
Evolutionary Transcriptomics of Cancer Development



Abstract
1. Introduction
2. Results
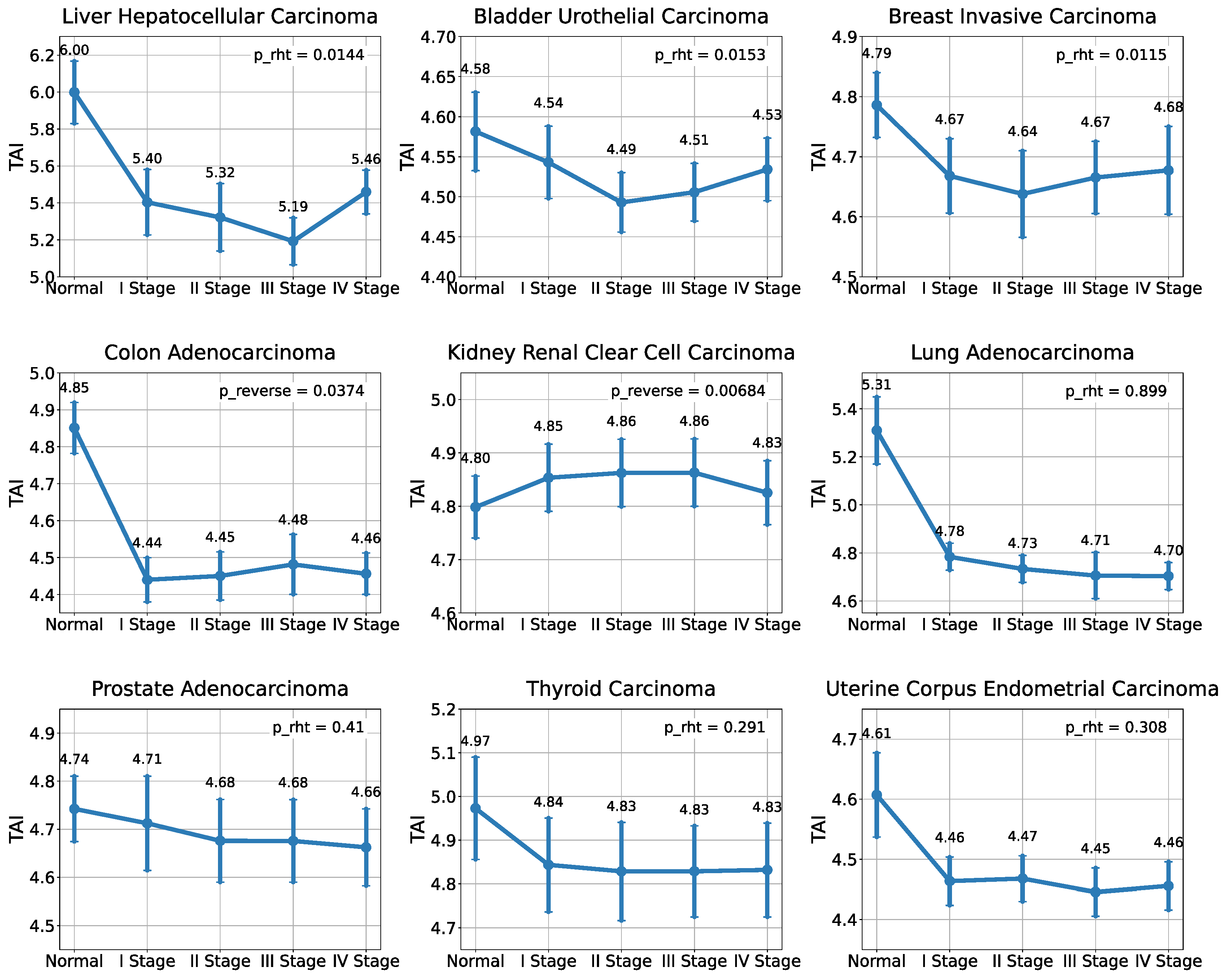
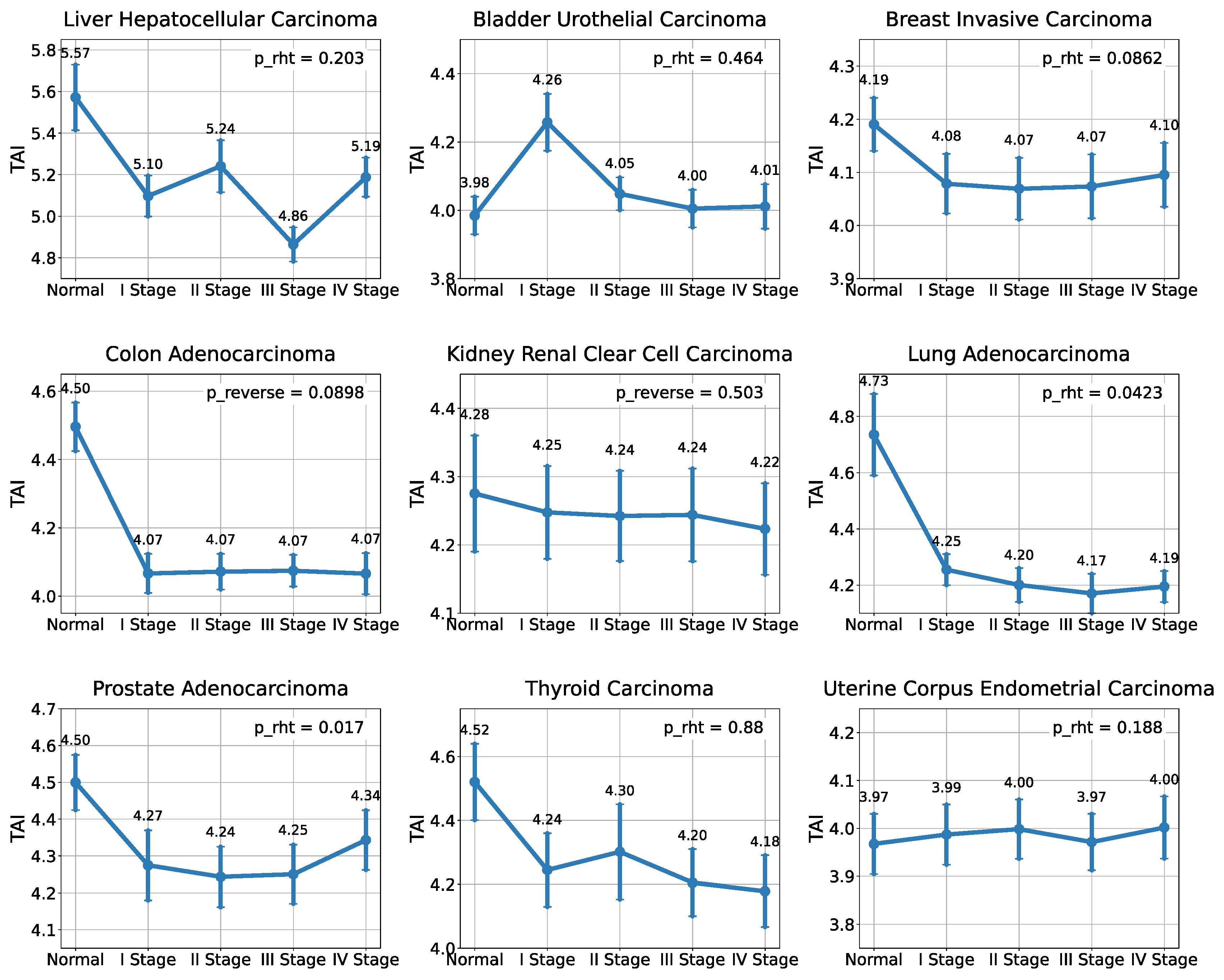
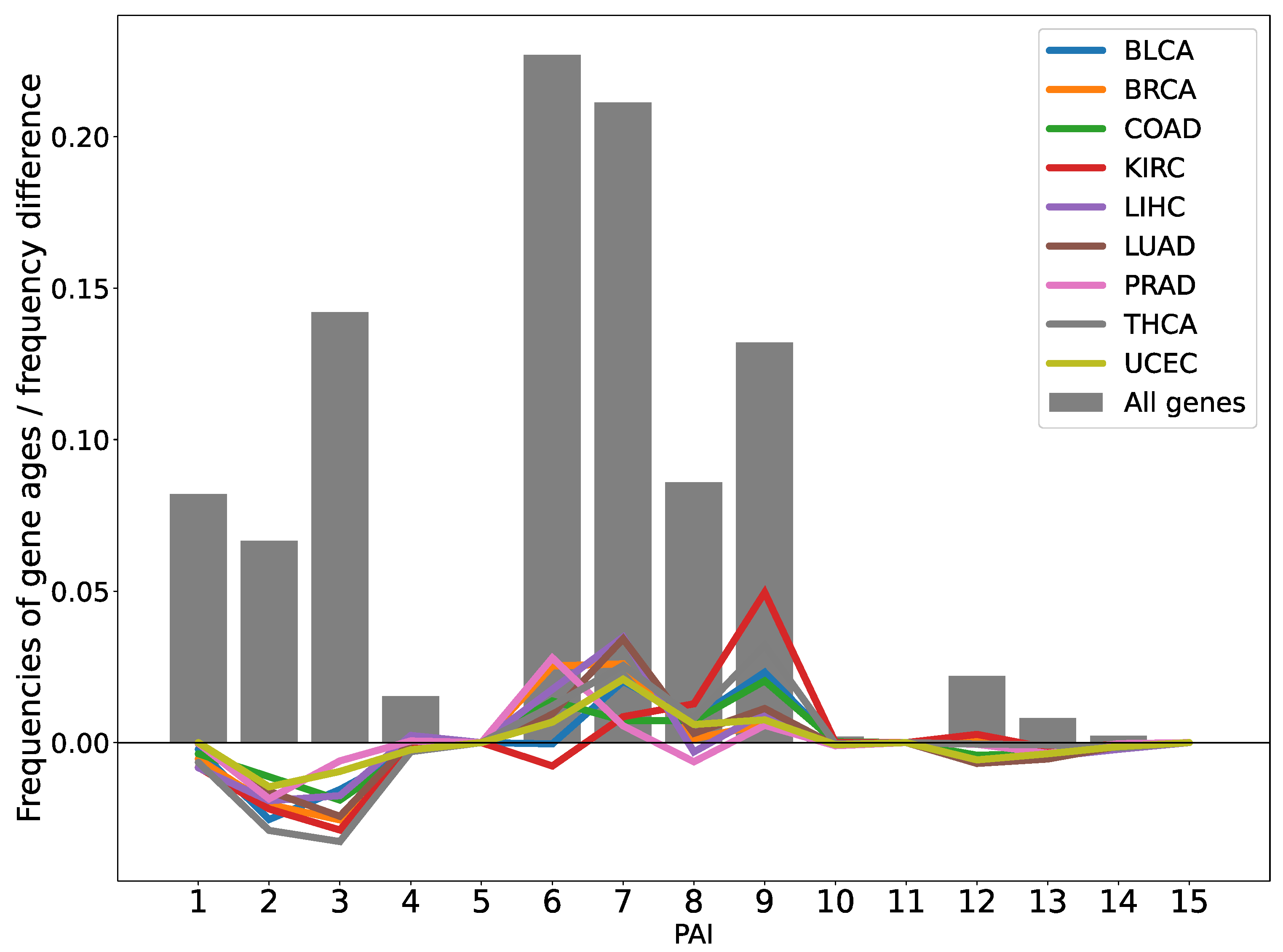
2.1. Analysis of the Evolutionary Characteristics of Transcriptomes
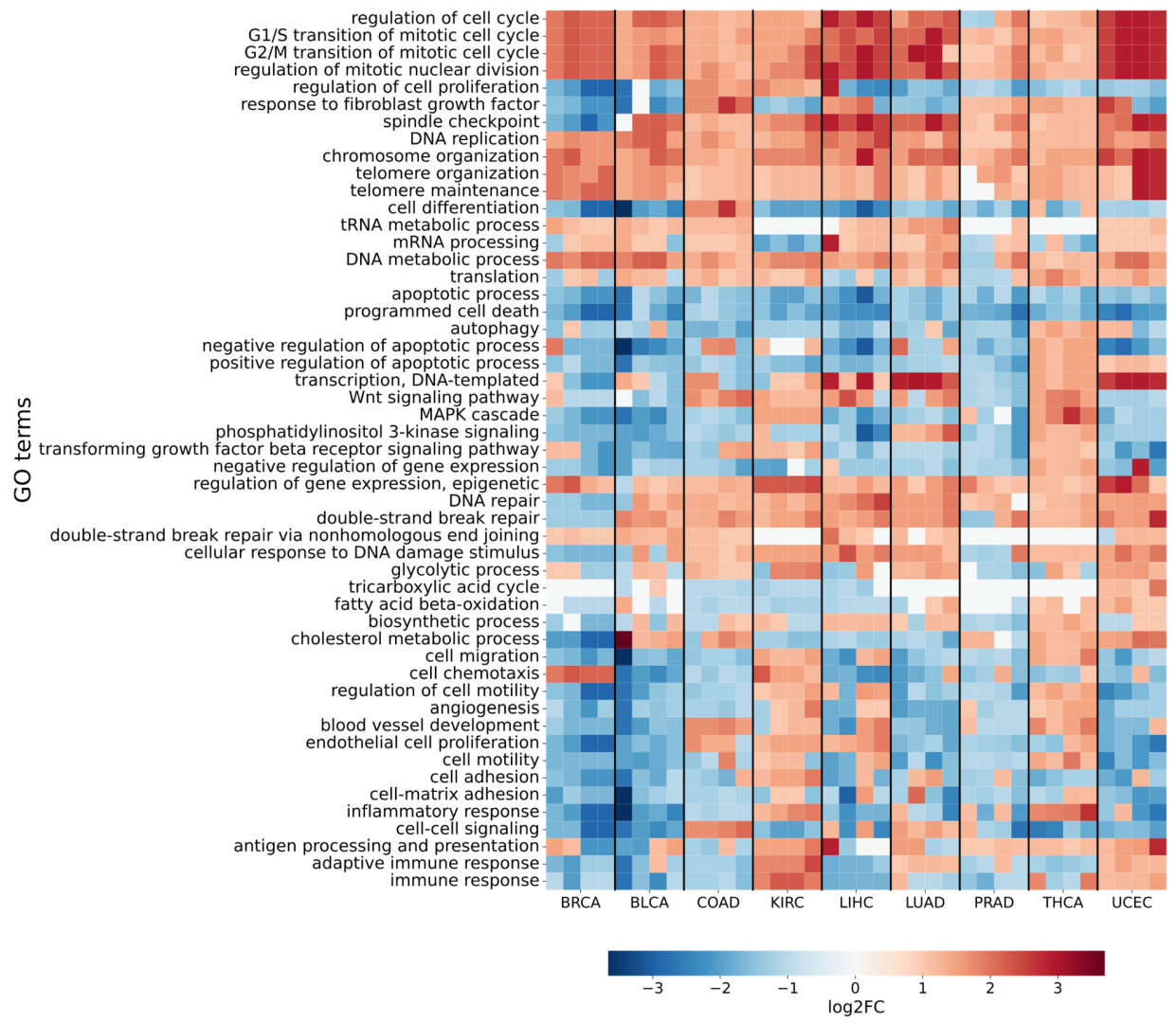
2.2. Differential Gene Expression Analysis in Malignant Tumors
3. Discussion
4. Materials and Methods
4.1. Expression Data and Tumor Samples
4.2. Differential Expression Analysis
4.3. Phylostratigraphic Analysis
4.4. Calculation of Transcriptome Age and Divergence Indices
4.5. Functional Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dujon, A.M.; Aktipis, A.; Alix-Panabières, C.; Amend, S.R.; Boddy, A.M.; Brown, J.S.; Capp, J.; DeGregori, J.; Ewald, P.; Gatenby, R.; et al. Identifying Key Questions in the Ecology and Evolution of Cancer. Evol. Appl. 2021, 14, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Verduzco, D.; Gatenby, R.A. Evolutionary Dynamics of Carcinogenesis and Why Targeted Therapy Does Not Work. Nat. Rev. Cancer 2012, 12, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal Evolution in Cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Maley, C.C.; Aktipis, A.; Graham, T.A.; Sottoriva, A.; Boddy, A.M.; Janiszewska, M.; Silva, A.S.; Gerlinger, M.; Yuan, Y.; Pienta, K.J.; et al. Classifying the Evolutionary and Ecological Features of Neoplasms. Nat. Rev. Cancer 2017, 17, 605–619. [Google Scholar] [CrossRef]
- Merlo, L.M.F.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an Evolutionary and Ecological Process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar] [CrossRef]
- Mina, M.; Raynaud, F.; Tavernari, D.; Battistello, E.; Sungalee, S.; Saghafinia, S.; Laessle, T.; Sanchez-Vega, F.; Schultz, N.; Oricchio, E.; et al. Conditional Selection of Genomic Alterations Dictates Cancer Evolution and Oncogenic Dependencies. Cancer Cell 2017, 32, 155–168.e6. [Google Scholar] [CrossRef]
- Pepper, J.W.; Scott Findlay, C.; Kassen, R.; Spencer, S.L.; Maley, C.C. SYNTHESIS: Cancer Research Meets Evolutionary Biology. Evol. Appl. 2009, 2, 62–70. [Google Scholar] [CrossRef]
- Davies, P.C.W.; Lineweaver, C.H. Cancer Tumors as Metazoa 1.0: Tapping Genes of Ancient Ancestors. Phys. Biol. 2011, 8, 015001. [Google Scholar] [CrossRef]
- Domazet-Lošo, T.; Brajković, J.; Tautz, D. A Phylostratigraphy Approach to Uncover the Genomic History of Major Adaptations in Metazoan Lineages. Trends Genet. 2007, 23, 533–539. [Google Scholar] [CrossRef]
- Domazet-Lošo, T.; Tautz, D. An Ancient Evolutionary Origin of Genes Associated with Human Genetic Diseases. Mol. Biol. Evol. 2008, 25, 2699–2707. [Google Scholar] [CrossRef]
- Domazet-Lošo, T.; Tautz, D. A Phylogenetically Based Transcriptome Age Index Mirrors Ontogenetic Divergence Patterns. Nature 2010, 468, 815–818. [Google Scholar] [CrossRef]
- Quint, M.; Drost, H.-G.; Gabel, A.; Ullrich, K.K.; Bönn, M.; Grosse, I. A Transcriptomic Hourglass in Plant Embryogenesis. Nature 2012, 490, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Cridge, A.G.; Dearden, P.K.; Brownfield, L.R. The Developmental Hourglass in the Evolution of Embryogenesis. In Evolutionary Developmental Biology; Springer International Publishing: Cham, Switzerland, 2019; pp. 1–10. [Google Scholar]
- Drost, H.-G.; Gabel, A.; Grosse, I.; Quint, M. Evidence for Active Maintenance of Phylotranscriptomic Hourglass Patterns in Animal and Plant Embryogenesis. Mol. Biol. Evol. 2015, 32, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Irie, N.; Kuratani, S. The Developmental Hourglass Model: A Predictor of the Basic Body Plan? Development 2014, 141, 4649–4655. [Google Scholar] [CrossRef]
- Zhang, L.; Tan, Y.; Fan, S.; Zhang, X.; Zhang, Z. Phylostratigraphic Analysis of Gene Co-Expression Network Reveals the Evolution of Functional Modules for Ovarian Cancer. Sci. Rep. 2019, 9, 2623. [Google Scholar] [CrossRef]
- Chen, H.; He, X. The Convergent Cancer Evolution toward a Single Cellular Destination. Mol. Biol. Evol. 2016, 33, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Altered Interactions between Unicellular and Multicellular Genes Drive Hallmarks of Transformation in a Diverse Range of Solid Tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 6406–6411. [Google Scholar] [CrossRef]
- Drost, H.-G.; Gabel, A.; Liu, J.; Quint, M.; Grosse, I. myTAI: Evolutionary Transcriptomics with R. Bioinformatics 2018, 34, 1589–1590. [Google Scholar] [CrossRef]
- Piovesan, A.; Antonaros, F.; Vitale, L.; Strippoli, P.; Pelleri, M.C.; Caracausi, M. Human Protein-Coding Genes and Gene Feature Statistics in 2019. BMC Res. Notes 2019, 12, 315. [Google Scholar] [CrossRef]
- Wiebe, D.S.; Omelyanchuk, N.A.; Mukhin, A.M.; Grosse, I.; Lashin, S.A.; Zemlyanskaya, E.V.; Mironova, V.V. Fold-Change-Specific Enrichment Analysis (FSEA): Quantification of Transcriptional Response Magnitude for Functional Gene Groups. Genes 2020, 11, 434. [Google Scholar] [CrossRef]
- Evan, G.I.; Vousden, K.H. Proliferation, Cell Cycle and Apoptosis in Cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Sicinski, P. Cell Cycle Proteins as Promising Targets in Cancer Therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Cancer Genes and the Pathways They Control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-Dependent Kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Y.; Xiong, J.; Lan, B.; Wang, X.; Liu, J.; Lin, J.; Fei, Z.; Zheng, X.; Chen, C. Role of PRKDC in Cancer Initiation, Progression, and Treatment. Cancer Cell Int. 2021, 21, 563. [Google Scholar] [CrossRef]
- Tan, K.T.; Yeh, C.-N.; Chang, Y.-C.; Cheng, J.-H.; Fang, W.-L.; Yeh, Y.-C.; Wang, Y.-C.; Hsu, D.S.-S.; Wu, C.-E.; Lai, J.-I.; et al. PRKDC: New Biomarker and Drug Target for Checkpoint Blockade Immunotherapy. J. Immunother. Cancer 2020, 8, e000485. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Y.; Guan, Y.; Huang, Y.; Lin, J.; Chen, L.; Li, J.; Chen, G.; Pan, L.K.; Xia, X.; et al. Prevalence of PRKDC Mutations and Association with Response to Immune Checkpoint Inhibitors in Solid Tumors. Mol. Oncol. 2020, 14, 2096–2110. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Chen, M.; Yu, X.; Gu, Z.; Qiu, H.; Qin, G.; Long, Q.; Fu, X.; Liu, T.; et al. MAD2L2 Inhibits Colorectal Cancer Growth by Promoting NCOA3 Ubiquitination and Degradation. Mol. Oncol. 2018, 12, 391–405. [Google Scholar] [CrossRef]
- Xu, K.; Zheng, X.; Shi, H.; Ou, J.; Ding, H. MAD2L2, a Key Regulator in Ovarian Cancer and Promoting Tumor Progression. Sci. Rep. 2024, 14, 130. [Google Scholar] [CrossRef]
- Wood, R.D.; Doublié, S. DNA Polymerase θ (POLQ), Double-Strand Break Repair, and Cancer. DNA Repair 2016, 44, 22–32. [Google Scholar] [CrossRef]
- Schrempf, A.; Slyskova, J.; Loizou, J.I. Targeting the DNA Repair Enzyme Polymerase θ in Cancer Therapy. Trends Cancer 2021, 7, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A Decade of Exploring the Cancer Epigenome—Biological and Translational Implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Morgan, M.A.; Shilatifard, A. Chromatin Signatures of Cancer. Genes Dev. 2015, 29, 238–249. [Google Scholar] [CrossRef]
- Truitt, M.L.; Ruggero, D. New Frontiers in Translational Control of the Cancer Genome. Nat. Rev. Cancer 2016, 16, 288–304. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting Apoptosis in Cancer Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Wong, R.S. Apoptosis in Cancer: From Pathogenesis to Treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Janiszewska, M.; Primi, M.C.; Izard, T. Cell Adhesion in Cancer: Beyond the Migration of Single Cells. J. Biol. Chem. 2020, 295, 2495–2505. [Google Scholar] [CrossRef]
- Le Bras, G.F.; Taubenslag, K.J.; Andl, C.D. The Regulation of Cell-Cell Adhesion during Epithelial-Mesenchymal Transition, Motility and Tumor Progression. Cell Adhes. Migr. 2012, 6, 365–373. [Google Scholar] [CrossRef]
- Paul, C.D.; Mistriotis, P.; Konstantopoulos, K. Cancer Cell Motility: Lessons from Migration in Confined Spaces. Nat. Rev. Cancer 2017, 17, 131–140. [Google Scholar] [CrossRef]
- Stuelten, C.H.; Parent, C.A.; Montell, D.J. Cell Motility in Cancer Invasion and Metastasis: Insights from Simple Model Organisms. Nat. Rev. Cancer 2018, 18, 296–312. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Saud, S.M.; Young, M.R.; Chen, G.; Hua, B. Targeting AMPK for Cancer Prevention and Treatment. Oncotarget 2015, 6, 7365–7378. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Saha, A.K.; Xiang, X.; Ruderman, N.B. AMPK, the Metabolic Syndrome and Cancer. Trends Pharmacol. Sci. 2005, 26, 69–76. [Google Scholar] [CrossRef]
- Edelbrock, M.A.; Kaliyaperumal, S.; Williams, K.J. Structural, Molecular and Cellular Functions of MSH2 and MSH6 during DNA Mismatch Repair, Damage Signaling and Other Noncanonical Activities. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2013, 743–744, 53–66. [Google Scholar] [CrossRef]
- Dong, G.; Mao, Q.; Xia, W.; Xu, Y.; Wang, J.; Xu, L.; Jiang, F. PKM2 and Cancer: The Function of PKM2 beyond Glycolysis. Oncol. Lett. 2016, 11, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; De Melo, J.; Tang, D. PKM2, a Central Point of Regulation in Cancer Metabolism. Int. J. Cell Biol. 2013, 2013, 242513. [Google Scholar] [CrossRef]
- Wu, S.; Le, H. Dual Roles of PKM2 in Cancer Metabolism. ABBS 2013, 45, 27–35. [Google Scholar] [CrossRef]
- Zhang, Z.; Deng, X.; Liu, Y.; Liu, Y.; Sun, L.; Chen, F. PKM2, Function and Expression and Regulation. Cell Biosci. 2019, 9, 52. [Google Scholar] [CrossRef]
- Ježek, P. 2-Hydroxyglutarate in Cancer Cells. Antioxid. Redox Signal. 2020, 33, 903–926. [Google Scholar] [CrossRef]
- Losman, J.-A.; Kaelin, W.G. What a Difference a Hydroxyl Makes: Mutant IDH, (R)-2-Hydroxyglutarate, and Cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- Gu, X.; Jiang, Y.; Xue, W.; Song, C.; Wang, Y.; Liu, Y.; Cui, B. SPNS 2 Promotes the Malignancy of Colorectal Cancer Cells via Regulating Akt and ERK Pathway. Clin. Exp. Pharma. Physio. 2019, 46, 861–871. [Google Scholar] [CrossRef]
- Le, T.N.U.; Nguyen, T.Q.; Kalailingam, P.; Nguyen, Y.T.K.; Sukumar, V.K.; Tan, C.K.H.; Tukijan, F.; Couty, L.; Hasan, Z.; Del Gaudio, I.; et al. Mfsd2b and Spns2 Are Essential for Maintenance of Blood Vessels during Development and in Anaphylactic Shock. Cell Rep. 2022, 40, 111208. [Google Scholar] [CrossRef]
- Pham, H.T.T.; Maurer, B.; Prchal-Murphy, M.; Grausenburger, R.; Grundschober, E.; Javaheri, T.; Nivarthi, H.; Boersma, A.; Kolbe, T.; Elabd, M.; et al. STAT5BN642H Is a Driver Mutation for T Cell Neoplasia. J. Clin. Investig. 2017, 128, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Rani, A.; Murphy, J.J. STAT5 in Cancer and Immunity. J. Interferon Cytokine Res. 2016, 36, 226–237. [Google Scholar] [CrossRef]
- Lolodi, O.; Wang, Y.-M.; Wright, W.C.; Chen, T. Differential Regulation of CYP3A4 and CYP3A5 and Its Implication in Drug Discovery. CDM 2018, 18, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, X.; Wang, Y.; Chen, Y.; Lu, H.; Meng, X.; Ye, X.; Chen, W. Activation/Inactivation of Anticancer Drugs by CYP3A4: Influencing Factors for Personalized Cancer Therapy. Drug Metab. Dispos. 2023, 51, 543–559. [Google Scholar] [CrossRef]
- Badenhorst, C.P.S.; Van Der Sluis, R.; Erasmus, E.; Van Dijk, A.A. Glycine Conjugation: Importance in Metabolism, the Role of Glycine N-Acyltransferase, and Factors That Influence Interindividual Variation. Expert Opin. Drug Metab. Toxicol. 2013, 9, 1139–1153. [Google Scholar] [CrossRef]
- Tian, X.; Wu, L.; Jiang, M.; Zhang, Z.; Wu, R.; Miao, J.; Liu, C.; Gao, S. Downregulation of GLYAT Facilitates Tumor Growth and Metastasis and Poor Clinical Outcomes Through the PI3K/AKT/Snail Pathway in Human Breast Cancer. Front. Oncol. 2021, 11, 641399. [Google Scholar] [CrossRef]
- Xia, Y.; Huang, W.; Jin, G.-Z. GLYAT Suppresses Liver Cancer and Clear Cell Renal Cell Carcinoma Progression by Downregulating ROCK1 Expression. Transl. Cancer Res. 2024, 13, 5097–5111. [Google Scholar] [CrossRef] [PubMed]
- Cengiz, B.; Yumrutas, O.; Bozgeyik, E.; Borazan, E.; Igci, Y.Z.; Bozgeyik, I.; Oztuzcu, S. Differential Expression of the UGT1A Family of Genes in Stomach Cancer Tissues. Tumor Biol. 2015, 36, 5831–5837. [Google Scholar] [CrossRef]
- Maruo, Y.; Iwai, M.; Mori, A.; Sato, H.; Takeuchi, Y. Polymorphism of UDP-Glucuronosyltransferase and Drug Metabolism. CDM 2005, 6, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Bussey, K.J.; Davies, P.C.W. Reverting to Single-Cell Biology: The Predictions of the Atavism Theory of Cancer. Prog. Biophys. Mol. Biol. 2021, 165, 49–55. [Google Scholar] [CrossRef]
- Chen, H.; Lin, F.; Xing, K.; He, X. The Reverse Evolution from Multicellularity to Unicellularity during Carcinogenesis. Nat. Commun. 2015, 6, 6367. [Google Scholar] [CrossRef]
- Thomas, F.; Ujvari, B.; Renaud, F.; Vincent, M. Cancer Adaptations: Atavism, de Novo Selection, or Something in Between? BioEssays 2017, 39, 1700039. [Google Scholar] [CrossRef]
- Vincent, M. Cancer: A De-repression of a Default Survival Program Common to All Cells?: A Life-history Perspective on the Nature of Cancer. BioEssays 2012, 34, 72–82. [Google Scholar] [CrossRef]
- Lambert, G.; Estévez-Salmeron, L.; Oh, S.; Liao, D.; Emerson, B.M.; Tlsty, T.D.; Austin, R.H. An Analogy between the Evolution of Drug Resistance in Bacterial Communities and Malignant Tissues. Nat. Rev. Cancer 2011, 11, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Przybycinski, J.; Nalewajska, M.; Marchelek-Mysliwiec, M.; Dziedziejko, V.; Pawlik, A. Poly-ADP-Ribose Polymerases (PARPs) as a Therapeutic Target in the Treatment of Selected Cancers. Expert Opin. Ther. Targets 2019, 23, 773–785. [Google Scholar] [CrossRef]
- Zhao, X.; Zhu, Y.; Hu, J.; Jiang, L.; Li, L.; Jia, S.; Zen, K. Shikonin Inhibits Tumor Growth in Mice by Suppressing Pyruvate Kinase M2-Mediated Aerobic Glycolysis. Sci. Rep. 2018, 8, 14517. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Wrenn, E.; Huang, Y.; Cheung, K. Collective Metastasis: Coordinating the Multicellular Voyage. Clin. Exp. Metastasis 2021, 38, 373–399. [Google Scholar] [CrossRef]
- Asano, K.; Nelson, C.M.; Nandadasa, S.; Aramaki-Hattori, N.; Lindner, D.J.; Alban, T.; Inagaki, J.; Ohtsuki, T.; Oohashi, T.; Apte, S.S.; et al. Stromal Versican Regulates Tumor Growth by Promoting Angiogenesis. Sci. Rep. 2017, 7, 17225. [Google Scholar] [CrossRef]
- Chang, A.C.-M.; Doherty, J.; Huschtscha, L.I.; Redvers, R.; Restall, C.; Reddel, R.R.; Anderson, R.L. STC1 Expression Is Associated with Tumor Growth and Metastasis in Breast Cancer. Clin. Exp. Metastasis 2015, 32, 15–27. [Google Scholar] [CrossRef]
- Cheng, Y.; Sun, H.; Wu, L.; Wu, F.; Tang, W.; Wang, X.; Lv, C. VUp-Regulation of VCAN Promotes the Proliferation, Invasion and Migration and Serves as a Biomarker in Gastric Cancer. OTT 2020, 13, 8665–8675. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Albitar, L.; LeBaron, R.; Welch, W.R.; Samimi, G.; Birrer, M.J.; Berkowitz, R.S.; Mok, S.C. Up-Regulation of Stromal Versican Expression in Advanced Stage Serous Ovarian Cancer. Gynecol. Oncol. 2010, 119, 114–120. [Google Scholar] [CrossRef]
- He, L.; Wang, T.; Gao, Q.; Zhao, G.; Huang, Y.; Yu, L.; Hou, Y. Stanniocalcin-1 Promotes Tumor Angiogenesis through up-Regulation of VEGF in Gastric Cancer Cells. J. Biomed. Sci. 2011, 18, 39. [Google Scholar] [CrossRef]
- Law, A.Y.S.; Wong, C.K.C. Stanniocalcin-1 and -2 Promote Angiogenic Sprouting in HUVECs via VEGF/VEGFR2 and Angiopoietin Signaling Pathways. Mol. Cell. Endocrinol. 2013, 374, 73–81. [Google Scholar] [CrossRef]
- Xu, C.; Sun, L.; Jiang, C.; Zhou, H.; Gu, L.; Liu, Y.; Xu, Q. SPP1, Analyzed by Bioinformatics Methods, Promotes the Metastasis in Colorectal Cancer by Activating EMT Pathway. Biomed. Pharmacother. 2017, 91, 1167–1177. [Google Scholar] [CrossRef]
- Zeng, B.; Zhou, M.; Wu, H.; Xiong, Z. SPP1 Promotes Ovarian Cancer Progression via Integrin β1/FAK/Akt Signaling Pathway. OncoTargets Ther. 2018, 11, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Raza, K.; Agrawal, A.K.; Vaidya, A. Therapy Targeting Angiogenic Potential of Tumor. In Nanotechnology Applications for Cancer Chemotherapy; Elsevier: Amsterdam, The Netherlands, 2021; pp. 113–139. ISBN 978-0-12-817846-1. [Google Scholar]
- Lin, X.; Kang, K.; Chen, P.; Zeng, Z.; Li, G.; Xiong, W.; Yi, M.; Xiang, B. Regulatory Mechanisms of PD-1/PD-L1 in Cancers. Mol. Cancer 2024, 23, 108. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to Build a Bridge from a Population-based to a More “Personalized” Approach to Cancer Staging. CA A Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Camarda, R.; Odegaard, J.; Paik, H.; Oskotsky, B.; Krings, G.; Goga, A.; Sirota, M.; Butte, A.J. Comprehensive Analysis of Normal Adjacent to Tumor Transcriptomes. Nat. Commun. 2017, 8, 1077. [Google Scholar] [CrossRef]
- Chandran, U.R.; Dhir, R.; Ma, C.; Michalopoulos, G.; Becich, M.; Gilbertson, J. Differences in Gene Expression in Prostate Cancer, Normal Appearing Prostate Tissue Adjacent to Cancer and Prostate Tissue from Cancer Free Organ Donors. BMC Cancer 2005, 5, 45. [Google Scholar] [CrossRef]
- Sanz-Pamplona, R.; Berenguer, A.; Cordero, D.; Molleví, D.G.; Crous-Bou, M.; Sole, X.; Paré-Brunet, L.; Guino, E.; Salazar, R.; Santos, C.; et al. Aberrant Gene Expression in Mucosa Adjacent to Tumor Reveals a Molecular Crosstalk in Colon Cancer. Mol. Cancer 2014, 13, 46. [Google Scholar] [CrossRef]
- Huang, X.; Stern, D.F.; Zhao, H. Transcriptional Profiles from Paired Normal Samples Offer Complementary Information on Cancer Patient Survival—Evidence from TCGA Pan-Cancer Data. Sci. Rep. 2016, 6, 20567. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Mustafin, Z.S.; Lashin, S.A.; Matushkin, Y.G. Phylostratigraphic Analysis of Gene Networks of Human Diseases. Vavilov J. Genet. Breed. 2021, 25, 46–56. [Google Scholar] [CrossRef]
- Ivanov, R.A.; Mukhin, A.M.; Kazantsev, F.V.; Mustafin, Z.S.; Afonnikov, D.A.; Matushkin, Y.G.; Lashin, S.A. Orthoweb: A Software Package for Evolutionary Analysis of Gene Networks. Vavilov J. Genet. Breed. 2025, 28, 874–881. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Waskom, M.L. seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCGA_id | Tumor Tissue | I Stage | II Stage | III Stage | IV Stage | NAT |
|---|---|---|---|---|---|---|
| LIHC | Liver adenocarcinoma | 255 | 3 | 79 | 3 | 50 |
| UCEC | Uterine carcinoma | 338 | 53 | 84 | 29 | 35 |
| COAD | Colon adenocarcinoma | 168 | 185 | 116 | 22 | 41 |
| BRCA | Breast carcinoma | 215 | 639 | 249 | 13 | 112 |
| PRAD | Prostate adenocarcinoma | 176 | 174 | 53 | 2 | 52 |
| KIRC | Kidney renal clear cell carcinoma | 417 | 123 | 124 | 84 | 72 |
| LUAD | Lung adenocarcinoma | 298 | 127 | 84 | 27 | 59 |
| BLCA | Bladder urothelial carcinoma | 4 | 130 | 142 | 138 | 18 |
| THCA | Thyroid carcinoma | 284 | 52 | 112 | 55 | 59 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanov, R.; Afonnikov, D.; Matushkin, Y.; Lashin, S. Evolutionary Transcriptomics of Cancer Development. Int. J. Mol. Sci. 2025, 26, 5041. https://doi.org/10.3390/ijms26115041
Ivanov R, Afonnikov D, Matushkin Y, Lashin S. Evolutionary Transcriptomics of Cancer Development. International Journal of Molecular Sciences. 2025; 26(11):5041. https://doi.org/10.3390/ijms26115041
Chicago/Turabian StyleIvanov, Roman, Dmitry Afonnikov, Yury Matushkin, and Sergey Lashin. 2025. "Evolutionary Transcriptomics of Cancer Development" International Journal of Molecular Sciences 26, no. 11: 5041. https://doi.org/10.3390/ijms26115041
APA StyleIvanov, R., Afonnikov, D., Matushkin, Y., & Lashin, S. (2025). Evolutionary Transcriptomics of Cancer Development. International Journal of Molecular Sciences, 26(11), 5041. https://doi.org/10.3390/ijms26115041