
The Splice Variant of the NCOR2 Gene BQ323636.1 Modulates ACSL4 Expression to Enhance Fatty Acid Metabolism and Support of Tumor Growth in Breast Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
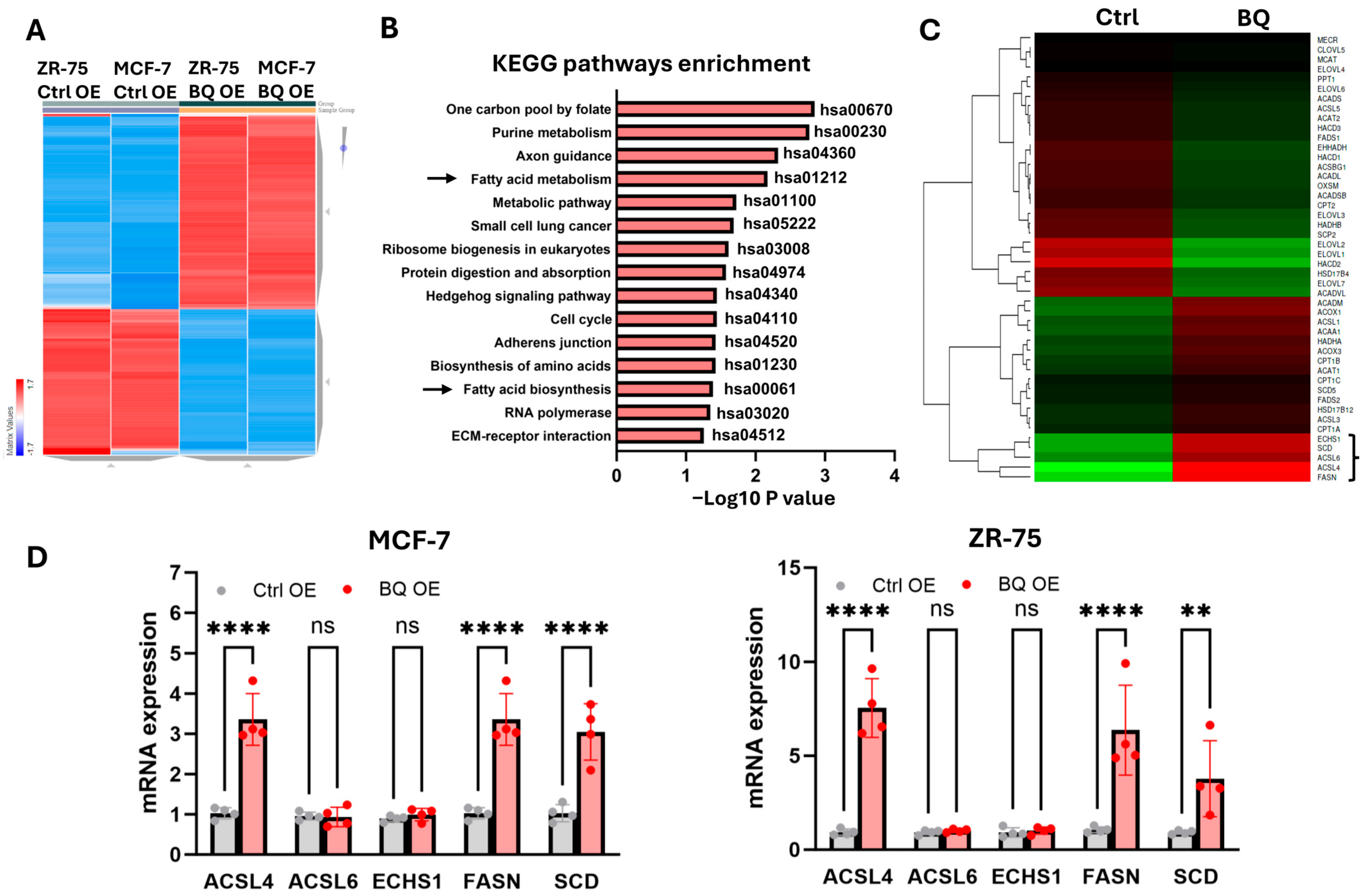
2.1. BQ Overexpression Enhanced Fatty Acid Metabolism to Promote Cancer Cell Proliferation
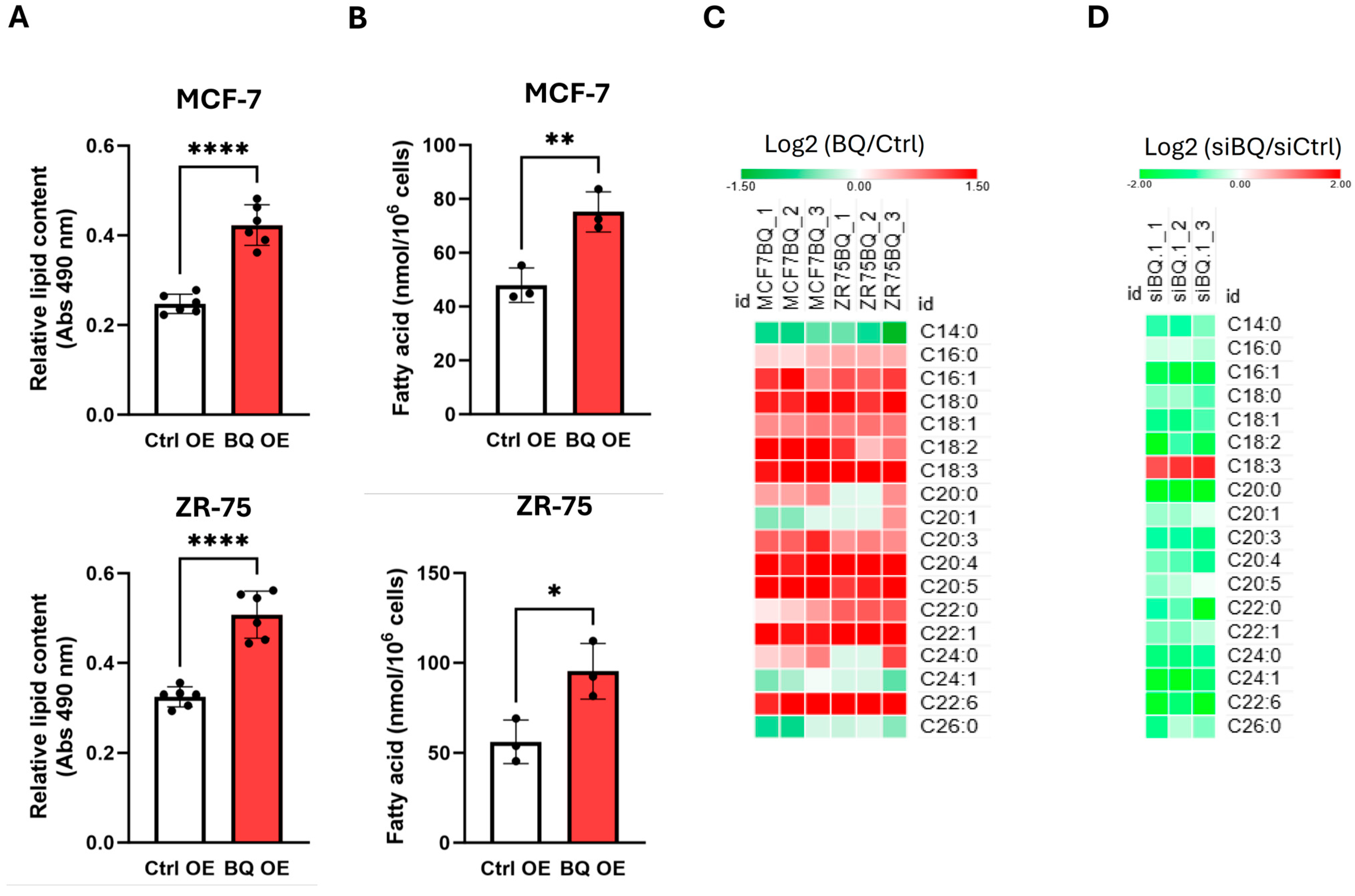
2.2. BQ Overexpression Modulated Fatty Acid Profiles in Breast Cancer Cells
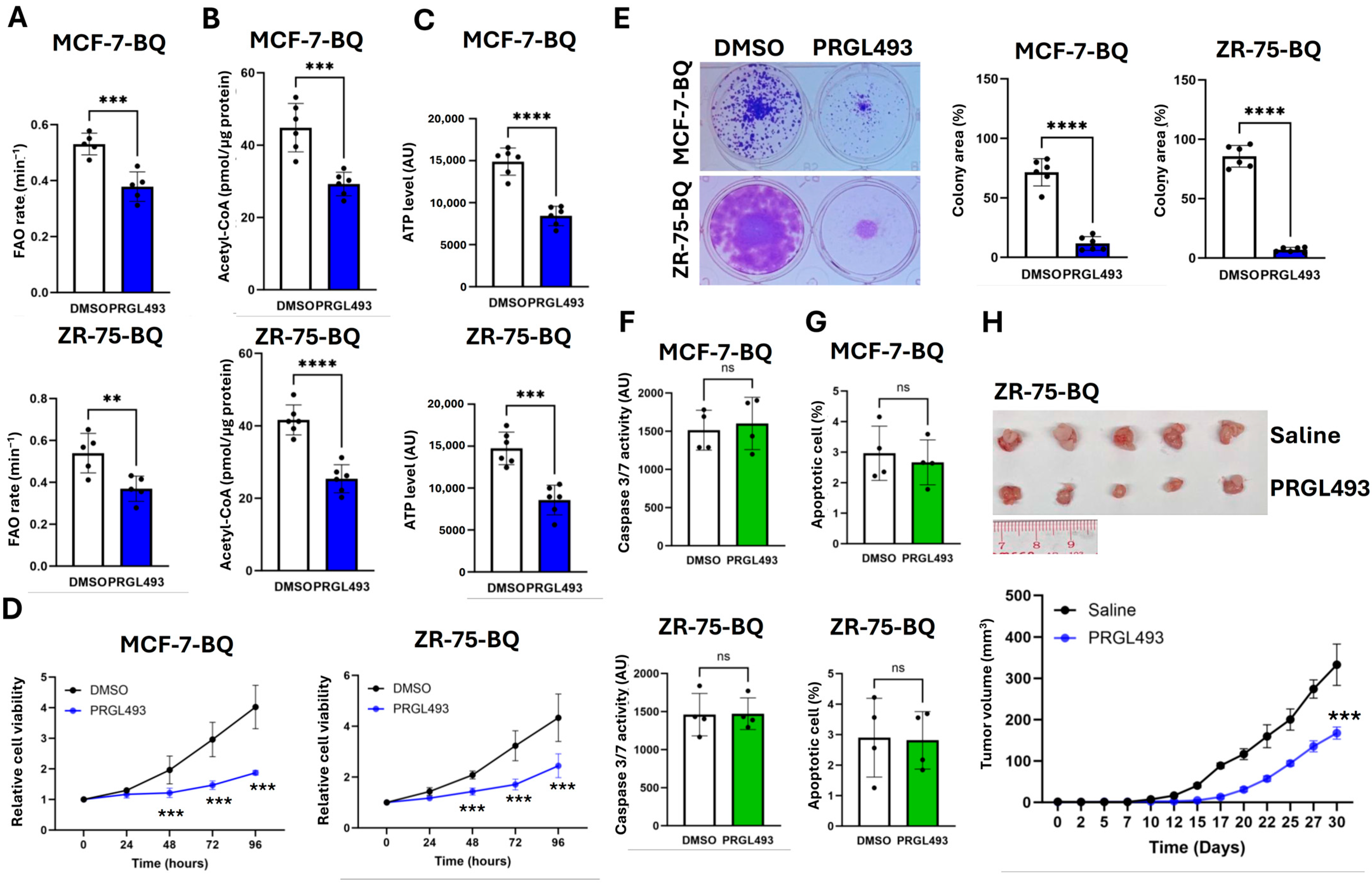
2.3. BQ Overexpression Employed ACSL4 to Enhance ATP Production via FAO
2.4. Suppression of ACLS4 Inhibited Tumor Growth
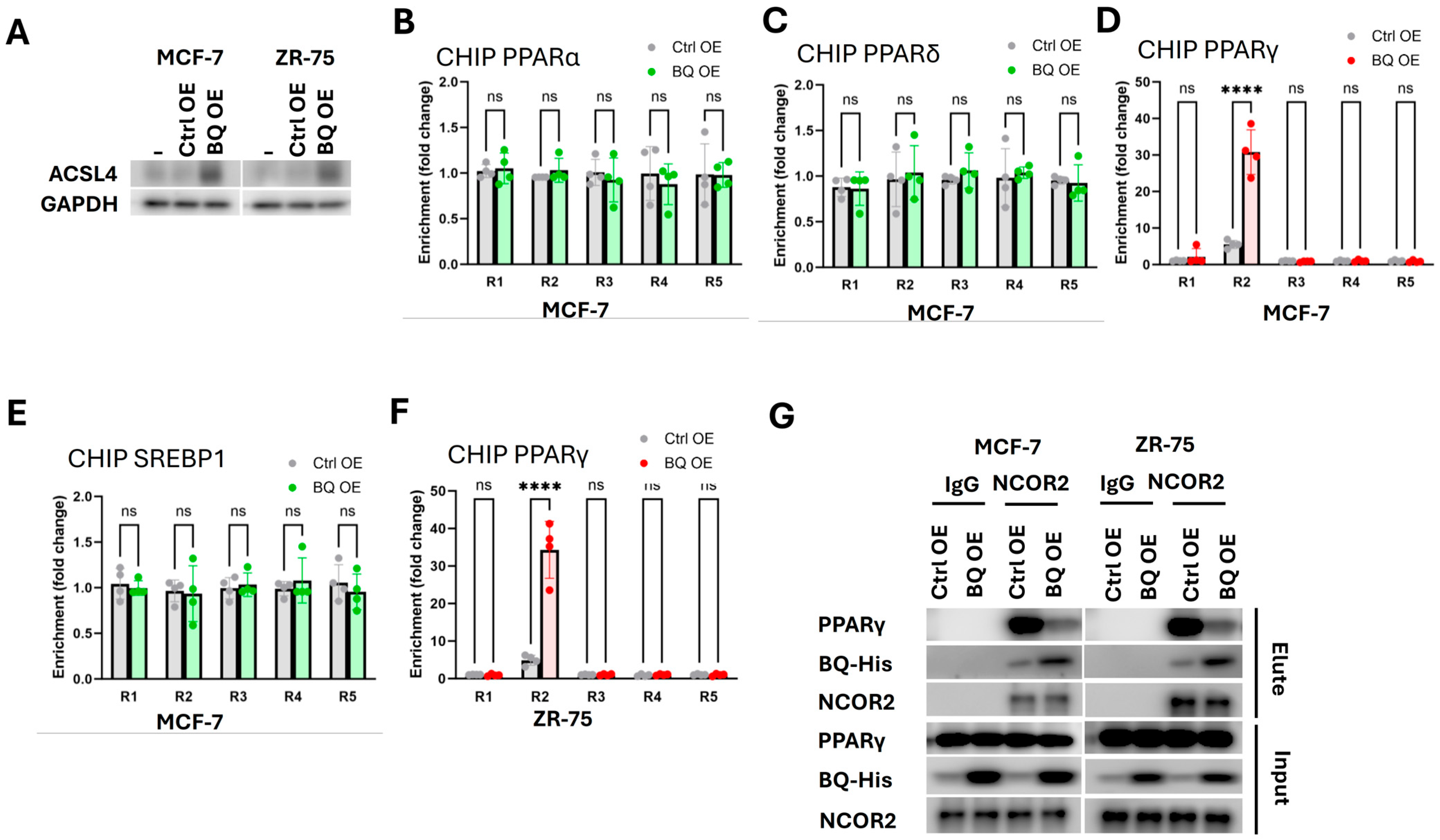
2.5. BQ Overexpression Disrupted the Interaction Between PPARγ and NCOR2
2.6. BQ Overexpression Employed NRF2 to Overcome High Energy Demand Associated Generation of ROS
3. Discussion
4. Material and Methods
4.1. Cell Culture, Transfection, siRNA, and Chemicals
4.2. RNA Sequencing and Pathway Enrichment Analysis
4.3. Targeted Quantitation of C14:0-C26:0 Fatty Acids Analysis
4.4. RNA Extraction and RT-qPCR
4.5. Chromatin Immunoprecipitation (ChIP)
4.6. Co-Immunoprecipitation (CoIP)
4.7. Western Blot
4.8. Lipid Content Measurement
4.9. Functional Assays
4.10. Xenograft
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, J.; Wu, S.G. Breast Cancer: An Overview of Current Therapeutic Strategies, Challenge, and Perspectives. Breast Cancer 2023, 15, 721–730. [Google Scholar] [CrossRef]
- Yoshitake, R.; Mori, H.; Ha, D.; Wu, X.; Wang, J.; Wang, X.; Saeki, K.; Chang, G.; Shim, H.J.; Chan, Y.; et al. Molecular features of luminal breast cancer defined through spatial and single-cell transcriptomics. Clin. Transl. Med. 2024, 14, e1548. [Google Scholar] [CrossRef]
- Marchio, C.; Annaratone, L.; Marques, A.; Casorzo, L.; Berrino, E.; Sapino, A. Evolving concepts in HER2 evaluation in breast cancer: Heterogeneity, HER2-low carcinomas and beyond. Semin. Cancer Biol. 2021, 72, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Mullen, N.J.; Singh, P.K. Nucleotide metabolism: A pan-cancer metabolic dependency. Nat. Rev. Cancer 2023, 23, 275–294. [Google Scholar] [CrossRef]
- Huff, S.E.; Winter, J.M.; Dealwis, C.G. Inhibitors of the Cancer Target Ribonucleotide Reductase, Past and Present. Biomolecules 2022, 12, 815. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Ma, H.; Wang, J.; Huang, M.; Fu, D.; Qin, L.; Yin, Q. Energy metabolism pathways in breast cancer progression: The reprogramming, crosstalk, and potential therapeutic targets. Transl. Oncol. 2022, 26, 101534. [Google Scholar] [CrossRef]
- Tran, T.Q.; Hanse, E.A.; Habowski, A.N.; Li, H.; Ishak Gabra, M.B.; Yang, Y.; Lowman, X.H.; Ooi, A.M.; Liao, S.Y.; Edwards, R.A.; et al. Alpha-Ketoglutarate attenuates Wnt signaling and drives differentiation in colorectal cancer. Nat. Cancer 2020, 1, 345–358. [Google Scholar] [CrossRef]
- Hilgemann, D.W.; Dai, G.; Collins, A.; Lariccia, V.; Magi, S.; Deisl, C.; Fine, M. Lipid signaling to membrane proteins: From second messengers to membrane domains and adapter-free endocytosis. J. Gen. Physiol. 2018, 150, 211–224. [Google Scholar] [CrossRef]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The Biochemistry and Physiology of Mitochondrial Fatty Acid beta-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef]
- Montaigne, D.; Butruille, L.; Staels, B. PPAR control of metabolism and cardiovascular functions. Nat. Rev. Cardiol. 2021, 18, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Li, X.; Ding, Y.; Liu, X.; Diggle, K.; Kisseleva, T.; Brenner, D.A. SREBP Regulation of Lipid Metabolism in Liver Disease, and Therapeutic Strategies. Biomedicines 2023, 11, 3280. [Google Scholar] [CrossRef]
- Lin, J.; Lai, Y.; Lu, F.; Wang, W. Targeting ACSLs to modulate ferroptosis and cancer immunity. Trends Endocrinol. Metab. 2024. [Google Scholar] [CrossRef]
- Grevengoed, T.J.; Klett, E.L.; Coleman, R.A. Acyl-CoA metabolism and partitioning. Annu. Rev. Nutr. 2014, 34, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Song, J.; He, Y.; Liu, Y.; Liu, Z.; Sun, W.; Hu, W.; Lei, Q.Y.; Hu, X.; Chen, Z.; et al. CRISPR/Cas9 Screens Reveal that Hexokinase 2 Enhances Cancer Stemness and Tumorigenicity by Activating the ACSL4-Fatty Acid beta-Oxidation Pathway. Adv. Sci. 2022, 9, e2105126. [Google Scholar] [CrossRef] [PubMed]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef]
- Ma, Y.; Zha, J.; Yang, X.; Li, Q.; Zhang, Q.; Yin, A.; Beharry, Z.; Huang, H.; Huang, J.; Bartlett, M.; et al. Long-chain fatty acyl-CoA synthetase 1 promotes prostate cancer progression by elevation of lipogenesis and fatty acid beta-oxidation. Oncogene 2021, 40, 1806–1820. [Google Scholar] [CrossRef]
- Quan, J.; Cheng, C.; Tan, Y.; Jiang, N.; Liao, C.; Liao, W.; Cao, Y.; Luo, X. Acyl-CoA synthetase long-chain 3-mediated fatty acid oxidation is required for TGFbeta1-induced epithelial-mesenchymal transition and metastasis of colorectal carcinoma. Int. J. Biol. Sci. 2022, 18, 2484–2496. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, P.; Liu, W.; Liu, G.; Zhang, J.; Yan, M.; Duan, Y.; Yang, N. A positive feedback loop between ZEB2 and ACSL4 regulates lipid metabolism to promote breast cancer metastasis. Elife 2023, 12, RP87510. [Google Scholar] [CrossRef]
- Wen, J.; Min, X.; Shen, M.; Hua, Q.; Han, Y.; Zhao, L.; Liu, L.; Huang, G.; Liu, J.; Zhao, X. ACLY facilitates colon cancer cell metastasis by CTNNB1. J. Exp. Clin. Cancer Res. 2019, 38, 401. [Google Scholar] [CrossRef]
- Rios Garcia, M.; Steinbauer, B.; Srivastava, K.; Singhal, M.; Mattijssen, F.; Maida, A.; Christian, S.; Hess-Stumpp, H.; Augustin, H.G.; Muller-Decker, K.; et al. Acetyl-CoA Carboxylase 1-Dependent Protein Acetylation Controls Breast Cancer Metastasis and Recurrence. Cell Metab. 2017, 26, 842–855. [Google Scholar] [CrossRef]
- Ferraro, G.B.; Ali, A.; Luengo, A.; Kodack, D.P.; Deik, A.; Abbott, K.L.; Bezwada, D.; Blanc, L.; Prideaux, B.; Jin, X.; et al. Fatty Acid Synthesis Is Required for Breast Cancer Brain Metastasis. Nat. Cancer 2021, 2, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.H.; Qiu, Y.; Stamatatos, O.; Janowitz, T.; Lukey, M.J. Enhancing the Efficacy of Glutamine Metabolism Inhibitors in Cancer Therapy. Trends. Cancer 2021, 7, 790–804. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Man, E.P.S.; Tsoi, H.; Lee, T.K.W.; Lee, P.; Ma, S.T.; Wong, L.S.; Luk, M.Y.; Rakha, E.A.; Green, A.R.; et al. BQ323636.1, a Novel Splice Variant to NCOR2, as a Predictor for Tamoxifen-Resistant Breast Cancer. Clin. Cancer Res. 2018, 24, 3681–3691. [Google Scholar] [CrossRef]
- Zhang, L.; Gong, C.; Lau, S.L.; Yang, N.; Wong, O.G.; Cheung, A.N.; Tsang, J.W.; Chan, K.Y.; Khoo, U.S. SpliceArray profiling of breast cancer reveals a novel variant of NCOR2/SMRT that is associated with tamoxifen resistance and control of ERalpha transcriptional activity. Cancer Res. 2013, 73, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, H.; Man, E.P.S.; Chau, K.M.; Khoo, U.S. Targeting the IL-6/STAT3 Signalling Cascade to Reverse Tamoxifen Resistance in Estrogen Receptor Positive Breast Cancer. Cancers 2021, 13, 1511. [Google Scholar] [CrossRef]
- Tsoi, H.; Lok, J.; Man, E.P.; Cheng, C.N.; Leung, M.H.; You, C.P.; Chan, S.Y.; Chan, W.L.; Khoo, U.S. Overexpression of BQ323636.1 contributes to anastrozole resistance in AR+ve/ER+ve breast cancer. J. Pathol. 2023, 261, 156–168. [Google Scholar] [CrossRef]
- Leung, M.H.; Tsoi, H.; Gong, C.; Man, E.P.; Zona, S.; Yao, S.; Lam, E.W.; Khoo, U.S. A Splice Variant of NCOR2, BQ323636.1, Confers Chemoresistance in Breast Cancer by Altering the Activity of NRF2. Cancers 2020, 12, 533. [Google Scholar] [CrossRef]
- Watson, P.J.; Fairall, L.; Schwabe, J.W. Nuclear hormone receptor co-repressors: Structure and function. Mol. Cell Endocrinol. 2012, 348, 440–449. [Google Scholar] [CrossRef]
- Tsoi, H.; Man, E.P.; Leung, M.H.; Mok, K.C.; Chau, K.M.; Wong, L.S.; Chan, W.L.; Chan, S.Y.; Luk, M.Y.; Cheng, C.N.; et al. KPNA1 regulates nuclear import of NCOR2 splice variant BQ323636.1 to confer tamoxifen resistance in breast cancer. Clin. Transl. Med. 2021, 11, e554. [Google Scholar] [CrossRef]
- Tsoi, H.; Shi, L.; Leung, M.H.; Man, E.P.S.; So, Z.Q.; Chan, W.L.; Khoo, U.S. Overexpression of BQ323636.1 Modulated AR/IL-8/CXCR1 Axis to Confer Tamoxifen Resistance in ER-Positive Breast Cancer. Life 2022, 12, 93. [Google Scholar] [CrossRef]
- Zhang, Y.K.; Qu, Y.Y.; Lin, Y.; Wu, X.H.; Chen, H.Z.; Wang, X.; Zhou, K.Q.; Wei, Y.; Guo, F.; Yao, C.F.; et al. Enoyl-CoA hydratase-1 regulates mTOR signaling and apoptosis by sensing nutrients. Nat. Commun. 2017, 8, 464. [Google Scholar] [CrossRef]
- Schroeder, B.; Vander Steen, T.; Espinoza, I.; Venkatapoorna, C.M.K.; Hu, Z.; Silva, F.M.; Regan, K.; Cuyas, E.; Meng, X.W.; Verdura, S.; et al. Fatty acid synthase (FASN) regulates the mitochondrial priming of cancer cells. Cell Death Dis. 2021, 12, 977. [Google Scholar] [CrossRef]
- AM, A.L.; Syed, D.N.; Ntambi, J.M. Insights into Stearoyl-CoA Desaturase-1 Regulation of Systemic Metabolism. Trends Endocrinol. Metab. 2017, 28, 831–842. [Google Scholar] [CrossRef]
- Castillo, A.F.; Orlando, U.D.; Maloberti, P.M.; Prada, J.G.; Dattilo, M.A.; Solano, A.R.; Bigi, M.M.; Rios Medrano, M.A.; Torres, M.T.; Indo, S.; et al. New inhibitor targeting Acyl-CoA synthetase 4 reduces breast and prostate tumor growth, therapeutic resistance and steroidogenesis. Cell. Mol. Life Sci. 2021, 78, 2893–2910. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, M.; Cao, J.; Wang, F.; Han, J.R.; Wu, T.W.; Li, L.; Yu, J.; Fan, Y.; Xie, G.; et al. ACSL4 and polyunsaturated lipids support metastatic extravasation and colonization. Cell 2025, 188, 412–429. [Google Scholar] [CrossRef] [PubMed]
- Bidault, G.; Virtue, S.; Petkevicius, K.; Jolin, H.E.; Dugourd, A.; Guenantin, A.C.; Leggat, J.; Mahler-Araujo, B.; Lam, B.Y.H.; Ma, M.K.; et al. SREBP1-induced fatty acid synthesis depletes macrophages antioxidant defences to promote their alternative activation. Nat. Metab. 2021, 3, 1150–1162. [Google Scholar] [CrossRef]
- Palma, F.R.; Gantner, B.N.; Sakiyama, M.J.; Kayzuka, C.; Shukla, S.; Lacchini, R.; Cunniff, B.; Bonini, M.G. ROS production by mitochondria: Function or dysfunction? Oncogene 2024, 43, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D. Thirty years of NRF2: Advances and therapeutic challenges. Nat. Rev. Drug. Discov. 2025. [Google Scholar] [CrossRef]
- Vogel, F.C.E.; Chaves-Filho, A.B.; Schulze, A. Lipids as mediators of cancer progression and metastasis. Nat. Cancer 2024, 5, 16–29. [Google Scholar] [CrossRef]
- Bergers, G.; Fendt, S.M. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef] [PubMed]
- Mok, K.C.; Tsoi, H.; Man, E.P.; Leung, M.H.; Chau, K.M.; Wong, L.S.; Chan, W.L.; Chan, S.Y.; Luk, M.Y.; Chan, J.Y.W.; et al. Repurposing hyperpolarization-activated cyclic nucleotide-gated channels as a novel therapy for breast cancer. Clin. Transl. Med. 2021, 11, e578. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Matsuura, Y.; Ishiguro-Watanabe, M. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res. 2025, 53, D672–D677. [Google Scholar] [CrossRef] [PubMed]
- Torre, D.; Lachmann, A.; Ma’ayan, A. BioJupies: Automated Generation of Interactive Notebooks for RNA-Seq Data Analysis in the Cloud. Cell Syst. 2018, 7, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Meylan, P.; Dreos, R.; Ambrosini, G.; Groux, R.; Bucher, P. EPD in 2020: Enhanced data visualization and extension to ncRNA promoters. Nucleic Acids Res. 2020, 48, D65–D69. [Google Scholar] [CrossRef]
- Kaczmarek, I.; Suchy, T.; Strnadova, M.; Thor, D. Qualitative and quantitative analysis of lipid droplets in mature 3T3-L1 adipocytes using oil red O. STAR Protoc. 2024, 5, 102977. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsoi, H.; You, C.-P.; Cheung, K.H.-L.; Tse, Y.-S.; Khoo, U.-S. The Splice Variant of the NCOR2 Gene BQ323636.1 Modulates ACSL4 Expression to Enhance Fatty Acid Metabolism and Support of Tumor Growth in Breast Cancer. Int. J. Mol. Sci. 2025, 26, 4989. https://doi.org/10.3390/ijms26114989
Tsoi H, You C-P, Cheung KH-L, Tse Y-S, Khoo U-S. The Splice Variant of the NCOR2 Gene BQ323636.1 Modulates ACSL4 Expression to Enhance Fatty Acid Metabolism and Support of Tumor Growth in Breast Cancer. International Journal of Molecular Sciences. 2025; 26(11):4989. https://doi.org/10.3390/ijms26114989
Chicago/Turabian StyleTsoi, Ho, Chan-Ping You, Koei Ho-Lam Cheung, Yin-Suen Tse, and Ui-Soon Khoo. 2025. "The Splice Variant of the NCOR2 Gene BQ323636.1 Modulates ACSL4 Expression to Enhance Fatty Acid Metabolism and Support of Tumor Growth in Breast Cancer" International Journal of Molecular Sciences 26, no. 11: 4989. https://doi.org/10.3390/ijms26114989
APA StyleTsoi, H., You, C.-P., Cheung, K. H.-L., Tse, Y.-S., & Khoo, U.-S. (2025). The Splice Variant of the NCOR2 Gene BQ323636.1 Modulates ACSL4 Expression to Enhance Fatty Acid Metabolism and Support of Tumor Growth in Breast Cancer. International Journal of Molecular Sciences, 26(11), 4989. https://doi.org/10.3390/ijms26114989