
A Novel Framework for the Design of Minimized Epigenetic Clocks Using the Analysis of DNA Methylation Heterogeneity

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
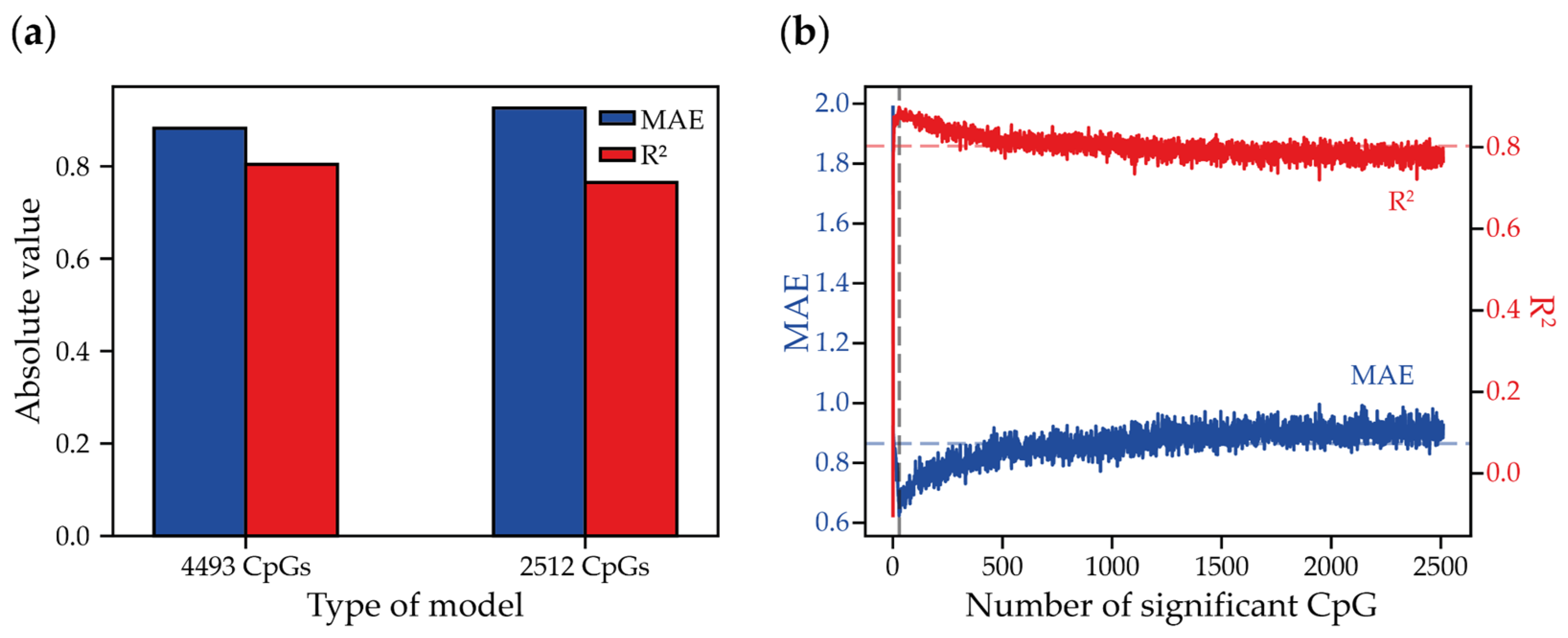
2.1. Simulation of the Design of Minimized Microarray-Based Cultural Age Clocks
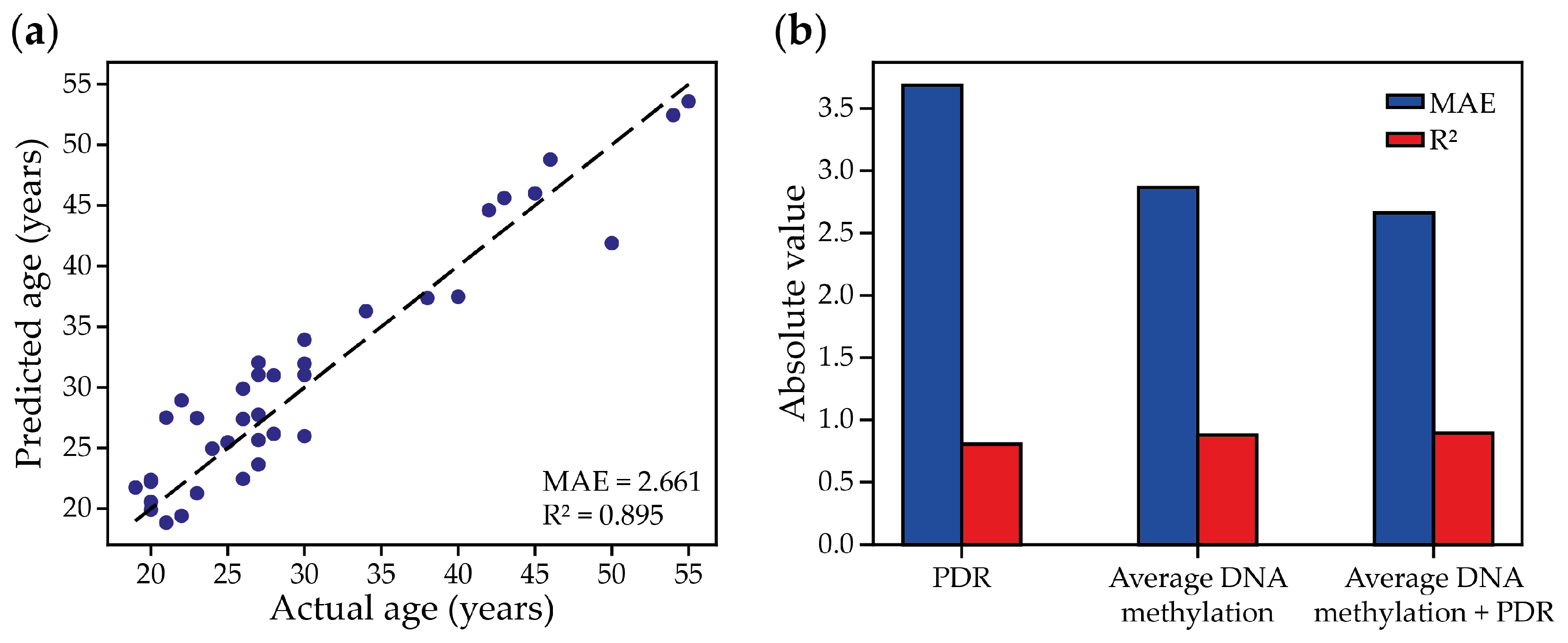
2.2. Using Targeted Bisulfite Sequencing to Build Minimized Epigenetic Clocks
2.3. Using DNA Methylation Heterogeneity Scores as a Predictor to Build BS-Seq-Based Minimized Epigenetic Clocks
2.4. Combining the Passage-Dependent Dynamics of WSH Scores and the DNA Methylation Level to Predict Cultural Passage
3. Discussion
4. Materials and Methods
4.1. Source Data
4.2. Donor MSCs
4.3. Targeted Bisulfite Sequencing
4.4. Microarray Data Analysis
4.5. Sequencing Reads Processing and Heterogeneity Calculation
4.6. Selection of Genomic Features for the Minimized Predictive Model
4.7. Building Predictive Models with RFR
4.8. Implementation of the Hybrid Model of Cultural Age
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Margiana, R.; Markov, A.; Zekiy, A.O.; Hamza, M.U.; Al-Dabbagh, K.A.; Al-Zubaidi, S.H.; Hameed, N.M.; Ahmad, I.; Sivaraman, R.; Kzar, H.H.; et al. Clinical Application of Mesenchymal Stem Cell in Regenerative Medicine: A Narrative Review. Stem Cell Res. Ther. 2022, 13, 366. [Google Scholar] [CrossRef]
- Galderisi, U.; Peluso, G.; Di Bernardo, G. Clinical Trials Based on Mesenchymal Stromal Cells Are Exponentially Increasing: Where Are We in Recent Years? Stem Cell Rev. Rep. 2022, 18, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; Andersen, J.K.; Kapahi, P.; Melov, S. Cellular Senescence: A Link between Cancer and Age-Related Degenerative Disease? Semin. Cancer Biol. 2011, 21, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, Z.; Su, H.; Chen, F.; Ma, M.; Yu, W.; Ye, G.; Cen, S.; Mi, R.; Wu, X.; et al. Effects of Long-Term Culture on the Biological Characteristics and RNA Profiles of Human Bone-Marrow-Derived Mesenchymal Stem Cells. Mol. Ther. Nucleic Acids 2021, 26, 557–574. [Google Scholar] [CrossRef]
- Zha, K.; Li, X.; Yang, Z.; Tian, G.; Sun, Z.; Sui, X.; Dai, Y.; Liu, S.; Guo, Q. Heterogeneity of Mesenchymal Stem Cells in Cartilage Regeneration: From Characterization to Application. NPJ Regen. Med. 2021, 6, 14. [Google Scholar] [CrossRef]
- González-Gualda, E.; Baker, A.G.; Fruk, L.; Muñoz-Espín, D. A Guide to Assessing Cellular Senescence in Vitro and in Vivo. FEBS J. 2021, 288, 56–80. [Google Scholar] [CrossRef]
- Carnero, A. Markers of Cellular Senescence; Springer: Berlin/Heidelberg, Germany, 2013; Volume 965, pp. 63–81. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef] [PubMed]
- Galvis, D.; Walsh, D.; Harries, L.W.; Latorre, E.; Rankin, J. A Dynamical Systems Model for the Measurement of Cellular Senescence. J. R. Soc. Interface 2019, 16, 20190311. [Google Scholar] [CrossRef]
- Gnani, D.; Crippa, S.; della Volpe, L.; Rossella, V.; Conti, A.; Lettera, E.; Rivis, S.; Ometti, M.; Fraschini, G.; Bernardo, M.E.; et al. An Early-senescence State in Aged Mesenchymal Stromal Cells Contributes to Hematopoietic Stem and Progenitor Cell Clonogenic Impairment through the Activation of a Pro-inflammatory Program. Aging Cell 2019, 18, e12933. [Google Scholar] [CrossRef]
- Kusumoto, D.; Seki, T.; Sawada, H.; Kunitomi, A.; Katsuki, T.; Kimura, M.; Ito, S.; Komuro, J.; Hashimoto, H.; Fukuda, K.; et al. Anti-Senescent Drug Screening by Deep Learning-Based Morphology Senescence Scoring. Nat. Commun. 2021, 12, 257. [Google Scholar] [CrossRef]
- Heckenbach, I.; Mkrtchyan, G.V.; Ezra, M.B.; Bakula, D.; Madsen, J.S.; Nielsen, M.H.; Oró, D.; Osborne, B.; Covarrubias, A.J.; Idda, M.L.; et al. Nuclear Morphology Is a Deep Learning Biomarker of Cellular Senescence. Nat. Aging 2022, 2, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Shokhirev, M.N.; Johnson, A.A. Modeling the Human Aging Transcriptome across Tissues, Health Status, and Sex. Aging Cell 2021, 20, e13280. [Google Scholar] [CrossRef]
- Simpson, D.J.; Chandra, T. Epigenetic Age Prediction. Aging Cell 2021, 20, e13452. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.V.C.; Bourc’his, D. The Diverse Roles of DNA Methylation in Mammalian Development and Disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Loyfer, N.; Magenheim, J.; Peretz, A.; Cann, G.; Bredno, J.; Klochendler, A.; Fox-Fisher, I.; Shabi-Porat, S.; Hecht, M.; Pelet, T.; et al. A DNA Methylation Atlas of Normal Human Cell Types. Nature 2023, 613, 355–364. [Google Scholar] [CrossRef]
- Yousefi, P.D.; Suderman, M.; Langdon, R.; Whitehurst, O.; Davey Smith, G.; Relton, C.L. DNA Methylation-Based Predictors of Health: Applications and Statistical Considerations. Nat. Rev. Genet. 2022, 23, 369–383. [Google Scholar] [CrossRef]
- Horvath, S. DNA Methylation Age of Human Tissues and Cell Types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA Methylation-Based Biomarkers and the Epigenetic Clock Theory of Ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An Epigenetic Biomarker of Aging for Lifespan and Healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Mayne, B.; Berry, O.; Jarman, S. Optimal Sample Size for Calibrating DNA Methylation Age Estimators. Mol. Ecol. Resour. 2021, 21, 2316–2323. [Google Scholar] [CrossRef]
- Le Clercq, L.; Kotzé, A.; Grobler, J.P.; Dalton, D.L. Biological Clocks as Age Estimation Markers in Animals: A Systematic Review and Meta-analysis. Biol. Rev. 2023, 98, 1972–2011. [Google Scholar] [CrossRef] [PubMed]
- Piferrer, F.; Anastasiadi, D. Age Estimation in Fishes Using Epigenetic Clocks: Applications to Fisheries Management and Conservation Biology. Front. Mar. Sci. 2023, 10, 1062151. [Google Scholar] [CrossRef]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S.; et al. Epigenetic Clock for Skin and Blood Cells Applied to Hutchinson Gilford Progeria Syndrome and Ex Vivo Studies. Aging 2018, 10, 1758–1775. [Google Scholar] [CrossRef] [PubMed]
- Cheishvili, D.; Do Carmo, S.; Caraci, F.; Grasso, M.; Cuello, A.C.; Szyf, M. EpiAge: A next-Generation Sequencing-Based ELOVL2 Epigenetic Clock for Biological Age Assessment in Saliva and Blood across Health and Disease. Aging 2025, 17, 131–160. [Google Scholar] [CrossRef]
- Franzen, J.; Georgomanolis, T.; Selich, A.; Kuo, C.-C.; Stöger, R.; Brant, L.; Mulabdić, M.S.; Fernandez-Rebollo, E.; Grezella, C.; Ostrowska, A.; et al. DNA Methylation Changes during Long-Term In Vitro Cell Culture Are Caused by Epigenetic Drift. Commun. Biol. 2021, 4, 598. [Google Scholar] [CrossRef]
- Franzen, J.; Zirkel, A.; Blake, J.; Rath, B.; Benes, V.; Papantonis, A.; Wagner, W. Senescence-associated DNA Methylation Is Stochastically Acquired in Subpopulations of Mesenchymal Stem Cells. Aging Cell 2017, 16, 183–191. [Google Scholar] [CrossRef]
- Scherer, M.; Nebel, A.; Franke, A.; Walter, J.; Lengauer, T.; Bock, C.; Müller, F.; List, M. Quantitative Comparison of Within-Sample Heterogeneity Scores for DNA Methylation Data. Nucleic Acids Res. 2020, 48, e46. [Google Scholar] [CrossRef]
- Guo, S.; Diep, D.; Plongthongkum, N.; Fung, H.-L.; Zhang, K.; Zhang, K. Identification of Methylation Haplotype Blocks Aids in Deconvolution of Heterogeneous Tissue Samples and Tumor Tissue-of-Origin Mapping from Plasma DNA. Nat. Genet. 2017, 49, 635–642. [Google Scholar] [CrossRef]
- Mo, S.; Dai, W.; Wang, H.; Lan, X.; Ma, C.; Su, Z.; Xiang, W.; Han, L.; Luo, W.; Zhang, L.; et al. Early Detection and Prognosis Prediction for Colorectal Cancer by Circulating Tumour DNA Methylation Haplotypes: A Multicentre Cohort Study. EClinicalMedicine 2023, 55, 101717. [Google Scholar] [CrossRef]
- Karetnikov, D.I.; Romanov, S.E.; Baklaushev, V.P.; Laktionov, P.P. Age Prediction Using DNA Methylation Heterogeneity Metrics. Int. J. Mol. Sci. 2024, 25, 4967. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-Wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA Methylation GrimAge Strongly Predicts Lifespan and Healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef] [PubMed]
- Kriukov, D.; Kuzmina, E.; Efimov, E.; Dylov, D.V.; Khrameeva, E.E. Epistemic Uncertainty Challenges Aging Clock Reliability in Predicting Rejuvenation Effects. Aging Cell 2024, 23, e14283. [Google Scholar] [CrossRef]
- Mei, X.; Blanchard, J.; Luellen, C.; Conboy, M.J.; Conboy, I.M. Fail-Tests of DNA Methylation Clocks, and Development of a Noise Barometer for Measuring Epigenetic Pressure of Aging and Disease. Aging 2023, 15, 8552–8575. [Google Scholar] [CrossRef]
- Pośpiech, E.; Pisarek, A.; Rudnicka, J.; Noroozi, R.; Boroń, M.; Masny, A.; Wysocka, B.; Migacz-Gruszka, K.; Lisman, D.; Pruszkowska-Przybylska, P.; et al. Introduction of a Multiplex Amplicon Sequencing Assay to Quantify DNA Methylation in Target Cytosine Markers Underlying Four Selected Epigenetic Clocks. Clin. Epigenet. 2023, 15, 128. [Google Scholar] [CrossRef]
- Shao, K.; Koch, C.; Gupta, M.K.; Lin, Q.; Lenz, M.; Laufs, S.; Denecke, B.; Schmidt, M.; Linke, M.; Hennies, H.C.; et al. Induced Pluripotent Mesenchymal Stromal Cell Clones Retain Donor-Derived Differences in DNA Methylation Profiles. Mol. Ther. 2013, 21, 240–250. [Google Scholar] [CrossRef]
- Fernández, A.F.; Bayón, G.F.; Urdinguio, R.G.; Toraño, E.G.; García, M.G.; Carella, A.; Petrus-Reurer, S.; Ferrero, C.; Martinez-Camblor, P.; Cubillo, I.; et al. H3K4me1 Marks DNA Regions Hypomethylated during Aging in Human Stem and Differentiated Cells. Genome Res. 2015, 25, 27–40. [Google Scholar] [CrossRef]
- Zhou, W.; Laird, P.W.; Shen, H. Comprehensive Characterization, Annotation and Innovative Use of Infinium DNA Methylation BeadChip Probes. Nucleic Acids Res. 2016, 45, e22. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A Web Server for Functional Enrichment Analysis and Functional Annotation of Gene Lists (2021 Update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Bacalini, M.G.; Deelen, J.; Pirazzini, C.; De Cecco, M.; Giuliani, C.; Lanzarini, C.; Ravaioli, F.; Marasco, E.; van Heemst, D.; Suchiman, H.E.D.; et al. Systemic Age-Associated DNA Hypermethylation of ELOVL2 Gene: In Vivo and In Vitro Evidences of a Cell Replication Process. J. Gerontol. Ser. A 2017, 72, 1015–1023. [Google Scholar] [CrossRef]
- Bekaert, B.; Kamalandua, A.; Zapico, S.C.; Van de Voorde, W.; Decorte, R. Improved Age Determination of Blood and Teeth Samples Using a Selected Set of DNA Methylation Markers. Epigenetics 2015, 10, 922–930. [Google Scholar] [CrossRef]
- Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Gori, D.; Giuliani, C.; Mari, D.; Di Blasio, A.M.; Gentilini, D.; Vitale, G.; Collino, S.; et al. Methylation of ELOVL2 Gene as a New Epigenetic Marker of Age. Aging Cell 2012, 11, 1132–1134. [Google Scholar] [CrossRef] [PubMed]
- Landan, G.; Cohen, N.M.; Mukamel, Z.; Bar, A.; Molchadsky, A.; Brosh, R.; Horn-Saban, S.; Zalcenstein, D.A.; Goldfinger, N.; Zundelevich, A.; et al. Epigenetic Polymorphism and the Stochastic Formation of Differentially Methylated Regions in Normal and Cancerous Tissues. Nat. Genet. 2012, 44, 1207–1214. [Google Scholar] [CrossRef]
- Landau, D.A.; Clement, K.; Ziller, M.J.; Boyle, P.; Fan, J.; Gu, H.; Stevenson, K.; Sougnez, C.; Wang, L.; Li, S.; et al. Locally Disordered Methylation Forms the Basis of Intratumor Methylome Variation in Chronic Lymphocytic Leukemia. Cancer Cell 2014, 26, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Wang, M.; de Andrade, A.; de F Bonaldo, M.; Galat, V.; Arndt, K.; Rajaram, V.; Goldman, S.; Tomita, T.; Soares, M.B. Genome-Wide Quantitative Assessment of Variation in DNA Methylation Patterns. Nucleic Acids Res. 2011, 39, 4099–4108. [Google Scholar] [CrossRef]
- Dabrowski, J.K.; Yang, E.J.; Crofts, S.J.C.; Hillary, R.F.; Simpson, D.J.; McCartney, D.L.; Marioni, R.E.; Kirschner, K.; Latorre-Crespo, E.; Chandra, T. Probabilistic Inference of Epigenetic Age Acceleration from Cellular Dynamics. Nat. Aging 2024, 4, 1493–1507. [Google Scholar] [CrossRef]
- Zhu, T.; Tong, H.; Du, Z.; Beck, S.; Teschendorff, A.E. An Improved Epigenetic Counter to Track Mitotic Age in Normal and Precancerous Tissues. Nat. Commun. 2024, 15, 4211. [Google Scholar] [CrossRef] [PubMed]
- Bhak, Y.; Jeong, H.; Cho, Y.S.; Jeon, S.; Cho, J.; Gim, J.-A.; Jeon, Y.; Blazyte, A.; Park, S.G.; Kim, H.-M.; et al. Depression and Suicide Risk Prediction Models Using Blood-Derived Multi-Omics Data. Transl. Psychiatry 2019, 9, 262. [Google Scholar] [CrossRef]
- Buckley, M.T.; Sun, E.D.; George, B.M.; Liu, L.; Schaum, N.; Xu, L.; Reyes, J.M.; Goodell, M.A.; Weissman, I.L.; Wyss-Coray, T.; et al. Cell-Type-Specific Aging Clocks to Quantify Aging and Rejuvenation in Neurogenic Regions of the Brain. Nat. Aging 2022, 3, 121–137. [Google Scholar] [CrossRef]
- Zindler, T.; Frieling, H.; Neyazi, A.; Bleich, S.; Friedel, E. Simulating ComBat: How Batch Correction Can Lead to the Systematic Introduction of False Positive Results in DNA Methylation Microarray Studies. BMC Bioinform. 2020, 21, 271. [Google Scholar] [CrossRef]
- Chervova, O.; Panteleeva, K.; Chernysheva, E.; Widayati, T.A.; Baronik, Ž.F.; Hrbková, N.; Schneider, J.L.; Bobak, M.; Beck, S.; Voloshin, V. Breaking New Ground on Human Health and Well-Being with Epigenetic Clocks: A Systematic Review and Meta-Analysis of Epigenetic Age Acceleration Associations. Ageing Res. Rev. 2024, 102, 102552. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Horvath, S. Epigenetic Ageing Clocks: Statistical Methods and Emerging Computational Challenges. Nat. Rev. Genet. 2025, 26, 350–368. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.J.; Zhao, Q.; Olova, N.N.; Dabrowski, J.; Xie, X.; Latorre-Crespo, E.; Chandra, T. Region-based Epigenetic Clock Design Improves RRBS-based Age Prediction. Aging Cell 2023, 22, e13866. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Franzen, J.; Stiehl, T.; Gobs, M.; Kuo, C.-C.; Nikolić, M.; Hapala, J.; Koop, B.E.; Strathmann, K.; Ritz-Timme, S.; et al. New Targeted Approaches for Epigenetic Age Predictions. BMC Biol. 2020, 18, 71. [Google Scholar] [CrossRef]
- Koch, C.M.; Joussen, S.; Schellenberg, A.; Lin, Q.; Zenke, M.; Wagner, W. Monitoring of Cellular Senescence by DNA-methylation at Specific CpG Sites. Aging Cell 2012, 11, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, J.; Osei-Owusu, I.A.; Avigdor, B.E.; Tupler, R.; Pevsner, J. Mosaicism in Human Health and Disease. Annu. Rev. Genet. 2020, 54, 487–510. [Google Scholar] [CrossRef]
- Kabacik, S.; Lowe, D.; Fransen, L.; Leonard, M.; Ang, S.-L.; Whiteman, C.; Corsi, S.; Cohen, H.; Felton, S.; Bali, R.; et al. The Relationship between Epigenetic Age and the Hallmarks of Aging in Human Cells. Nat. Aging 2022, 2, 484–493. [Google Scholar] [CrossRef]
- Kwiatkowska, K.M.; Mavrogonatou, E.; Papadopoulou, A.; Sala, C.; Calzari, L.; Gentilini, D.; Bacalini, M.G.; Dall’Olio, D.; Castellani, G.; Ravaioli, F.; et al. Heterogeneity of Cellular Senescence: Cell Type-Specific and Senescence Stimulus-Dependent Epigenetic Alterations. Cells 2023, 12, 927. [Google Scholar] [CrossRef]
- Endicott, J.L.; Nolte, P.A.; Shen, H.; Laird, P.W. Cell Division Drives DNA Methylation Loss in Late-Replicating Domains in Primary Human Cells. Nat. Commun. 2022, 13, 6659. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal Criteria for Defining Multipotent Mesenchymal Stromal Cells. The International Society for Cellular Therapy Position Statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to Detect Senescence-Associated Beta-Galactosidase (SA-Βgal) Activity, a Biomarker of Senescent Cells in Culture and In Vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Krueger, F. TrimGalore: A Wrapper around Cutadapt and FastQC to Consistently Apply Adapter and Quality Trimming to FastQ Files, with Extra Functionality for RRBS Data. Available online: https://github.com/FelixKrueger/TrimGalore (accessed on 22 March 2025).
- Krueger, F.; Andrews, S.R. Bismark: A Flexible Aligner and Methylation Caller for Bisulfite-Seq Applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Lee, D.; Koo, B.; Yang, J.; Kim, S. Metheor: Ultrafast DNA Methylation Heterogeneity Calculation from Bisulfite Read Alignments. PLoS Comput. Biol. 2023, 19, e1010946. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | R2 | MAE |
|---|---|---|
| Average methylation in 24 CpG | 1.207 | 0.885 |
| FDRP | 2.252 | 0.616 |
| ME | 2.232 | 0.565 |
| MHL | 1.461 | 0.828 |
| PDR | 1.726 | 0.694 |
| PM | 1.973 | 0.662 |
| qFDRP | 2.412 | 0.499 |
| Average + MHL | 1.094 | 0.897 |
| Average + PDR | 1.183 | 0.884 |
| Average + MHL + PDR | 1.119 | 0.888 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanov, S.E.; Karetnikov, D.I.; Kalashnikova, D.A.; Polivcev, D.E.; Osipov, Y.A.; Maksimov, D.A.; Antoshina, P.A.; Shloma, V.V.; Samoilova, E.M.; Ivanova, A.A.; et al. A Novel Framework for the Design of Minimized Epigenetic Clocks Using the Analysis of DNA Methylation Heterogeneity. Int. J. Mol. Sci. 2025, 26, 5051. https://doi.org/10.3390/ijms26115051
Romanov SE, Karetnikov DI, Kalashnikova DA, Polivcev DE, Osipov YA, Maksimov DA, Antoshina PA, Shloma VV, Samoilova EM, Ivanova AA, et al. A Novel Framework for the Design of Minimized Epigenetic Clocks Using the Analysis of DNA Methylation Heterogeneity. International Journal of Molecular Sciences. 2025; 26(11):5051. https://doi.org/10.3390/ijms26115051
Chicago/Turabian StyleRomanov, Stanislav E., Dmitry I. Karetnikov, Darya A. Kalashnikova, Denis E. Polivcev, Yakov A. Osipov, Daniil A. Maksimov, Polina A. Antoshina, Viktor V. Shloma, Ekaterina M. Samoilova, Alina A. Ivanova, and et al. 2025. "A Novel Framework for the Design of Minimized Epigenetic Clocks Using the Analysis of DNA Methylation Heterogeneity" International Journal of Molecular Sciences 26, no. 11: 5051. https://doi.org/10.3390/ijms26115051
APA StyleRomanov, S. E., Karetnikov, D. I., Kalashnikova, D. A., Polivcev, D. E., Osipov, Y. A., Maksimov, D. A., Antoshina, P. A., Shloma, V. V., Samoilova, E. M., Ivanova, A. A., Karimov, R. F., Tkalin, A. N., Shevchenko, A. A., Kalsin, V. A., Baklaushev, V. P., & Laktionov, P. P. (2025). A Novel Framework for the Design of Minimized Epigenetic Clocks Using the Analysis of DNA Methylation Heterogeneity. International Journal of Molecular Sciences, 26(11), 5051. https://doi.org/10.3390/ijms26115051