chem. 2018, 130, 14078–14082. [Google Scholar] [CrossRef]
- Sun, J.; Wang, K.; Ma, K.; Park, J.Y.; Lin, Z.Y.; Savoie, B.M.; Dou, L. Emerging two-dimensional organic semiconductor-incorporated perovskites─ a fascinating family of hybrid electronic materials. J. Am. Chem. Soc. 2023, 145, 20694–20715. [Google Scholar] [CrossRef]
- Hong, Y.; Lam, J.W.; Tang, B.Z. Aggregation-induced emission. Chem. Soc. Rev. 2011, 40, 5361–5388. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, H.; Lam, J.W.; Tang, B.Z. Aggregation-induced emission: New vistas at the aggregate level. Angew. Chem. Int. Ed. 2020, 59, 9888–9907. [Google Scholar] [CrossRef]
- Peng, Q.; Shuai, Z. Molecular mechanism of aggregation-induced emission. Aggregate 2021, 2, e91. [Google Scholar] [CrossRef]
- Lim, C.K.; Maldonado, M.; Zalesny, R.; Valiev, R.; Ågren, H.; Gomes, A.S.; Jiang, J.; Pachter, R.; Prasad, P.N. Interlayer-sensitized linear and nonlinear photoluminescence of quasi-2D hybrid perovskites using aggregation-induced enhanced emission active organic cation layers. Adv. Funct. Mater. 2020, 30, 1909375. [Google Scholar] [CrossRef]
- Kim, J.H.; An, B.K.; Yoon, S.J.; Park, S.K.; Kwon, J.E.; Lim, C.K.; Park, S.Y. Highly Fluorescent and Color-Tunable Exciplex Emission from Poly (N-vinylcarbazole) Film Containing Nanostructured Supramolecular Acceptors. Adv. Funct. Mater. 2014, 24, 2746–2753. [Google Scholar] [CrossRef]
- Zhang, M.; Guo, X.; Zhang, S.; Hou, J. Synergistic effect of fluorination on molecular energy level modulation in highly efficient photovoltaic polymers. Adv. Mater. 2014, 26, 1118–1123. [Google Scholar] [CrossRef]
- An, B.K.; Gierschner, J.; Park, S.Y. π-Conjugated cyanostilbene derivatives: A unique self-assembly motif for molecular nanostructures with enhanced emission and transport. Acc. Chem. Res. 2012, 45, 544–554. [Google Scholar] [CrossRef]
- Pan, F.; Li, J.; Ma, X.; Nie, Y.; Liu, B.; Ye, H. Free and self-trapped exciton emission in perovskite CsPbBr 3 microcrystals. RSC Adv. 2022, 12, 1035–1042. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beisenbayev, A.R.; Ivanov-Prianichnikov, I.; Peshkov, A.; Adil, T.; Hayrapetyan, D.; Lim, C.-K. Triple-Band Warm White-Light Emission from Type II Band-Aligned Aggregation-Induced Enhanced Emission Organic Cation-Incorporated Two-Dimensional Lead Iodide Perovskite. Int. J. Mol. Sci. 2025, 26, 5054. https://doi.org/10.3390/ijms26115054
Beisenbayev AR, Ivanov-Prianichnikov I, Peshkov A, Adil T, Hayrapetyan D, Lim C-K. Triple-Band Warm White-Light Emission from Type II Band-Aligned Aggregation-Induced Enhanced Emission Organic Cation-Incorporated Two-Dimensional Lead Iodide Perovskite. International Journal of Molecular Sciences. 2025; 26(11):5054. https://doi.org/10.3390/ijms26115054
Chicago/Turabian StyleBeisenbayev, Almaz R., Igor Ivanov-Prianichnikov, Anatoly Peshkov, Tangsulu Adil, Davit Hayrapetyan, and Chang-Keun Lim. 2025. "Triple-Band Warm White-Light Emission from Type II Band-Aligned Aggregation-Induced Enhanced Emission Organic Cation-Incorporated Two-Dimensional Lead Iodide Perovskite" International Journal of Molecular Sciences 26, no. 11: 5054. https://doi.org/10.3390/ijms26115054
APA StyleBeisenbayev, A. R., Ivanov-Prianichnikov, I., Peshkov, A., Adil, T., Hayrapetyan, D., & Lim, C.-K. (2025). Triple-Band Warm White-Light Emission from Type II Band-Aligned Aggregation-Induced Enhanced Emission Organic Cation-Incorporated Two-Dimensional Lead Iodide Perovskite. International Journal of Molecular Sciences, 26(11), 5054. https://doi.org/10.3390/ijms26115054