
Induced Pluripotent Stem Cells in Cardiomyopathy: Advancing Disease Modeling, Therapeutic Development, and Regenerative Therapy

 ,
,  , , and
, , and {kind=link}
{kind=link}
Abstract
1. Introduction
2. Generation of iPSCs for Cardiomyopathy Models
2.1. Reprogramming Somatic Cells into iPSCs
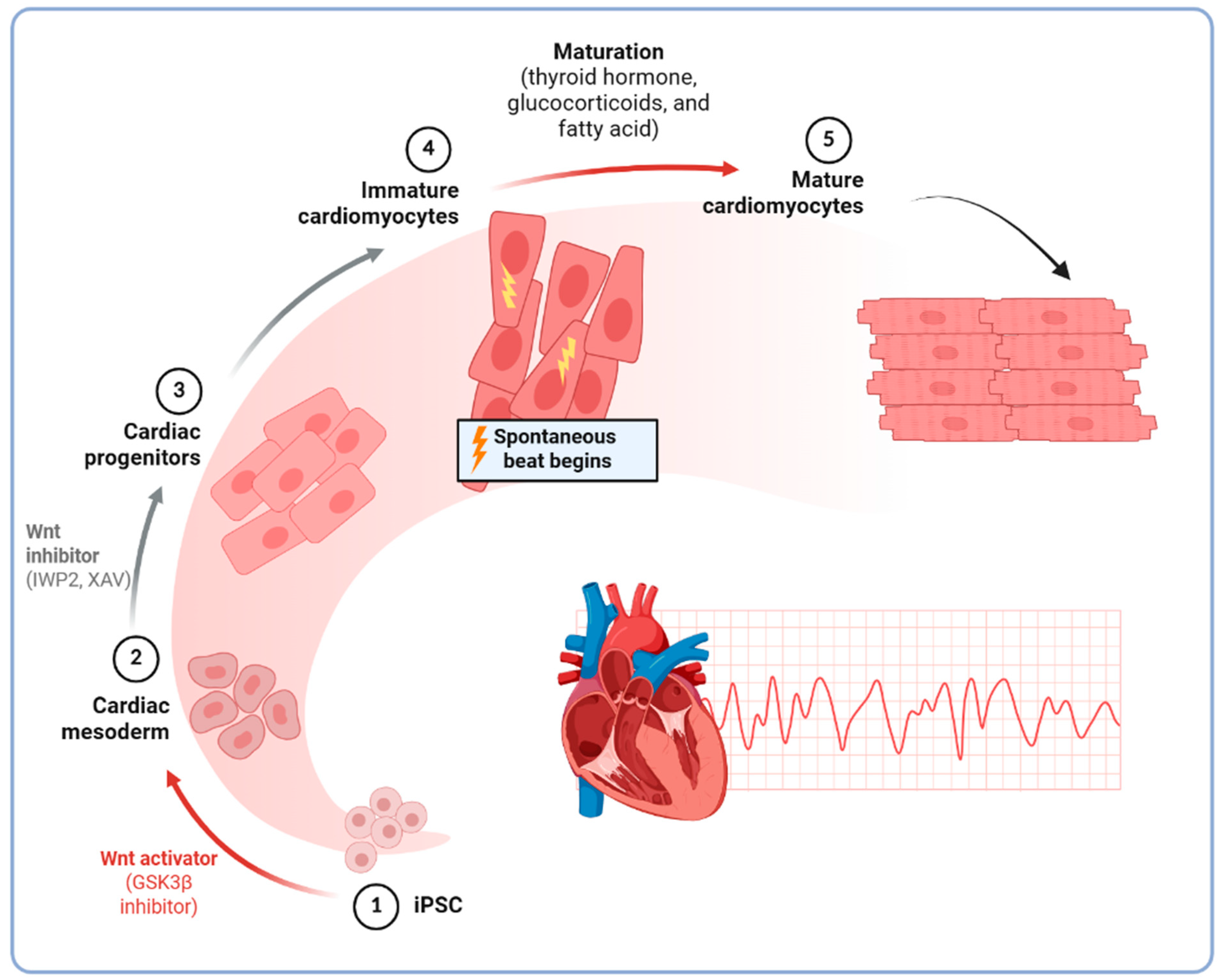
2.2. Differentiation into Cardiomyocytes
2.3. Characterization of iPSC-Derived Cardiomyocytes
3. Applications of iPSCs in Cardiomyopathy Research
3.1. Disease Modeling
3.2. Drug Screening and Personalized Medicine
3.3. Gene Editing and Therapeutic Approaches
4. Challenges and Limitations of iPSC-CM Models in Cardiomyopathy Research and Therapy
4.1. Immaturity of iPSC-CMs
4.2. Variability and Modeling Limitations
4.3. Cost and Technical Expertise
4.4. Ethical Considerations
5. Future Perspectives and Emerging Trends
5.1. Advances in Maturation Techniques
5.2. Integration of Multi-Omics Approaches
5.3. AI in iPSC-Based Research
6. Regenerative Medicine and Future Prospect
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| ACM | Arrhythmogenic cardiomyopathy |
| AI | Artificial intelligence |
| ARVC | Right ventricular cardiomyopathy |
| CiPA | Comprehensive in vitro Proarrhythmia Assay |
| CPVT | Catecholaminergic polymorphic ventricular tachycardia |
| DCM | Dilated cardiomyopathy |
| DL | Deep learning |
| DMD | Duchenne muscular dystrophy |
| HCM | Hypertrophic cardiomyopathy |
| HTS | High-throughput screening |
| iPSC | Induced pluripotent stem cell |
| iPS-CM | iPSC-derived cardiomyocyte |
| LVNC | Left ventricular noncompaction |
| ML | Machine learning |
| RCM | Restrictive cardiomyopathy |
| SCD | Sudden cardiac death |
References
- Polovina, M.; Tschöpe, C.; Rosano, G.; Metra, M.; Crea, F.; Mullens, W.; Bauersachs, J.; Sliwa, K.; de Boer, R.A.; Farmakis, D.; et al. Incidence, risk assessment and prevention of sudden cardiac death in cardiomyopathies. Eur. J. Heart Fail. 2023, 25, 2144–2163. [Google Scholar] [CrossRef] [PubMed]
- Seferović, P.M.; Polovina, M.; Bauersachs, J.; Arad, M.; Ben Gal, T.; Lund, L.H.; Felix, S.B.; Arbustini, E.; Caforio, A.L.P.; Farmakis, D.; et al. Heart failure in cardiomyopathies: A position paper from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 553–576. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.F.; Oakley, C.M. The cardiomyopathies. Br. Heart J. 1972, 34, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Graziano, F.; Bauce, B.; Bueno Marinas, M.; Calore, C.; Celeghin, R.; Cipriani, A.; De Gaspari, M.; De Lazzari, M.; Migliore, F.; et al. The ‘Padua classification’ of cardiomyopathies into three groups: Hypertrophic/restrictive, dilated/hypokinetic, and scarring/arrhythmogenic. Eur. Heart J. Suppl. 2025, 27, i73–i82. [Google Scholar] [CrossRef]
- Maron, B.J.; Spirito, P.; Roman, M.J.; Paranicas, M.; Okin, P.M.; Best, L.G.; Lee, E.T.; Devereux, R.B. Prevalence of hypertrophic cardiomyopathy in a population-based sample of American Indians aged 51 to 77 years (the Strong Heart Study). Am. J. Cardiol. 2004, 93, 1510–1514. [Google Scholar] [CrossRef]
- Tudurachi, B.S.; Zăvoi, A.; Leonte, A.; Țăpoi, L.; Ureche, C.; Bîrgoan, S.G.; Chiuariu, T.; Anghel, L.; Radu, R.; Sascău, R.A.; et al. An Update on MYBPC3 Gene Mutation in Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 10510. [Google Scholar] [CrossRef]
- Franke, M.; Książczyk, T.M.; Dux, M.; Chmielewski, P.; Truszkowska, G.; Czapczak, D.; Pietrzak, R.; Bilinska, Z.T.; Demkow, U.; Werner, B. A MYH7 variant in a five-generation-family with hypertrophic cardiomyopathy. Front. Genet. 2024, 15, 1306333. [Google Scholar] [CrossRef]
- Anfinson, M.; Fitts, R.H.; Lough, J.W.; James, J.M.; Simpson, P.M.; Handler, S.S.; Mitchell, M.E.; Tomita-Mitchell, A. Significance of α-Myosin Heavy Chain (MYH6) Variants in Hypoplastic Left Heart Syndrome and Related Cardiovascular Diseases. J. Cardiovasc. Dev. Dis. 2022, 9, 144. [Google Scholar] [CrossRef]
- Reichart, D.; Magnussen, C.; Zeller, T.; Blankenberg, S. Dilated cardiomyopathy: From epidemiologic to genetic phenotypes: A translational review of current literature. J. Intern. Med. 2019, 286, 362–372. [Google Scholar] [CrossRef]
- Chmielewski, P.; Truszkowska, G.; Kowalik, I.; Rydzanicz, M.; Michalak, E.; Sobieszczańska-Małek, M.; Franaszczyk, M.; Stawiński, P.; Stępień-Wojno, M.; Oręziak, A.; et al. Titin-Related Dilated Cardiomyopathy: The Clinical Trajectory and the Role of Circulating Biomarkers in the Clinical Assessment. Diagnostics 2021, 12, 13. [Google Scholar] [CrossRef]
- Zaragoza, M.V.; Bui, T.A.; Widyastuti, H.P.; Mehrabi, M.; Cang, Z.; Sha, Y.; Grosberg, A.; Nie, Q. LMNA-Related Dilated Cardiomyopathy: Single-Cell Transcriptomics during Patient-Derived iPSC Differentiation Support Cell Type and Lineage-Specific Dysregulation of Gene Expression and Development for Cardiomyocytes and Epicardium-Derived Cells with Lamin A/C Haploinsufficiency. Cells 2024, 13, 1479. [Google Scholar] [CrossRef]
- Brodehl, A.; Gerull, B. Genetic Insights into Primary Restrictive Cardiomyopathy. J. Clin. Med. 2022, 11, 2094. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, J.; Kubo, T.; Duque, M.; Uribe, W.; Shaw, A.; Murphy, R.; Gimeno, J.R.; Elliott, P.; McKenna, W.J. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J. Clin. Investig. 2003, 111, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Peddy, S.B.; Vricella, L.A.; Crosson, J.E.; Oswald, G.L.; Cohn, R.D.; Cameron, D.E.; Valle, D.; Loeys, B.L. Infantile restrictive cardiomyopathy resulting from a mutation in the cardiac troponin T gene. Pediatrics 2006, 117, 1830–1833. [Google Scholar] [CrossRef]
- Ploski, R.; Rydzanicz, M.; Ksiazczyk, T.M.; Franaszczyk, M.; Pollak, A.; Kosinska, J.; Michalak, E.; Stawinski, P.; Ziolkowska, L.; Bilinska, Z.T.; et al. Evidence for troponin C (TNNC1) as a gene for autosomal recessive restrictive cardiomyopathy with fatal outcome in infancy. Am. J. Med. Genet. A 2016, 170, 3241–3248. [Google Scholar] [CrossRef]
- Caleshu, C.; Sakhuja, R.; Nussbaum, R.L.; Schiller, N.B.; Ursell, P.C.; Eng, C.; De Marco, T.; McGlothlin, D.; Burchard, E.G.; Rame, J.E. Furthering the link between the sarcomere and primary cardiomyopathies: Restrictive cardiomyopathy associated with multiple mutations in genes previously associated with hypertrophic or dilated cardiomyopathy. Am. J. Med. Genet. A 2011, 155, 2229–2235. [Google Scholar] [CrossRef]
- Kaski, J.P.; Syrris, P.; Burch, M.; Tomé-Esteban, M.T.; Fenton, M.; Christiansen, M.; Andersen, P.S.; Sebire, N.; Ashworth, M.; Deanfield, J.E.; et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart 2008, 94, 1478–1484. [Google Scholar] [CrossRef]
- Wu, W.; Lu, C.X.; Wang, Y.N.; Liu, F.; Chen, W.; Liu, Y.T.; Han, Y.C.; Cao, J.; Zhang, S.Y.; Zhang, X. Novel Phenotype-Genotype Correlations of Restrictive Cardiomyopathy With Myosin-Binding Protein C (MYBPC3) Gene Mutations Tested by Next-Generation Sequencing. J. Am. Heart Assoc. 2015, 4, e001879. [Google Scholar] [CrossRef]
- Hager, S.; Mahrholdt, H.; Goldfarb, L.G.; Goebel, H.H.; Sechtem, U. Images in cardiovascular medicine. Giant right atrium in the setting of desmin-related restrictive cardiomyopathy. Circulation 2006, 113, e53–e55. [Google Scholar] [CrossRef]
- Brodehl, A.; Ferrier, R.A.; Hamilton, S.J.; Greenway, S.C.; Brundler, M.A.; Yu, W.; Gibson, W.T.; McKinnon, M.L.; McGillivray, B.; Alvarez, N.; et al. Mutations in FLNC are Associated with Familial Restrictive Cardiomyopathy. Hum. Mutat. 2016, 37, 269–279. [Google Scholar] [CrossRef]
- Paller, M.S.; Martin, C.M.; Pierpont, M.E. Restrictive cardiomyopathy: An unusual phenotype of a lamin A variant. ESC Heart Fail. 2018, 5, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.C.; Coughlin, C.R.; Geiger, E.A.; Salvador, B.J.; Elias, E.R.; Cavanaugh, J.L.; Chatfield, K.C.; Miyamoto, S.D.; Shaikh, T.H. Discovery of a potentially deleterious variant in TMEM87B in a patient with a hemizygous 2q13 microdeletion suggests a recessive condition characterized by congenital heart disease and restrictive cardiomyopathy. Cold Spring Harb. Mol. Case Stud. 2016, 2, a000844. [Google Scholar] [CrossRef]
- Alhamoudi, K.M.; Barhoumi, T.; Al-Eidi, H.; Asiri, A.; Nashabat, M.; Alaamery, M.; Alharbi, M.; Alhaidan, Y.; Tabarki, B.; Umair, M.; et al. A homozygous nonsense mutation in DCBLD2 is a candidate cause of developmental delay, dysmorphic features and restrictive cardiomyopathy. Sci. Rep. 2021, 11, 12861. [Google Scholar] [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Klauke, B.; Grewe, S.A.; Schirmer, I.; Peterschröder, A.; Faber, L.; Vorgerd, M.; Gummert, J.; Anselmetti, D.; et al. The novel αB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum. Mutat. 2017, 38, 947–952. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef]
- Norman, M.; Simpson, M.; Mogensen, J.; Shaw, A.; Hughes, S.; Syrris, P.; Sen-Chowdhry, S.; Rowland, E.; Crosby, A.; McKenna, W.J. Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation 2005, 112, 636–642. [Google Scholar] [CrossRef]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef]
- Gerull, B.; Brodehl, A. Insights Into Genetics and Pathophysiology of Arrhythmogenic Cardiomyopathy. Curr. Heart Fail. Rep. 2021, 18, 378–390. [Google Scholar] [CrossRef]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef]
- Brodehl, A.; Meshkov, A.; Myasnikov, R.; Kiseleva, A.; Kulikova, O.; Klauke, B.; Sotnikova, E.; Stanasiuk, C.; Divashuk, M.; Pohl, G.M.; et al. Hemi- and Homozygous Loss-of-Function Mutations in DSG2 (Desmoglein-2) Cause Recessive Arrhythmogenic Cardiomyopathy with an Early Onset. Int. J. Mol. Sci. 2021, 22, 3786. [Google Scholar] [CrossRef]
- Brodehl, A.; Weiss, J.; Debus, J.D.; Stanasiuk, C.; Klauke, B.; Deutsch, M.A.; Fox, H.; Bax, J.; Ebbinghaus, H.; Gärtner, A.; et al. A homozygous DSC2 deletion associated with arrhythmogenic cardiomyopathy is caused by uniparental isodisomy. J. Mol. Cell. Cardiol. 2020, 141, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Heuser, A.; Plovie, E.R.; Ellinor, P.T.; Grossmann, K.S.; Shin, J.T.; Wichter, T.; Basson, C.T.; Lerman, B.B.; Sasse-Klaassen, S.; Thierfelder, L.; et al. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2006, 79, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padrón-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef] [PubMed]
- van Hengel, J.; Calore, M.; Bauce, B.; Dazzo, E.; Mazzotti, E.; De Bortoli, M.; Lorenzon, A.; Li Mura, I.E.; Beffagna, G.; Rigato, I.; et al. Mutations in the area composita protein αT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 201–210. [Google Scholar] [CrossRef]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.; Connors, S.; French, V.M.; Drenckhahn, J.D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef]
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right ventricular cardiomyopathy and sudden death in young people. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef]
- Perlman, R.L. Mouse models of human disease: An evolutionary perspective. Evol. Med. Public Health 2016, 2016, 170–176. [Google Scholar] [CrossRef]
- Jain, A.; Norton, N.; Bruno, K.A.; Cooper, L.T., Jr.; Atwal, P.S.; Fairweather, D. Sex Differences, Genetic and Environmental Influences on Dilated Cardiomyopathy. J. Clin. Med. 2021, 10, 2289. [Google Scholar] [CrossRef]
- Hartiala, J.A.; Hilser, J.R.; Biswas, S.; Lusis, A.J.; Allayee, H. Gene-Environment Interactions for Cardiovascular Disease. Curr. Atheroscler. Rep. 2021, 23, 75. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Bedada, F.B.; Wheelwright, M.; Metzger, J.M. Maturation status of sarcomere structure and function in human iPSC-derived cardiac myocytes. Biochim. Biophys. Acta 2016, 1863, 1829–1838. [Google Scholar] [CrossRef]
- Clancy, C.E.; Santana, L.F. Advances in induced pluripotent stem cell-derived cardiac myocytes: Technological breakthroughs, key discoveries and new applications. J. Physiol. 2024, 602, 3871–3892. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Li, Y.; Tchao, J.; Kaplan, A.D.; Lin, B.; Li, Y.; Mich-Basso, J.; Lis, A.; Hassan, N.; London, B.; et al. Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc. Res. 2014, 104, 258–269. [Google Scholar] [CrossRef]
- Wang, L.; Kim, K.; Parikh, S.; Cadar, A.G.; Bersell, K.R.; He, H.; Pinto, J.R.; Kryshtal, D.O.; Knollmann, B.C. Hypertrophic cardiomyopathy-linked mutation in troponin T causes myofibrillar disarray and pro-arrhythmic action potential changes in human iPSC cardiomyocytes. J. Mol. Cell. Cardiol. 2018, 114, 320–327. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Lowry, W.E.; Richter, L.; Yachechko, R.; Pyle, A.D.; Tchieu, J.; Sridharan, R.; Clark, A.T.; Plath, K. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc. Natl. Acad. Sci. USA 2008, 105, 2883–2888. [Google Scholar] [CrossRef]
- Seki, T.; Yuasa, S.; Oda, M.; Egashira, T.; Yae, K.; Kusumoto, D.; Nakata, H.; Tohyama, S.; Hashimoto, H.; Kodaira, M.; et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell 2010, 7, 11–14. [Google Scholar] [CrossRef]
- Seki, T.; Yuasa, S.; Fukuda, K. Generation of induced pluripotent stem cells from a small amount of human peripheral blood using a combination of activated T cells and Sendai virus. Nat. Protoc. 2012, 7, 718–728. [Google Scholar] [CrossRef]
- Vlahos, K.; Sourris, K.; Mayberry, R.; McDonald, P.; Bruveris, F.F.; Schiesser, J.V.; Bozaoglu, K.; Lockhart, P.J.; Stanley, E.G.; Elefanty, A.G. Generation of iPSC lines from peripheral blood mononuclear cells from 5 healthy adults. Stem Cell Res. 2019, 34, 101380. [Google Scholar] [CrossRef]
- Zhou, T.; Benda, C.; Dunzinger, S.; Huang, Y.; Ho, J.C.; Yang, J.; Wang, Y.; Zhang, Y.; Zhuang, Q.; Li, Y.; et al. Generation of human induced pluripotent stem cells from urine samples. Nat. Protoc. 2012, 7, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Afzal, M.Z.; Gartz, M.; Klyachko, E.A.; Khan, S.S.; Shah, S.J.; Gupta, S.; Shapiro, A.D.; Vaughan, D.E.; Strande, J.L. Generation of human iPSCs from urine derived cells of a non-affected control subject. Stem Cell Res. 2017, 18, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Wu, W.S. Gene-delivery systems for iPS cell generation. Expert Opin. Biol. Ther. 2010, 10, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2015, 33, 58–63. [Google Scholar] [CrossRef]
- Pozner, T.; Grandizio, C.; Mitchell, M.W.; Turan, N.; Scheinfeldt, L. Human iPSC Reprogramming Success: The Impact of Approaches and Source Materials. Stem Cells Int. 2025, 2025, 2223645. [Google Scholar] [CrossRef]
- Sullivan, S.; Stacey, G.N.; Akazawa, C.; Aoyama, N.; Baptista, R.; Bedford, P.; Bennaceur Griscelli, A.; Chandra, A.; Elwood, N.; Girard, M.; et al. Quality control guidelines for clinical-grade human induced pluripotent stem cell lines. Regen. Med. 2018, 13, 859–866. [Google Scholar] [CrossRef]
- Novoa, J.; Westra, I.; Steeneveld, E.; Neves, N.F.; Daleman, L.; Asensio, A.B.; Davis, R.P.; Carlotti, F.; Freund, C.; Rabelink, T.; et al. Validating human induced pluripotent stem cell-specific quality control tests for the release of an intermediate drug product in a Good Manufacturing Practice quality system. Cytotherapy 2024, 26, 1105–1117. [Google Scholar] [CrossRef]
- Teo, A.K.; Ali, Y.; Wong, K.Y.; Chipperfield, H.; Sadasivam, A.; Poobalan, Y.; Tan, E.K.; Wang, S.T.; Abraham, S.; Tsuneyoshi, N.; et al. Activin and BMP4 synergistically promote formation of definitive endoderm in human embryonic stem cells. Stem Cells 2012, 30, 631–642. [Google Scholar] [CrossRef]
- Kattman, S.J.; Witty, A.D.; Gagliardi, M.; Dubois, N.C.; Niapour, M.; Hotta, A.; Ellis, J.; Keller, G. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 2011, 8, 228–240. [Google Scholar] [CrossRef]
- Kadari, A.; Mekala, S.; Wagner, N.; Malan, D.; Köth, J.; Doll, K.; Stappert, L.; Eckert, D.; Peitz, M.; Matthes, J.; et al. Robust Generation of Cardiomyocytes from Human iPS Cells Requires Precise Modulation of BMP and WNT Signaling. Stem Cell Rev. Rep. 2015, 11, 560–569. [Google Scholar] [CrossRef]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Buikema, J.W.; Lee, S.; Goodyer, W.R.; Maas, R.G.; Chirikian, O.; Li, G.; Miao, Y.; Paige, S.L.; Lee, D.; Wu, H.; et al. Wnt Activation and Reduced Cell-Cell Contact Synergistically Induce Massive Expansion of Functional Human iPSC-Derived Cardiomyocytes. Cell Stem Cell 2020, 27, 50–63.e55. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Holmström, A.; Wu, J.C. Chemically Defined Culture and Cardiomyocyte Differentiation of Human Pluripotent Stem Cells. Curr. Protoc. Hum. Genet. 2015, 87, 21.3.1–21.3.15. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zou, J. Differentiation of Cardiomyocytes from Human Pluripotent Stem Cells in Fully Chemically Defined Conditions. STAR Protoc. 2020, 1, 100015. [Google Scholar] [CrossRef]
- Roshanravan, N.; Ghaffari, S.; Bastani, S.; Pahlavan, S.; Asghari, S.; Doustvandi, M.A.; Jalilzadeh-Razin, S.; Dastouri, M. Human cardiac organoids: A recent revolution in disease modeling and regenerative medicine. J. Cardiovasc. Thorac. Res. 2023, 15, 68–72. [Google Scholar] [CrossRef]
- Kurokawa, Y.K.; George, S.C. Tissue engineering the cardiac microenvironment: Multicellular microphysiological systems for drug screening. Adv. Drug Deliv. Rev. 2016, 96, 225–233. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef]
- Tohyama, S.; Hattori, F.; Sano, M.; Hishiki, T.; Nagahata, Y.; Matsuura, T.; Hashimoto, H.; Suzuki, T.; Yamashita, H.; Satoh, Y.; et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 2013, 12, 127–137. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Burridge, P.W.; Kropp, E.M.; Chuppa, S.L.; Kwok, W.M.; Wu, J.C.; Boheler, K.R.; Gundry, R.L. High efficiency differentiation of human pluripotent stem cells to cardiomyocytes and characterization by flow cytometry. J. Vis. Exp. 2014, 91, 52010. [Google Scholar] [CrossRef]
- Wolnik, J.; Adamska, P.; Oleksy, A.; Dulak, J.; Biniecka, M. Enriching Cardiomyocytes Derived from hiPSCs by Magnetic-Activated Cell Sorting (MACS). Methods Mol. Biol. 2024, 2835, 83–98. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, R.; Huang, W.; Wang, J.; Jiang, C.; Li, A.; Huang, C.L.H.; Zhang, Y. Gene Expression, Morphology, and Electrophysiology During the Dynamic Development of Human Induced Pluripotent Stem Cell-Derived Atrial- and Ventricular-Like Cardiomyocytes. Biologics 2024, 18, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Lundy, S.D.; Zhu, W.Z.; Regnier, M.; Laflamme, M.A. Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev. 2013, 22, 1991–2002. [Google Scholar] [CrossRef]
- Gherghiceanu, M.; Barad, L.; Novak, A.; Reiter, I.; Itskovitz-Eldor, J.; Binah, O.; Popescu, L.M. Cardiomyocytes derived from human embryonic and induced pluripotent stem cells: Comparative ultrastructure. J. Cell. Mol. Med. 2011, 15, 2539–2551. [Google Scholar] [CrossRef]
- Hatani, T.; Funakoshi, S.; Deerinck, T.J.; Bushong, E.A.; Kimura, T.; Takeshima, H.; Ellisman, M.H.; Hoshijima, M.; Yoshida, Y. Nano-structural analysis of engrafted human induced pluripotent stem cell-derived cardiomyocytes in mouse hearts using a genetic-probe APEX2. Biochem. Biophys. Res. Commun. 2018, 505, 1251–1256. [Google Scholar] [CrossRef]
- Yoshinaga, D.; Wuriyanghai, Y.; Makiyama, T. Multielectrode Array Assays Using Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Methods Mol. Biol. 2021, 2320, 111–119. [Google Scholar] [CrossRef]
- Astro, V.; Ramirez-Calderon, G.; Adamo, A. Protocol to measure calcium spikes in cardiomyocytes obtained from human pluripotent stem cells using a ready-to-use media. STAR Protoc. 2023, 4, 102252. [Google Scholar] [CrossRef]
- Koivumäki, J.T.; Naumenko, N.; Tuomainen, T.; Takalo, J.; Oksanen, M.; Puttonen, K.A.; Lehtonen, Š.; Kuusisto, J.; Laakso, M.; Koistinaho, J.; et al. Structural Immaturity of Human iPSC-Derived Cardiomyocytes: In Silico Investigation of Effects on Function and Disease Modeling. Front. Physiol. 2018, 9, 80. [Google Scholar] [CrossRef]
- Goversen, B.; van der Heyden, M.A.G.; van Veen, T.A.B.; de Boer, T.P. The immature electrophysiological phenotype of iPSC-CMs still hampers in vitro drug screening: Special focus on IK1. Pharmacol. Ther. 2018, 183, 127–136. [Google Scholar] [CrossRef]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flügel, L.; Dorn, T.; Goedel, A.; Höhnke, C.; Hofmann, F.; et al. Patient-Specific Induced Pluripotent Stem-Cell Models for Long-QT Syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef]
- Garfinkel, A.C.; Seidman, J.G.; Seidman, C.E. Genetic Pathogenesis of Hypertrophic and Dilated Cardiomyopathy. Heart Fail. Clin. 2018, 14, 139–146. [Google Scholar] [CrossRef]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef] [PubMed]
- Duncker, D.J.; Bakkers, J.; Brundel, B.J.; Robbins, J.; Tardiff, J.C.; Carrier, L. Animal and in silico models for the study of sarcomeric cardiomyopathies. Cardiovasc. Res. 2015, 105, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Lee, A.S.; Liang, P.; Sanchez-Freire, V.; Nguyen, P.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Sun, N.; et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 2013, 12, 101–113. [Google Scholar] [CrossRef]
- Bhagwan, J.R.; Mosqueira, D.; Chairez-Cantu, K.; Mannhardt, I.; Bodbin, S.E.; Bakar, M.; Smith, J.G.W.; Denning, C. Isogenic models of hypertrophic cardiomyopathy unveil differential phenotypes and mechanism-driven therapeutics. J. Mol. Cell. Cardiol. 2020, 145, 43–53. [Google Scholar] [CrossRef]
- Dainis, A.; Zaleta-Rivera, K.; Ribeiro, A.; Chang, A.C.H.; Shang, C.; Lan, F.; Burridge, P.W.; Liu, W.R.; Wu, J.C.; Chang, A.C.Y.; et al. Silencing of MYH7 ameliorates disease phenotypes in human iPSC-cardiomyocytes. Physiol. Genom. 2020, 52, 293–303. [Google Scholar] [CrossRef]
- Shemer, Y.; Mekies, L.N.; Ben Jehuda, R.; Baskin, P.; Shulman, R.; Eisen, B.; Regev, D.; Arbustini, E.; Gerull, B.; Gherghiceanu, M.; et al. Investigating LMNA-Related Dilated Cardiomyopathy Using Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Int. J. Mol. Sci. 2021, 22, 7874. [Google Scholar] [CrossRef]
- Korover, N.; Etzion, S.; Cherniak, A.; Rabinski, T.; Levitas, A.; Etzion, Y.; Ofir, R.; Parvari, R.; Cohen, S. Functional defects in hiPSCs-derived cardiomyocytes from patients with a PLEKHM2-mutation associated with dilated cardiomyopathy and left ventricular non-compaction. Biol. Res. 2023, 56, 34. [Google Scholar] [CrossRef]
- Schick, R.; Mekies, L.N.; Shemer, Y.; Eisen, B.; Hallas, T.; Ben Jehuda, R.; Ben-Ari, M.; Szantai, A.; Willi, L.; Shulman, R.; et al. Functional abnormalities in induced Pluripotent Stem Cell-derived cardiomyocytes generated from titin-mutated patients with dilated cardiomyopathy. PLoS ONE 2018, 13, e0205719. [Google Scholar] [CrossRef]
- Mosqueira, D.; Mannhardt, I.; Bhagwan, J.R.; Lis-Slimak, K.; Katili, P.; Scott, E.; Hassan, M.; Prondzynski, M.; Harmer, S.C.; Tinker, A.; et al. CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur. Heart J. 2018, 39, 3879–3892. [Google Scholar] [CrossRef]
- Seeger, T.; Shrestha, R.; Lam, C.K.; Chen, C.; McKeithan, W.L.; Lau, E.; Wnorowski, A.; McMullen, G.; Greenhaw, M.; Lee, J.; et al. A Premature Termination Codon Mutation in MYBPC3 Causes Hypertrophic Cardiomyopathy via Chronic Activation of Nonsense-Mediated Decay. Circulation 2019, 139, 799–811. [Google Scholar] [CrossRef]
- Toepfer, C.N.; Garfinkel, A.C.; Venturini, G.; Wakimoto, H.; Repetti, G.; Alamo, L.; Sharma, A.; Agarwal, R.; Ewoldt, J.K.; Cloonan, P.; et al. Myosin Sequestration Regulates Sarcomere Function, Cardiomyocyte Energetics, and Metabolism, Informing the Pathogenesis of Hypertrophic Cardiomyopathy. Circulation 2020, 141, 828–842. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Yuasa, S.; Mearini, G.; Egashira, T.; Seki, T.; Kodaira, M.; Kusumoto, D.; Kuroda, Y.; Okata, S.; Suzuki, T.; et al. Endothelin-1 induces myofibrillar disarray and contractile vector variability in hypertrophic cardiomyopathy-induced pluripotent stem cell-derived cardiomyocytes. J. Am. Heart Assoc. 2014, 3, e001263. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yazawa, M.; Liu, J.; Han, L.; Sanchez-Freire, V.; Abilez, O.J.; Navarrete, E.G.; Hu, S.; Wang, L.; Lee, A.; et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 2012, 4, 130ra47. [Google Scholar] [CrossRef]
- Tse, H.F.; Ho, J.C.; Choi, S.W.; Lee, Y.K.; Butler, A.W.; Ng, K.M.; Siu, C.W.; Simpson, M.A.; Lai, W.H.; Chan, Y.C.; et al. Patient-specific induced-pluripotent stem cells-derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Hum. Mol. Genet. 2013, 22, 1395–1403. [Google Scholar] [CrossRef]
- Dai, Y.; Amenov, A.; Ignatyeva, N.; Koschinski, A.; Xu, H.; Soong, P.L.; Tiburcy, M.; Linke, W.A.; Zaccolo, M.; Hasenfuss, G.; et al. Troponin destabilization impairs sarcomere-cytoskeleton interactions in iPSC-derived cardiomyocytes from dilated cardiomyopathy patients. Sci. Rep. 2020, 10, 209. [Google Scholar] [CrossRef]
- Lee, Y.K.; Lau, Y.M.; Cai, Z.J.; Lai, W.H.; Wong, L.Y.; Tse, H.F.; Ng, K.M.; Siu, C.W. Modeling Treatment Response for Lamin A/C Related Dilated Cardiomyopathy in Human Induced Pluripotent Stem Cells. J. Am. Heart Assoc. 2017, 6, e005677. [Google Scholar] [CrossRef]
- Lee, J.; Termglinchan, V.; Diecke, S.; Itzhaki, I.; Lam, C.K.; Garg, P.; Lau, E.; Greenhaw, M.; Seeger, T.; Wu, H.; et al. Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 2019, 572, 335–340. [Google Scholar] [CrossRef]
- Takasaki, A.; Hirono, K.; Hata, Y.; Wang, C.; Takeda, M.; Yamashita, J.K.; Chang, B.; Nakaoka, H.; Okabe, M.; Miyao, N.; et al. Sarcomere gene variants act as a genetic trigger underlying the development of left ventricular noncompaction. Pediatr. Res. 2018, 84, 733–742. [Google Scholar] [CrossRef]
- Kodo, K.; Ong, S.G.; Jahanbani, F.; Termglinchan, V.; Hirono, K.; InanlooRahatloo, K.; Ebert, A.D.; Shukla, P.; Abilez, O.J.; Churko, J.M.; et al. iPSC-derived cardiomyocytes reveal abnormal TGF-β signalling in left ventricular non-compaction cardiomyopathy. Nat. Cell Biol. 2016, 18, 1031–1042. [Google Scholar] [CrossRef]
- Ebrahim, M.A.; Ali, N.M.; Albash, B.Y.; Al Sayegh, A.H.; Ahmad, N.B.; Voß, S.; Klag, F.; Groß, J.; Holler, S.; Walhorn, V.; et al. Phenotypic Diversity Caused by the DES Missense Mutation p.R127P (c.380G>C) Contributing to Significant Cardiac Mortality and Morbidity Associated With a Desmin Filament Assembly Defect. Circ. Genom. Precis. Med. 2025, e004896. [Google Scholar] [CrossRef]
- Hovhannisyan, Y.; Li, Z.; Callon, D.; Suspène, R.; Batoumeni, V.; Canette, A.; Blanc, J.; Hocini, H.; Lefebvre, C.; El-Jahrani, N.; et al. Critical contribution of mitochondria in the development of cardiomyopathy linked to desmin mutation. Stem Cell Res. Ther. 2024, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Kulikova, O.; Brodehl, A.; Kiseleva, A.; Myasnikov, R.; Meshkov, A.; Stanasiuk, C.; Gärtner, A.; Divashuk, M.; Sotnikova, E.; Koretskiy, S.; et al. The Desmin (DES) Mutation p.A337P Is Associated with Left-Ventricular Non-Compaction Cardiomyopathy. Genes 2021, 12, 121. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Wei, H.; Lu, J.; Ho, S.; Zhang, G.; Sun, X.; Oh, Y.; Tan, S.H.; Ng, M.L.; Shim, W.; et al. Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Caspi, O.; Huber, I.; Gepstein, A.; Arbel, G.; Maizels, L.; Boulos, M.; Gepstein, L. Modeling of arrhythmogenic right ventricular cardiomyopathy with human induced pluripotent stem cells. Circ. Cardiovasc. Genet. 2013, 6, 557–568. [Google Scholar] [CrossRef]
- Wen, J.Y.; Wei, C.Y.; Shah, K.; Wong, J.; Wang, C.; Chen, H.S. Maturation-Based Model of Arrhythmogenic Right Ventricular Dysplasia Using Patient-Specific Induced Pluripotent Stem Cells. Circ. J. 2015, 79, 1402–1408. [Google Scholar] [CrossRef]
- Dorn, T.; Kornherr, J.; Parrotta, E.I.; Zawada, D.; Ayetey, H.; Santamaria, G.; Iop, L.; Mastantuono, E.; Sinnecker, D.; Goedel, A.; et al. Interplay of cell-cell contacts and RhoA/MRTF-A signaling regulates cardiomyocyte identity. EMBO J. 2018, 37, e98133. [Google Scholar] [CrossRef]
- Chen, P.; Xiao, Y.; Wang, Y.; Zheng, Z.; Chen, L.; Yang, X.; Li, J.; Wu, W.; Zhang, S. Intracellular calcium current disorder and disease phenotype in OBSCN mutant iPSC-based cardiomyocytes in arrhythmogenic right ventricular cardiomyopathy. Theranostics 2020, 10, 11215–11229. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Cyganek, L.; Tombers, C.; Li, X.; Buljubasic, F.; Lang, S.; Tiburcy, M.; Zimmermann, W.H.; et al. Electrical dysfunctions in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with an arrhythmogenic right ventricular cardiomyopathy. Europace 2018, 20, f46–f56. [Google Scholar] [CrossRef]
- Ng, R.; Manring, H.; Papoutsidakis, N.; Albertelli, T.; Tsai, N.; See, C.J.; Li, X.; Park, J.; Stevens, T.L.; Bobbili, P.J.; et al. Patient mutations linked to arrhythmogenic cardiomyopathy enhance calpain-mediated desmoplakin degradation. JCI Insight 2019, 5, e128643. [Google Scholar] [CrossRef]
- Ng, K.M.; Mok, P.Y.; Butler, A.W.; Ho, J.C.; Choi, S.W.; Lee, Y.K.; Lai, W.H.; Au, K.W.; Lau, Y.M.; Wong, L.Y.; et al. Amelioration of X-Linked Related Autophagy Failure in Danon Disease With DNA Methylation Inhibitor. Circulation 2016, 134, 1373–1389. [Google Scholar] [CrossRef]
- Gao, L.; Lu, Z.; Zhang, Y.; Liu, L.; Sun, J.; Fu, H.; Mao, J.; Hu, L. Clinical characteristics and induced pluripotent stem cells (iPSCs) disease model of Fabry disease caused by a novel GLA mutation. QJM Int. J. Med. 2024, 117, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Raval, K.K.; Tao, R.; White, B.E.; De Lange, W.J.; Koonce, C.H.; Yu, J.; Kishnani, P.S.; Thomson, J.A.; Mosher, D.F.; Ralphe, J.C.; et al. Pompe disease results in a Golgi-based glycosylation deficit in human induced pluripotent stem cell-derived cardiomyocytes. J. Biol. Chem. 2015, 290, 3121–3136. [Google Scholar] [CrossRef]
- Bonilauri, B.; Shin, H.S.; Htet, M.; Yan, C.D.; Witteles, R.M.; Sallam, K.; Wu, J.C. Generation of two induced pluripotent stem cell lines from patients with cardiac amyloidosis carrying heterozygous transthyretin (TTR) mutation. Stem Cell Res. 2023, 72, 103215. [Google Scholar] [CrossRef]
- Montero-Calle, P.; Flandes-Iparraguirre, M.; Kuebler, B.; Arán, B.; Larequi, E.; Anaut, I.; Coppiello, G.; Aranguren, X.L.; Veiga, A.; Elorz, M.T.B.; et al. Generation of an induced pluripotent stem cell line (ESi107-A) from a transthyretin amyloid cardiomyopathy (ATTR-CM) patient carrying a p.Ser43Asn mutation in the TTR gene. Stem Cell Res. 2023, 71, 103189. [Google Scholar] [CrossRef]
- Haupt, L.P.; Rebs, S.; Maurer, W.; Hübscher, D.; Tiburcy, M.; Pabel, S.; Maus, A.; Köhne, S.; Tappu, R.; Haas, J.; et al. Doxorubicin induces cardiotoxicity in a pluripotent stem cell model of aggressive B cell lymphoma cancer patients. Basic. Res. Cardiol. 2022, 117, 13. [Google Scholar] [CrossRef]
- Rampoldi, A.; Singh, M.; Wu, Q.; Duan, M.; Jha, R.; Maxwell, J.T.; Bradner, J.M.; Zhang, X.; Saraf, A.; Miller, G.W.; et al. Cardiac Toxicity From Ethanol Exposure in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Toxicol. Sci. 2019, 169, 280–292. [Google Scholar] [CrossRef]
- Liu, R.; Sun, F.; Armand, L.C.; Wu, R.; Xu, C. Chronic Ethanol Exposure Induces Deleterious Changes in Cardiomyocytes Derived from Human Induced Pluripotent Stem Cells. Stem Cell Rev. Rep. 2021, 17, 2314–2331. [Google Scholar] [CrossRef]
- Gintant, G.; Burridge, P.; Gepstein, L.; Harding, S.; Herron, T.; Hong, C.; Jalife, J.; Wu, J.C. Use of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes in Preclinical Cancer Drug Cardiotoxicity Testing: A Scientific Statement From the American Heart Association. Circ. Res. 2019, 125, e75–e92. [Google Scholar] [CrossRef]
- van Doorn, E.C.H.; Amesz, J.H.; Sadeghi, A.H.; de Groot, N.M.S.; Manintveld, O.C.; Taverne, Y.J.H.J. Preclinical Models of Cardiac Disease: A Comprehensive Overview for Clinical Scientists. Cardiovasc. Eng. Technol. 2024, 15, 232–249. [Google Scholar] [CrossRef]
- Burnett, S.D.; Blanchette, A.D.; Chiu, W.A.; Rusyn, I. Human induced pluripotent stem cell (iPSC)-derived cardiomyocytes as an in vitro model in toxicology: Strengths and weaknesses for hazard identification and risk characterization. Expert Opin. Drug Metab. Toxicol. 2021, 17, 887–902. [Google Scholar] [CrossRef]
- US Food and Drug Administration (FDA). Available online: https://www.fda.gov/drugs/regulatory-science-action/impact-story-improved-assessment-cardiotoxic-risk-drug-candidates-comprehensive-in-vitro-proarrhythmia (accessed on 18 May 2025).
- Pan, D.; Li, B.; Wang, S. Establishment and validation of a torsade de pointes prediction model based on human iPSC—Derived cardiomyocytes. Exp. Ther. Med. 2023, 25, 61. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, D.; Baba, S.; Makiyama, T.; Shibata, H.; Hirata, T.; Akagi, K.; Matsuda, K.; Kohjitani, H.; Wuriyanghai, Y.; Umeda, K.; et al. Phenotype-Based High-Throughput Classification of Long QT Syndrome Subtypes Using Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2019, 13, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Goetz, L.H.; Schork, N.J. Personalized medicine: Motivation, challenges, and progress. Fertil. Steril. 2018, 109, 952–963. [Google Scholar] [CrossRef]
- Oh, J.; Kwon, O.B.; Park, S.W.; Kim, J.W.; Lee, H.; Kim, Y.K.; Choi, E.J.; Jung, H.; Choi, D.K.; Oh, B.J.; et al. Advancing Cardiovascular Drug Screening Using Human Pluripotent Stem Cell-Derived Cardiomyocytes. Int. J. Mol. Sci. 2024, 25, 7971. [Google Scholar] [CrossRef]
- Wu, X.; Swanson, K.; Yildirim, Z.; Liu, W.; Liao, R.; Wu, J.C. Clinical trials in-a-dish for cardiovascular medicine. Eur. Heart J. 2024, 45, 4275–4290. [Google Scholar] [CrossRef]
- Ferdinandy, P.; Baczkó, I.; Bencsik, P.; Giricz, Z.; Görbe, A.; Pacher, P.; Varga, Z.V.; Varró, A.; Schulz, R. Definition of hidden drug cardiotoxicity: Paradigm change in cardiac safety testing and its clinical implications. Eur. Heart J. 2019, 40, 1771–1777. [Google Scholar] [CrossRef]
- Lee, T.Y.T.; Coles, J.G.; Maynes, J.T. iPSC-cardiomyocytes in the preclinical prediction of candidate pharmaceutical toxicity. Front. Pharmacol. 2024, 15, 1308217. [Google Scholar] [CrossRef]
- Sharma, A.; Burridge, P.W.; McKeithan, W.L.; Serrano, R.; Shukla, P.; Sayed, N.; Churko, J.M.; Kitani, T.; Wu, H.; Holmström, A.; et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci. Transl. Med. 2017, 9, eaaf2584. [Google Scholar] [CrossRef]
- Grafton, F.; Ho, J.; Ranjbarvaziri, S.; Farshidfar, F.; Budan, A.; Steltzer, S.; Maddah, M.; Loewke, K.E.; Green, K.; Patel, S.; et al. Deep learning detects cardiotoxicity in a high-content screen with induced pluripotent stem cell-derived cardiomyocytes. eLife 2021, 10, e68714. [Google Scholar] [CrossRef]
- Pölönen, R.P.; Swan, H.; Aalto-Setälä, K. Mutation-specific differences in arrhythmias and drug responses in CPVT patients: Simultaneous patch clamp and video imaging of iPSC derived cardiomyocytes. Mol. Biol. Rep. 2020, 47, 1067–1077. [Google Scholar] [CrossRef]
- Kinnear, C.; Said, A.; Meng, G.; Zhao, Y.; Wang, E.Y.; Rafatian, N.; Parmar, N.; Wei, W.; Billia, F.; Simmons, C.A.; et al. Myosin inhibitor reverses hypertrophic cardiomyopathy in genotypically diverse pediatric iPSC-cardiomyocytes to mirror variant correction. Cell Rep. Med. 2024, 5, 101520. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Deschenes, I.; Zhao, M.T. Precision medicine for long QT syndrome: Patient-specific iPSCs take the lead. Expert Rev. Mol. Med. 2023, 25, e5. [Google Scholar] [CrossRef] [PubMed]
- Oikonomopoulos, A.; Kitani, T.; Wu, J.C. Pluripotent Stem Cell-Derived Cardiomyocytes as a Platform for Cell Therapy Applications: Progress and Hurdles for Clinical Translation. Mol. Ther. 2018, 26, 1624–1634. [Google Scholar] [CrossRef]
- Hinson, J.T.; Chopra, A.; Nafissi, N.; Polacheck, W.J.; Benson, C.C.; Swist, S.; Gorham, J.; Yang, L.; Schafer, S.; Sheng, C.C.; et al. HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015, 349, 982–986. [Google Scholar] [CrossRef]
- McDermott-Roe, C.; Lv, W.; Maximova, T.; Wada, S.; Bukowy, J.; Marquez, M.; Lai, S.; Shehu, A.; Benjamin, I.; Geurts, A.; et al. Investigation of a dilated cardiomyopathy-associated variant in BAG3 using genome-edited iPSC-derived cardiomyocytes. JCI Insight 2019, 4, e128799. [Google Scholar] [CrossRef]
- Te Riele, A.S.; Agullo-Pascual, E.; James, C.A.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Sobreira, N.L.; Amat-Alarcon, N.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef]
- Jaffré, F.; Miller, C.L.; Schänzer, A.; Evans, T.; Roberts, A.E.; Hahn, A.; Kontaridis, M.I. Inducible Pluripotent Stem Cell-Derived Cardiomyocytes Reveal Aberrant Extracellular Regulated Kinase 5 and Mitogen-Activated Protein Kinase Kinase 1/2 Signaling Concomitantly Promote Hypertrophic Cardiomyopathy in RAF1-Associated Noonan Syndrome. Circulation 2019, 140, 207–224. [Google Scholar] [CrossRef]
- Chai, A.C.; Cui, M.; Chemello, F.; Li, H.; Chen, K.; Tan, W.; Atmanli, A.; McAnally, J.R.; Zhang, Y.; Xu, L.; et al. Base editing correction of hypertrophic cardiomyopathy in human cardiomyocytes and humanized mice. Nat. Med. 2023, 29, 401–411. [Google Scholar] [CrossRef]
- Nishiyama, T.; Zhang, Y.; Cui, M.; Li, H.; Sanchez-Ortiz, E.; McAnally, J.R.; Tan, W.; Kim, J.; Chen, K.; Xu, L.; et al. Precise genomic editing of pathogenic mutations in RBM20 rescues dilated cardiomyopathy. Sci. Transl. Med. 2022, 14, eade1633. [Google Scholar] [CrossRef]
- Yang, L.; Liu, Z.; Sun, J.; Chen, Z.; Gao, F.; Guo, Y. Adenine base editor-based correction of the cardiac pathogenic Lmna c.1621C > T mutation in murine hearts. J. Cell. Mol. Med. 2024, 28, e18145. [Google Scholar] [CrossRef]
- Ma, S.; Jiang, W.; Liu, X.; Lu, W.J.; Qi, T.; Wei, J.; Wu, F.; Chang, Y.; Zhang, S.; Song, Y.; et al. Efficient Correction of a Hypertrophic Cardiomyopathy Mutation by ABEmax-NG. Circ. Res. 2021, 129, 895–908. [Google Scholar] [CrossRef]
- Pan, X.; Philippen, L.; Lahiri, S.K.; Lee, C.; Park, S.H.; Word, T.A.; Li, N.; Jarrett, K.E.; Gupta, R.; Reynolds, J.O.; et al. In Vivo Ryr2 Editing Corrects Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Res. 2018, 123, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, H.; Budan, A.; Zhang, L.; Majetic, M.; Shenwai, R.; Levinson, A.; Cisne-Thompson, O.; Farshidfar, F.; Tsui, J.; Figarska, S.; et al. Reduced Expression of MTSS1 Increases Sarcomere Number and Improves Contractility in Select Forms of Monogenic DCM. medRxiv 2024, 24311020. Available online: https://www.medrxiv.org/content/10.1101/2024.08.14.24311020v1 (accessed on 18 May 2025).
- Greer-Short, A.; Greenwood, A.; Leon, E.C.; Qureshi, T.N.; von Kraut, K.; Wong, J.; Tsui, J.H.; Reid, C.A.; Cheng, Z.; Easter, E.; et al. AAV9-mediated MYBPC3 gene therapy with optimized expression cassette enhances cardiac function and survival in MYBPC3 cardiomyopathy models. Nat. Commun. 2025, 16, 2196. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.-L.; Li, H.; Rodriguez-Caycedo, C.; Mireault, A.A.; Huang, J.; Shelton, J.M.; McAnally, J.R.; Amoasii, L.; Mammen, P.P.A.; Bassel-Duby, R.; et al. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 2019, 5, eaav4324. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, H.; Zhu, M.; Han, R.Y.; Guo, S.; Han, R. Correction of DMD in human iPSC-derived cardiomyocytes by base-editing-induced exon skipping. Mol. Ther. Methods Clin. Dev. 2023, 28, 40–50. [Google Scholar] [CrossRef]
- Brooks, I.R.; Garrone, C.M.; Kerins, C.; Kiar, C.S.; Syntaka, S.; Xu, J.Z.; Spagnoli, F.M.; Watt, F.M. Functional genomics and the future of iPSCs in disease modeling. Stem Cell Rep. 2022, 17, 1033–1047. [Google Scholar] [CrossRef]
- Su, J.; Song, Y.; Zhu, Z.; Huang, X.; Fan, J.; Qiao, J.; Mao, F. Cell–cell communication: New insights and clinical implications. Signal Transduct. Target. Ther. 2024, 9, 196. [Google Scholar] [CrossRef]
- Denning, C.; Borgdorff, V.; Crutchley, J.; Firth, K.S.; George, V.; Kalra, S.; Kondrashov, A.; Hoang, M.D.; Mosqueira, D.; Patel, A.; et al. Cardiomyocytes from human pluripotent stem cells: From laboratory curiosity to industrial biomedical platform. Biochim. Biophys. Acta 2016, 1863, 1728–1748. [Google Scholar] [CrossRef]
- Ivashchenko, C.Y.; Pipes, G.C.; Lozinskaya, I.M.; Lin, Z.; Xiaoping, X.; Needle, S.; Grygielko, E.T.; Hu, E.; Toomey, J.R.; Lepore, J.J.; et al. Human-induced pluripotent stem cell-derived cardiomyocytes exhibit temporal changes in phenotype. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H913–H922. [Google Scholar] [CrossRef]
- Pesl, M.; Pribyl, J.; Caluori, G.; Cmiel, V.; Acimovic, I.; Jelinkova, S.; Dvorak, P.; Starek, Z.; Skladal, P.; Rotrekl, V. Phenotypic assays for analyses of pluripotent stem cell-derived cardiomyocytes. J. Mol. Recognit. 2017, 30, e2602. [Google Scholar] [CrossRef]
- Chen, H.S.; Kim, C.; Mercola, M. Electrophysiological challenges of cell-based myocardial repair. Circulation 2009, 120, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Vitale, A.M.; Matigian, N.A.; Ravishankar, S.; Bellette, B.; Wood, S.A.; Wolvetang, E.J.; Mackay-Sim, A. Variability in the generation of induced pluripotent stem cells: Importance for disease modeling. Stem Cells Transl. Med. 2012, 1, 641–650. [Google Scholar] [CrossRef]
- Scesa, G.; Adami, R.; Bottai, D. iPSC Preparation and Epigenetic Memory: Does the Tissue Origin Matter? Cells 2021, 10, 1470. [Google Scholar] [CrossRef]
- Yoshihara, M.; Hayashizaki, Y.; Murakawa, Y. Genomic Instability of iPSCs: Challenges Towards Their Clinical Applications. Stem Cell Rev. Rep. 2017, 13, 7–16. [Google Scholar] [CrossRef]
- Liu, C.; Feng, X.; Li, G.; Gokulnath, P.; Xiao, J. Generating 3D human cardiac constructs from pluripotent stem cells. eBioMedicine 2022, 76, 103813. [Google Scholar] [CrossRef]
- Jin, L.; Hwang, B.; Rezapourdamanab, S.; Sridhar, V.; Nandwani, R.; Amoli, M.S.; Serpooshan, V. Bioengineering Approaches to In Vitro Modeling of Genetic and Acquired Cardiac Diseases. Curr. Cardiol. Rep. 2025, 27, 72. [Google Scholar] [CrossRef]
- Omole, A.E.; Fakoya, A.O.J. Ten years of progress and promise of induced pluripotent stem cells: Historical origins, characteristics, mechanisms, limitations, and potential applications. PeerJ 2018, 6, e4370. [Google Scholar] [CrossRef]
- Zheng, Y.L. Some Ethical Concerns About Human Induced Pluripotent Stem Cells. Sci. Eng. Ethics. 2016, 22, 1277–1284. [Google Scholar] [CrossRef]
- Cornetta, K.; Patel, K.; Wanjiku, C.M.; Busakhala, N. Equitable Access to Gene Therapy: A Call to Action for the American Society of Gene and Cell Therapy. Mol. Ther. 2018, 26, 2715–2716. [Google Scholar] [CrossRef]
- Steeg, R.; Mueller, S.C.; Mah, N.; Holst, B.; Cabrera-Socorro, A.; Stacey, G.N.; De Sousa, P.A.; Courtney, A.; Zimmermann, H. EBiSC best practice: How to ensure optimal generation, qualification, and distribution of iPSC lines. Stem Cell Rep. 2021, 16, 1853–1867. [Google Scholar] [CrossRef]
- Teixeira, T.; Kweder, S.L.; Saint-Raymond, A. Are the European Medicines Agency, US Food and Drug Administration, and Other International Regulators Talking to Each Other? Clin. Pharmacol. Ther. 2020, 107, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.L.; Tulloch, N.L.; Razumova, M.V.; Saiget, M.; Muskheli, V.; Pabon, L.; Reinecke, H.; Regnier, M.; Murry, C.E. Mechanical Stress Conditioning and Electrical Stimulation Promote Contractility and Force Maturation of Induced Pluripotent Stem Cell-Derived Human Cardiac Tissue. Circulation 2016, 134, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 2018, 556, 239–243. [Google Scholar] [CrossRef]
- Lyra-Leite, D.M.; Gutiérrez-Gutiérrez, Ó.; Wang, M.; Zhou, Y.; Cyganek, L.; Burridge, P.W. A review of protocols for human iPSC culture, cardiac differentiation, subtype-specification, maturation, and direct reprogramming. STAR Protoc. 2022, 3, 101560. [Google Scholar] [CrossRef]
- Kistamás, K.; Müller, A.; Muenthaisong, S.; Lamberto, F.; Zana, M.; Dulac, M.; Leal, F.; Maziz, A.; Costa, P.; Bernotiene, E.; et al. Multifactorial approaches to enhance maturation of human iPSC-derived cardiomyocytes. J. Mol. Liq. 2023, 387, 122668. [Google Scholar] [CrossRef]
- Besser, R.R.; Ishahak, M.; Mayo, V.; Carbonero, D.; Claure, I.; Agarwal, A. Engineered Microenvironments for Maturation of Stem Cell Derived Cardiac Myocytes. Theranostics 2018, 8, 124–140. [Google Scholar] [CrossRef]
- Li, L.; Li, D.; Wang, J.; Dai, Y. Single-cell RNA sequencing reveals key regulators and differentiation trajectory of iPSC-derived cardiomyocytes. Sci. Rep. 2024, 14, 29268. [Google Scholar] [CrossRef]
- Wang, J.; Morgan, W.; Saini, A.; Liu, T.; Lough, J.; Han, L. Single-cell transcriptomic profiling reveals specific maturation signatures in human cardiomyocytes derived from LMNB2-inactivated induced pluripotent stem cells. Front. Cell Dev. Biol. 2022, 10, 895162. [Google Scholar] [CrossRef]
- Venkatesh, S.; Baljinnyam, E.; Tong, M.; Kashihara, T.; Yan, L.; Liu, T.; Li, H.; Xie, L.H.; Nakamura, M.; Oka, S.I.; et al. Proteomic analysis of mitochondrial biogenesis in cardiomyocytes differentiated from human induced pluripotent stem cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2021, 320, R547–R562. [Google Scholar] [CrossRef]
- Hunter, B.; Li, M.; Parker, B.L.; Koay, Y.C.; Harney, D.J.; Pearson, E.; Cao, J.; Chen, G.T.; Guneratne, O.; Smyth, G.K.; et al. Proteomic and metabolomic analyses of the human adult myocardium reveal ventricle-specific regulation in end-stage cardiomyopathies. Commun. Biol. 2024, 7, 1666. [Google Scholar] [CrossRef]
- Perrino, C.; Barabási, A.L.; Condorelli, G.; Davidson, S.M.; De Windt, L.; Dimmeler, S.; Engel, F.B.; Hausenloy, D.J.; Hill, J.A.; Van Laake, L.W.; et al. Epigenomic and transcriptomic approaches in the post-genomic era: Path to novel targets for diagnosis and therapy of the ischaemic heart? Position Paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. Cardiovasc. Res. 2017, 113, 725–736. [Google Scholar] [CrossRef]
- Johansson, M.; Ulfenborg, B.; Andersson, C.X.; Heydarkhan-Hagvall, S.; Jeppsson, A.; Sartipy, P.; Synnergren, J. Multi-Omics Characterization of a Human Stem Cell-Based Model of Cardiac Hypertrophy. Life 2022, 12, 293. [Google Scholar] [CrossRef]
- Tan, W.L.W.; Seow, W.Q.; Zhang, A.; Rhee, S.; Wong, W.H.; Greenleaf, W.J.; Wu, J.C. Current and future perspectives of single-cell multi-omics technologies in cardiovascular research. Nat. Cardiovasc. Res. 2023, 2, 20–34. [Google Scholar] [CrossRef]
- Kernik, D.C.; Morotti, S.; Wu, H.; Garg, P.; Duff, H.J.; Kurokawa, J.; Jalife, J.; Wu, J.C.; Grandi, E.; Clancy, C.E. A computational model of induced pluripotent stem-cell derived cardiomyocytes incorporating experimental variability from multiple data sources. J. Physiol. 2019, 597, 4533–4564. [Google Scholar] [CrossRef]
- Kusumoto, D.; Lachmann, M.; Kunihiro, T.; Yuasa, S.; Kishino, Y.; Kimura, M.; Katsuki, T.; Itoh, S.; Seki, T.; Fukuda, K. Automated Deep Learning-Based System to Identify Endothelial Cells Derived from Induced Pluripotent Stem Cells. Stem Cell Rep. 2018, 10, 1687–1695. [Google Scholar] [CrossRef]
- Drouard, G.; Mykkänen, J.; Heiskanen, J.; Pohjonen, J.; Ruohonen, S.; Pahkala, K.; Lehtimäki, T.; Wang, X.; Ollikainen, M.; Ripatti, S.; et al. Exploring machine learning strategies for predicting cardiovascular disease risk factors from multi-omic data. BMC Med. Inform. Decis. Mak. 2024, 24, 116. [Google Scholar] [CrossRef]
- Rahman, S.M.; Lan, J.; Kaeli, D.; Dy, J.; Alshawabkeh, A.; Gu, A.Z. Machine learning-based biomarkers identification from toxicogenomics—Bridging to regulatory relevant phenotypic endpoints. J. Hazard. Mater. 2022, 423, 127141. [Google Scholar] [CrossRef]
- Williams, B.; Halloin, C.; Löbel, W.; Finklea, F.; Lipke, E.; Zweigerdt, R.; Cremaschi, S. Data-Driven Model Development for Cardiomyocyte Production Experimental Failure Prediction. In Computer Aided Chemical Engineering; Pierucci, S., Manenti, F., Bozzano, G.L., Manca, D., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 48, pp. 1639–1644. [Google Scholar]
- Schork, N.J. Artificial Intelligence and Personalized Medicine. Cancer Treat Res. 2019, 178, 265–283. [Google Scholar] [CrossRef]
- Vo, Q.D.; Saito, Y.; Ida, T.; Nakamura, K.; Yuasa, S. The use of artificial intelligence in induced pluripotent stem cell-based technology over 10-year period: A systematic scoping review. PLoS ONE 2024, 19, e0302537. [Google Scholar] [CrossRef]
- Bettini, A.; Camelliti, P.; Stuckey, D.J.; Day, R.M. Injectable biodegradable microcarriers for iPSC expansion and cardiomyocyte differentiation. Adv. Sci. 2024, 11, 2404355. [Google Scholar] [CrossRef]
- Liu, N.; Ye, X.; Yao, B.; Zhao, M.; Wu, P.; Liu, G.; Zhuang, D.; Jiang, H.; Chen, X.; He, Y.; et al. Advances in 3D bioprinting technology for cardiac tissue engineering and regeneration. Bioact. Mater. 2021, 6, 1388–1401. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Ito, Y.; Ito, E.; Takeda, M.; Mikami, T.; Taguchi, T.; Mochizuki-Oda, N.; Sasai, M.; Shimamoto, T.; Nitta, Y.; et al. Safety confirmation of induced pluripotent stem cell-derived cardiomyocyte patch transplantation for ischemic cardiomyopathy: First three case reports. Front. Cardiovasc. Med. 2023, 10, 1182209. [Google Scholar] [CrossRef] [PubMed]
- Kirkeby, A.; Main, H.; Carpenter, M. Pluripotent stem-cell-derived therapies in clinical trial: A 2025 update. Cell Stem Cell 2025, 32, 10–37. [Google Scholar] [CrossRef]
- Selvakumar, D.; Reyes, L.; Chong, J.J.H. Cardiac Cell Therapy with Pluripotent Stem Cell-Derived Cardiomyocytes: What Has Been Done and What Remains to Do? Curr. Cardiol. Rep. 2022, 24, 445–461. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vo, Q.D.; Nakamura, K.; Saito, Y.; Akagi, S.; Miyoshi, T.; Yuasa, S. Induced Pluripotent Stem Cells in Cardiomyopathy: Advancing Disease Modeling, Therapeutic Development, and Regenerative Therapy. Int. J. Mol. Sci. 2025, 26, 4984. https://doi.org/10.3390/ijms26114984
Vo QD, Nakamura K, Saito Y, Akagi S, Miyoshi T, Yuasa S. Induced Pluripotent Stem Cells in Cardiomyopathy: Advancing Disease Modeling, Therapeutic Development, and Regenerative Therapy. International Journal of Molecular Sciences. 2025; 26(11):4984. https://doi.org/10.3390/ijms26114984
Chicago/Turabian StyleVo, Quan Duy, Kazufumi Nakamura, Yukihiro Saito, Satoshi Akagi, Toru Miyoshi, and Shinsuke Yuasa. 2025. "Induced Pluripotent Stem Cells in Cardiomyopathy: Advancing Disease Modeling, Therapeutic Development, and Regenerative Therapy" International Journal of Molecular Sciences 26, no. 11: 4984. https://doi.org/10.3390/ijms26114984
APA StyleVo, Q. D., Nakamura, K., Saito, Y., Akagi, S., Miyoshi, T., & Yuasa, S. (2025). Induced Pluripotent Stem Cells in Cardiomyopathy: Advancing Disease Modeling, Therapeutic Development, and Regenerative Therapy. International Journal of Molecular Sciences, 26(11), 4984. https://doi.org/10.3390/ijms26114984