
Signs of Alzheimer’s Disease: Tied to Aging
Abstract
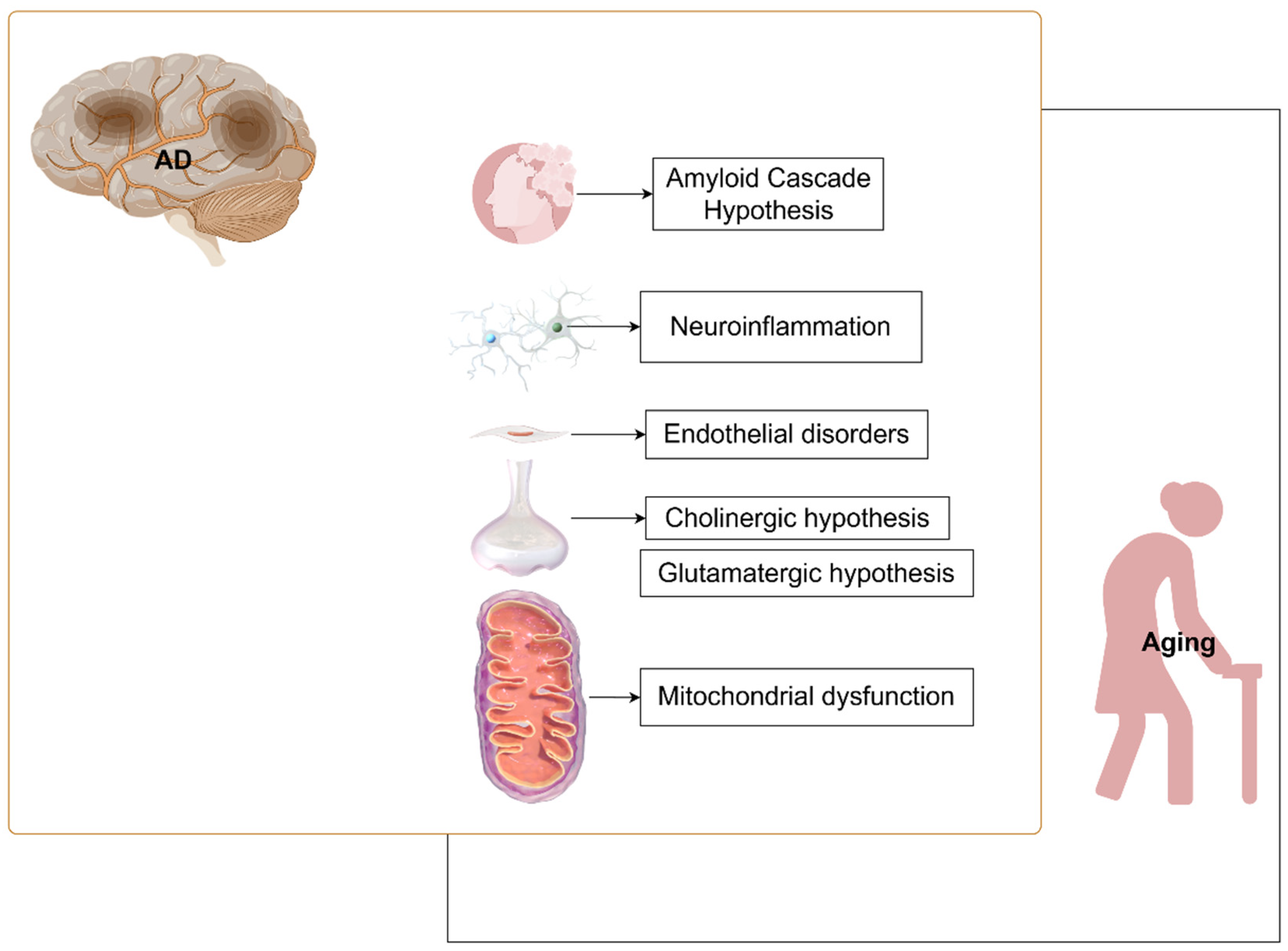
1. Introduction
2. Amyloid Cascade Hypothesis
2.1. Aβ
2.2. Tau
2.3. AD Deteriorates with Aging
3. Nerve Damage
3.1. Neuroinflammation
3.2. Endothelial Dysfunction
3.3. Cholinergic Hypothesis
3.4. Glutamatergic Hypothesis
4. Mitochondrial Dysfunction
4.1. Metabolic Dysregulation
4.2. Other Factors About Mitochondrial Homeostasis
5. Therapeutic Strategies for Aging and AD
5.1. Animal Models
5.1.1. Animal Models of Aging
5.1.2. Animal Models of AD
5.2. Therapeutic Strategies
5.2.1. Preclinical Strategies
5.2.2. Interventions
5.2.3. Prevention
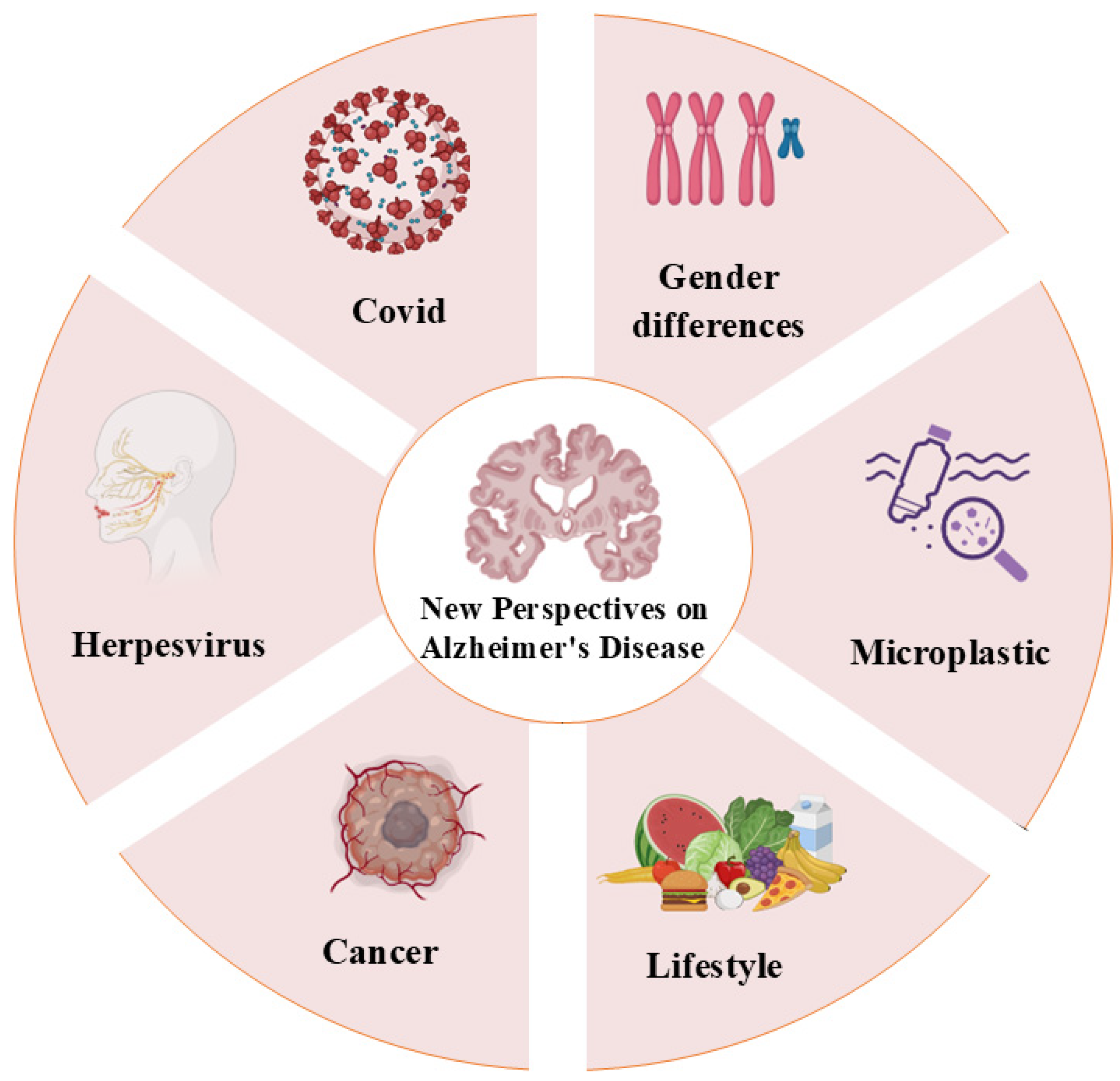
6. New Perspectives on AD
7. Conclusions and Future Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Liu, R.-M. Aging, Cellular Senescence, and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 1989. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of Aging: An Expanding Universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics Improve Physical Function and Increase Lifespan in Old Age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Robbins, P.D.; Jurk, D.; Khosla, S.; Kirkland, J.L.; LeBrasseur, N.K.; Miller, J.D.; Passos, J.F.; Pignolo, R.J.; Tchkonia, T.; Niedernhofer, L.J. Senolytic Drugs: Reducing Senescent Cell Viability to Extend Health Span. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 779–803. [Google Scholar] [CrossRef]
- Zhang, L.; Pitcher, L.E.; Yousefzadeh, M.J.; Niedernhofer, L.J.; Robbins, P.D.; Zhu, Y. Cellular Senescence: A Key Therapeutic Target in Aging and Diseases. J. Clin. Investig. 2022, 132, e158450. [Google Scholar] [CrossRef]
- Kritsilis, M.; Rizou, S.V.; Koutsoudaki, P.N.; Evangelou, K.; Gorgoulis, V.G.; Papadopoulos, D. Ageing, Cellular Senescence and Neurodegenerative Disease. Int. J. Mol. Sci. 2018, 19, 2937. [Google Scholar] [CrossRef]
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173. [Google Scholar] [CrossRef] [PubMed]
- Twarowski, B.; Herbet, M. Inflammatory Processes in Alzheimer’s Disease—Pathomechanism, Diagnosis and Treatment: A Review. Int. J. Mol. Sci. 2023, 24, 6518. [Google Scholar] [CrossRef]
- Grant, W.B.; Blake, S.M. Diet’s Role in Modifying Risk of Alzheimer’s Disease: History and Present Understanding. J. Alzheimers Dis. 2023, 96, 1353–1382. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Bayer, T.A.; Cappai, R.; Masters, C.L.; Beyreuther, K.; Multhaup, G. It All Sticks Together--the APP-Related Family of Proteins and Alzheimer’s Disease. Mol. Psychiatry 1999, 4, 524. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Estus, S.; Younkin, L.H.; Selkoe, D.J.; Younkin, S.G. Processing of the Amyloid Protein Precursor to Potentially Amyloidogenic Derivatives. Science 1992, 255, 728–730. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Gouras, G.K.; Olsson, T.T.; Hansson, O. β-Amyloid Peptides and Amyloid Plaques in Alzheimer’s Disease. Neurotherapeutics 2015, 12, 3–11. [Google Scholar] [CrossRef]
- Hardy, J.; Allsop, D. Amyloid Deposition as the Central Event in the Aetiology of Alzheimer’s Disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Wildsmith, K.R.; Holley, M.; Savage, J.C.; Skerrett, R.; Landreth, G.E. Evidence for Impaired Amyloid β Clearance in Alzheimer’s Disease. Alzheimers Res. Ther. 2013, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gamache, E.; Rosenman, D.J.; Xie, J.; Lopez, M.M.; Li, Y.-M.; Wang, C. Familial Alzheimer’s Mutations within APPTM Increase Aβ42 Production by Enhancing Accessibility of ε-Cleavage Site. Nat. Commun. 2014, 5, 3037. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, X.; Xia, W.; Zhang, Y.; Wang, C. Targeting Amyloidogenic Processing of APP in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 137. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, H.; Zhao, Y.; Liang, Z.; Gong, X.; Yu, J.; Huang, T.; Yang, C.; Wu, M.; Xiao, Y.; et al. BACE1 SUMOylation Deregulates Phosphorylation and Ubiquitination in Alzheimer’s Disease Pathology. J. Neurochem. 2023, 166, 318–327. [Google Scholar] [CrossRef]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Doré, V.; Fowler, C.; Li, Q.-X.; Martins, R.; Rowe, C.; et al. High Performance Plasma Amyloid-β Biomarkers for Alzheimer’s Disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Levites, Y.; Dammer, E.B.; Ran, Y.; Tsering, W.; Duong, D.; Abreha, M.; Gadhavi, J.; Lolo, K.; Trejo-Lopez, J.; Phillips, J.; et al. Integrative Proteomics Identifies a Conserved Aβ Amyloid Responsome, Novel Plaque Proteins, and Pathology Modifiers in Alzheimer’s Disease. Cell Rep. Med. 2024, 5, 101669. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, D. Tau Protein and Tauopathies: Exploring Tau Protein–Protein and Microtubule Interactions, Cross-Interactions and Therapeutic Strategies. ChemMedChem 2024, 19, e202400180. [Google Scholar] [CrossRef]
- Hernández, F.; Ferrer, I.; Pérez, M.; Zabala, J.C.; del Rio, J.A.; Avila, J. Tau Aggregation. Neuroscience 2023, 518, 64–69. [Google Scholar] [CrossRef]
- Morishima-Kawashima, M.; Hasegawa, M.; Takio, K.; Suzuki, M.; Yoshida, H.; Titani, K.; Ihara, Y. Proline-Directed and Non-Proline-Directed Phosphorylation of PHF-Tau. J. Biol. Chem. 1995, 270, 823–829. [Google Scholar] [CrossRef]
- Montalto, G.; Ricciarelli, R. Tau, Tau Kinases, and Tauopathies: An Updated Overview. BioFactors 2023, 49, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Ferreira, A. The Generation of a 17 kDa Neurotoxic Fragment: An Alternative Mechanism by Which Tau Mediates β-Amyloid-Induced Neurodegeneration. J. Neurosci. 2005, 25, 5365–5375. [Google Scholar] [CrossRef]
- Rajendran, K.; Krishnan, U.M. Mechanistic Insights and Emerging Therapeutic Stratagems for Alzheimer’s Disease. Ageing Res. Rev. 2024, 97, 102309. [Google Scholar] [CrossRef]
- D’Souza, I.; Poorkaj, P.; Hong, M.; Nochlin, D.; Lee, V.M.-Y.; Bird, T.D.; Schellenberg, G.D. Missense and Silent Tau Gene Mutations Cause Frontotemporal Dementia with Parkinsonism-Chromosome 17 Type, by Affecting Multiple Alternative RNA Splicing Regulatory Elements. Proc. Natl. Acad. Sci. USA 1999, 96, 5598–5603. [Google Scholar] [CrossRef]
- Hong, M.; Zhukareva, V.; Vogelsberg-Ragaglia, V.; Wszolek, Z.; Reed, L.; Miller, B.I.; Geschwind, D.H.; Bird, T.D.; McKeel, D.; Goate, A.; et al. Mutation-Specific Functional Impairments in Distinct Tau Isoforms of Hereditary FTDP-17. Science 1998, 282, 1914–1917. [Google Scholar] [CrossRef]
- Tesseur, I.; Van Dorpe, J.; Spittaels, K.; Van den Haute, C.; Moechars, D.; Van Leuven, F. Expression of Human Apolipoprotein E4 in Neurons Causes Hyperphosphorylation of Protein Tau in the Brains of Transgenic Mice. Am. J. Pathol. 2000, 156, 951–964. [Google Scholar] [CrossRef]
- Thijssen, E.H.; La Joie, R.; Wolf, A.; Strom, A.; Wang, P.; Iaccarino, L.; Bourakova, V.; Cobigo, Y.; Heuer, H.; Spina, S.; et al. Diagnostic Value of Plasma Phosphorylated Tau181 in Alzheimer’s Disease and Frontotemporal Lobar Degeneration. Nat. Med. 2020, 26, 387–397. [Google Scholar] [CrossRef]
- Janelidze, S.; Mattsson, N.; Palmqvist, S.; Smith, R.; Beach, T.G.; Serrano, G.E.; Chai, X.; Proctor, N.K.; Eichenlaub, U.; Zetterberg, H.; et al. Plasma P-Tau181 in Alzheimer’s Disease: Relationship to Other Biomarkers, Differential Diagnosis, Neuropathology and Longitudinal Progression to Alzheimer’s Dementia. Nat. Med. 2020, 26, 379–386. [Google Scholar] [CrossRef]
- Socualaya, K.M.Q.; Therriault, J.; Aliaga, A.; Macedo, A.C.; Rahmouni, N.; Arias, J.F.; Stevenson, J.; Wang, Y.-T.; Tissot, C.; Benedet, A.L.; et al. Plasma P-Tau217 Outperforms [18F]FDG-PET in Identifying Biological Alzheimer’s Disease in Atypical and Early-Onset Dementia. Alzheimers Dement. 2025, 20, e093116. [Google Scholar] [CrossRef]
- Horie, K.; Salvadó, G.; Koppisetti, R.K.; Janelidze, S.; Barthélemy, N.R.; He, Y.; Sato, C.; Gordon, B.A.; Jiang, H.; Benzinger, T.L.S.; et al. Plasma MTBR-Tau243 Biomarker Identifies Tau Tangle Pathology in Alzheimer’s Disease. Nat. Med. 2025, 1–10. [Google Scholar] [CrossRef]
- Depp, C.; Sun, T.; Sasmita, A.O.; Spieth, L.; Berghoff, S.A.; Nazarenko, T.; Overhoff, K.; Steixner-Kumar, A.A.; Subramanian, S.; Arinrad, S.; et al. Myelin Dysfunction Drives Amyloid-β Deposition in Models of Alzheimer’s Disease. Nature 2023, 618, 349–357. [Google Scholar] [CrossRef]
- Makhlouf, H. Tau-induced Astrocyte Senescence and Dysfunction in AD Tauopathy. Alzheimers Dement. 2025, 20 (Suppl. 1), e093341. [Google Scholar] [CrossRef]
- Edler, M.K.; Mhatre-Winters, I.; Richardson, J.R. Microglia in Aging and Alzheimer’s Disease: A Comparative Species Review. Cells 2021, 10, 1138. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a Central Mechanism in Alzheimer’s Disease. Alzheimers Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Anwar, M.M. The Orchestrating Role of Deteriorating Neurons and TREM-1 in Crosstalk with SYK in Alzheimer’s Disease Progression and Neuroinflammation. Inflammopharmacology 2023, 31, 2303–2310. [Google Scholar] [CrossRef]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An Early and Late Peak in Microglial Activation in Alzheimer’s Disease Trajectory. Brain J. Neurol. 2017, 140, 792–803. [Google Scholar] [CrossRef]
- Sheng, J.G.; Mrak, R.E.; Griffin, W.S. Glial-Neuronal Interactions in Alzheimer Disease: Progressive Association of IL-1alpha+ Microglia and S100beta+ Astrocytes with Neurofibrillary Tangle Stages. J. Neuropathol. Exp. Neurol. 1997, 56, 285–290. [Google Scholar] [CrossRef]
- Xing, C.; Li, W.; Deng, W.; Ning, M.; Lo, E.H. A Potential Gliovascular Mechanism for Microglial Activation: Differential Phenotypic Switching of Microglia by Endothelium versus Astrocytes. J. Neuroinflamm. 2018, 15, 143. [Google Scholar] [CrossRef]
- Sudduth, T.L.; Schmitt, F.A.; Nelson, P.T.; Wilcock, D.M. Neuroinflammatory Phenotype in Early Alzheimer’s Disease. Neurobiol. Aging 2013, 34, 1051–1059. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Itagaki, S.; McGeer, P.L.; Akiyama, H.; Zhu, S.; Selkoe, D. Relationship of Microglia and Astrocytes to Amyloid Deposits of Alzheimer Disease. J. Neuroimmunol. 1989, 24, 173–182. [Google Scholar] [CrossRef]
- Kumar, H.; Chakrabarti, A.; Sarma, P.; Modi, M.; Banerjee, D.; Radotra, B.D.; Bhatia, A.; Medhi, B. Novel Therapeutic Mechanism of Action of Metformin and Its Nanoformulation in Alzheimer’s Disease and Role of AKT/ERK/GSK Pathway. Eur. J. Pharm. Sci. 2023, 181, 106348. [Google Scholar] [CrossRef]
- Fernandez, C.G.; Hamby, M.E.; McReynolds, M.L.; Ray, W.J. The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 14. [Google Scholar] [CrossRef]
- Malpetti, M.; Franzmeier, N.; Brendel, M. PET Imaging to Measure Neuroinflammation In Vivo. In Biomarkers for Alzheimer’s Disease Drug Development; Perneczky, R., Ed.; Springer: New York, NY, USA, 2024; pp. 177–193. [Google Scholar] [CrossRef]
- Appleton, J.; Finn, Q.; Zanotti-Fregonara, P.; Yu, M.; Faridar, A.; Nakawah, M.O.; Zarate, C.; Carrillo, M.C.; Dickerson, B.C.; Rabinovici, G.D.; et al. Brain Inflammation Co-Localizes Highly with Tau in Mild Cognitive Impairment Due to Early-Onset Alzheimer’s Disease. Brain 2025, 148, 119–132. [Google Scholar] [CrossRef]
- Fuh, J.-L.; Lin, Y.-S.; Yeh, S.H.-H.; Lee, W.-J.; Chang, W.-Y.; Chang, C.-W.; Yang, B.-H.; Peng, N.-J.; Huang, W.-S. Positron Emission Tomography (PET) Imaging of Neuroinflammation in Healthy and Alzheimer Disease Participants Using TSPO Tracers. Alzheimers Dement. 2023, 19 (Suppl. 16), e073250. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular Senescence: From Physiology to Pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Cuollo, L.; Antonangeli, F.; Santoni, A.; Soriani, A. The Senescence-Associated Secretory Phenotype (SASP) in the Challenging Future of Cancer Therapy and Age-Related Diseases. Biology 2020, 9, 485. [Google Scholar] [CrossRef]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD+ Supplementation Reduces Neuroinflammation and Cell Senescence in a Transgenic Mouse Model of Alzheimer’s Disease via cGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired Immune Surveillance Accelerates Accumulation of Senescent Cells and Aging. Nat. Commun. 2018, 9, 5435. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Flores, R.R.; Zhu, Y.; Schmiechen, Z.C.; Brooks, R.W.; Trussoni, C.E.; Cui, Y.; Angelini, L.; Lee, K.-A.; McGowan, S.J.; et al. An Aged Immune System Drives Senescence and Ageing of Solid Organs. Nature 2021, 594, 100–105. [Google Scholar] [CrossRef]
- Rabin, J.S.; Schultz, A.P.; Hedden, T.; Viswanathan, A.; Marshall, G.A.; Kilpatrick, E.; Klein, H.; Buckley, R.F.; Yang, H.-S.; Properzi, M.; et al. Interactive Associations of Vascular Risk and β-Amyloid Burden with Cognitive Decline in Clinically Normal Elderly Individuals. JAMA Neurol. 2018, 75, 1124–1131. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral Amyloid Angiopathy and Alzheimer Disease—One Peptide, Two Pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef]
- Bennett, R.E.; Robbins, A.B.; Hu, M.; Cao, X.; Betensky, R.A.; Clark, T.; Das, S.; Hyman, B.T. Tau Induces Blood Vessel Abnormalities and Angiogenesis-Related Gene Expression in P301L Transgenic Mice and Human Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1289–E1298. [Google Scholar] [CrossRef]
- Asunción-Alvarez, D.; Palacios, J.; Ybañez-Julca, R.O.; Rodriguez-Silva, C.N.; Nwokocha, C.; Cifuentes, F.; Greensmith, D.J. Calcium Signaling in Endothelial and Vascular Smooth Muscle Cells: Sex Differences and the Influence of Estrogens and Androgens. Am. J. Physiol. Heart Circ. Physiol. 2024, 326, H950–H970. [Google Scholar] [CrossRef]
- Wang, X.; He, B. Endothelial Dysfunction: Molecular Mechanisms and Clinical Implications. MedComm 2024, 5, e651. [Google Scholar] [CrossRef]
- Tousoulis, D.; Kampoli, A.-M.; Papageorgiou, C.T.N.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef]
- Schaeffer, S.; Iadecola, C. Revisiting the Neurovascular Unit. Nat. Neurosci. 2021, 24, 1198–1209. [Google Scholar] [CrossRef]
- Saunders, A.; Macosko, E.; Wysoker, A.; Goldman, M.; Krienen, F.; de Rivera, H.; Bien, E.; Baum, M.; Wang, S.; Goeva, A.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, 1015–1030.e16. [Google Scholar] [CrossRef]
- Bunch, T.J. Atrial Fibrillation and Dementia. Circulation 2020, 142, 618–620. [Google Scholar] [CrossRef]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood–Brain Barrier Breakdown Is an Early Biomarker of Human Cognitive Dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Alkhalifa, A.E.; Al-Ghraiybah, N.F.; Odum, J.; Shunnarah, J.G.; Austin, N.; Kaddoumi, A. Blood–Brain Barrier Breakdown in Alzheimer’s Disease: Mechanisms and Targeted Strategies. Int. J. Mol. Sci. 2023, 24, 16288. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the Neurovascular Unit: Key Functions and Signaling Pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes Are Required for Blood–Brain Barrier Integrity during Embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, S.; Yang, D.; Zheng, W.; Xia, H.; Zhu, Q.; Wang, Z.; Hu, B.; Jiang, X.; Qin, X.; et al. Inhibition of IFITM3 in Cerebrovascular Endothelium Alleviates Alzheimer’s-related Phenotypes. Alzheimers Dement. 2025, 21, e14543. [Google Scholar] [CrossRef]
- Jaeger, L.B.; Dohgu, S.; Sultana, R.; Lynch, J.L.; Owen, J.B.; Erickson, M.A.; Shah, G.N.; Price, T.O.; Fleegal-Demotta, M.A.; Butterfiled, D.A.; et al. Lipopolysaccharide Alters the Blood–Brain Barrier Transport of Amyloid β Protein: A Mechanism for Inflammation in the Progression of Alzheimer’s Disease. Brain. Behav. Immun. 2009, 23, 507–517. [Google Scholar] [CrossRef]
- Takeda, S.; Sato, N.; Ikimura, K.; Nishino, H.; Rakugi, H.; Morishita, R. Increased Blood–Brain Barrier Vulnerability to Systemic Inflammation in an Alzheimer Disease Mouse Model. Neurobiol. Aging 2013, 34, 2064–2070. [Google Scholar] [CrossRef]
- Askenazi, M.; Kavanagh, T.; Pires, G.; Ueberheide, B.; Wisniewski, T.; Drummond, E. Compilation of Reported Protein Changes in the Brain in Alzheimer’s Disease. Nat. Commun. 2023, 14, 4466. [Google Scholar] [CrossRef]
- Bai, R.; Guo, J.; Ye, X.-Y.; Xie, Y.; Xie, T. Oxidative Stress: The Core Pathogenesis and Mechanism of Alzheimer’s Disease. Ageing Res. Rev. 2022, 77, 101619. [Google Scholar] [CrossRef] [PubMed]
- Muneer, P.M.A.; Chandra, N.; Haorah, J. Interactions of Oxidative Stress and Neurovascular Inflammation in the Pathogenesis of Traumatic Brain Injury. Mol. Neurobiol. 2015, 51, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jung, U.J.; Kim, S.R. Role of Oxidative Stress in Blood–Brain Barrier Disruption and Neurodegenerative Diseases. Antioxidants 2024, 13, 1462. [Google Scholar] [CrossRef]
- Bloom, S.I.; Islam, M.T.; Lesniewski, L.A.; Donato, A.J. Mechanisms and Consequences of Endothelial Cell Senescence. Nat. Rev. Cardiol. 2023, 20, 38–51. [Google Scholar] [CrossRef]
- Hwang, H.J.; Kim, N.; Herman, A.B.; Gorospe, M.; Lee, J.-S. Factors and Pathways Modulating Endothelial Cell Senescence in Vascular Aging. Int. J. Mol. Sci. 2022, 23, 10135. [Google Scholar] [CrossRef]
- Singh, V.; Kaur, R.; Kumari, P.; Pasricha, C.; Singh, R. ICAM-1 and VCAM-1: Gatekeepers in Various Inflammatory and Cardiovascular Disorders. Clin. Chim. Acta 2023, 548, 117487. [Google Scholar] [CrossRef]
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a Neuromodulator: Cholinergic Signaling Shapes Nervous System Function and Behavior. Neuron 2012, 76, 116–129. [Google Scholar] [CrossRef]
- Wurtman, R.J. Choline Metabolism as a Basis for the Selective Vulnerability of Cholinergic Neurons. Trends Neurosci. 1992, 15, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Bekdash, R.A. The Cholinergic System, the Adrenergic System and the Neuropathology of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 1273. [Google Scholar] [CrossRef]
- Chen, Z.-R.; Huang, J.-B.; Yang, S.-L.; Hong, F.-F. Role of Cholinergic Signaling in Alzheimer’s Disease. Molecules 2022, 27, 1816. [Google Scholar] [CrossRef] [PubMed]
- Hasselmo, M.E. The Role of Acetylcholine in Learning and Memory. Curr. Opin. Neurobiol. 2006, 16, 710–715. [Google Scholar] [CrossRef]
- Davies, P.; Maloney, A.J. Selective Loss of Central Cholinergic Neurons in Alzheimer’s Disease. Lancet Lond. Engl. 1976, 2, 1403. [Google Scholar] [CrossRef]
- Chatterjee, S.; Mudher, A. Alzheimer’s Disease and Type 2 Diabetes: A Critical Assessment of the Shared Pathological Traits. Front. Neurosci. 2018, 12, 383. [Google Scholar] [CrossRef] [PubMed]
- T.den Heijer, T.; Vermeer, S.E.; van Dijk, E.J.; Prins, N.D.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M.B. Type 2 Diabetes and Atrophy of Medial Temporal Lobe Structures on Brain MRI. Diabetologia 2003, 46, 1604–1610. [Google Scholar] [CrossRef]
- Yonamine, C.Y.; Michalani, M.L.E.; Moreira, R.J.; Machado, U.F. Glucose Transport and Utilization in the Hippocampus: From Neurophysiology to Diabetes-Related Development of Dementia. Int. J. Mol. Sci. 2023, 24, 16480. [Google Scholar] [CrossRef]
- Dutta, B.J.; Singh, S.; Seksaria, S.; Das Gupta, G.; Singh, A. Inside the Diabetic Brain: Insulin Resistance and Molecular Mechanism Associated with Cognitive Impairment and Its Possible Therapeutic Strategies. Pharmacol. Res. 2022, 182, 106358. [Google Scholar] [CrossRef]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The Diagnosis of Mild Cognitive Impairment Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimers Dement. J. Alzheimers Assoc. 2011, 7, 270–279. [Google Scholar] [CrossRef]
- Drzezga, A.; Grimmer, T.; Riemenschneider, M.; Lautenschlager, N.; Siebner, H.; Alexopoulus, P.; Minoshima, S.; Schwaiger, M.; Kurz, A. Prediction of Individual Clinical Outcome in MCI by Means of Genetic Assessment and 18F-FDG PET. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2005, 46, 1625–1632. [Google Scholar]
- Chételat, G.; Arbizu, J.; Barthel, H.; Garibotto, V.; Law, I.; Morbelli, S.; van de Giessen, E.; Agosta, F.; Barkhof, F.; Brooks, D.J.; et al. Amyloid-PET and 18F-FDG-PET in the Diagnostic Investigation of Alzheimer’s Disease and Other Dementias. Lancet Neurol. 2020, 19, 951–962. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. JAD 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.H.Y.; Walby, J.L.; Palpagama, T.H.; Turner, C.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Glutamatergic Receptor Expression Changes in the Alzheimer’s Disease Hippocampus and Entorhinal Cortex. Brain Pathol. 2021, 31, e13005. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Wang, J.; Xia, Y.; Zhang, J.; Chen, L. Recent Advances in Alzheimer’s Disease: Mechanisms, Clinical Trials and New Drug Development Strategies. Signal Transduct. Target. Ther. 2024, 9, 211. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.G.M. NMDA Receptors and Memory Encoding. Neuropharmacology 2013, 74, 32–40. [Google Scholar] [CrossRef]
- Conway, M.E.; Hutson, S.M. BCAA Metabolism and NH3 Homeostasis. Adv. Neurobiol. 2016, 13, 99–132. [Google Scholar] [CrossRef]
- Yudkoff, M. Interactions in the Metabolism of Glutamate and the Branched-Chain Amino Acids and Ketoacids in the CNS. Neurochem. Res. 2017, 42, 10–18. [Google Scholar] [CrossRef]
- Conway, M.E. Alzheimer’s Disease: Targeting the Glutamatergic System. Biogerontology 2020, 21, 257–274. [Google Scholar] [CrossRef]
- Clements, J.D.; Lester, R.A.J.; Tong, G.; Jahr, C.E.; Westbrook, G.L. The Time Course of Glutamate in the Synaptic Cleft. Science 1992, 258, 1498–1501. [Google Scholar] [CrossRef]
- Tahiri, E.; Corti, E.; Duarte, C.B. Regulation of Synaptic NMDA Receptor Activity by Post-Translational Modifications. Neurochem. Res. 2025, 50, 110. [Google Scholar] [CrossRef] [PubMed]
- Koffie, R.M.; Hashimoto, T.; Tai, H.-C.; Kay, K.R.; Serrano-Pozo, A.; Joyner, D.; Hou, S.; Kopeikina, K.J.; Frosch, M.P.; Lee, V.M.; et al. Apolipoprotein E4 Effects in Alzheimer’s Disease Are Mediated by Synaptotoxic Oligomeric Amyloid-β. Brain 2012, 135, 2155–2168. [Google Scholar] [CrossRef] [PubMed]
- Pickett, E.K.; Herrmann, A.G.; McQueen, J.; Abt, K.; Dando, O.; Tulloch, J.; Jain, P.; Dunnett, S.; Sohrabi, S.; Fjeldstad, M.P.; et al. Amyloid Beta and Tau Cooperate to Cause Reversible Behavioral and Transcriptional Deficits in a Model of Alzheimer’s Disease. Cell Rep. 2019, 29, 3592–3604.e5. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.F.; Hascup, E.R.; Bartke, A.; Hascup, K.N. Friend or Foe? Defining the Role of Glutamate in Aging and Alzheimer’s Disease. Front. Aging 2022, 3, 929474. [Google Scholar] [CrossRef]
- Li, D.; Gao, X.; Ma, X.; Wang, M.; Cheng, C.; Xue, T.; Gao, F.; Shen, Y.; Zhang, J.; Liu, Q. Aging-Induced tRNAGlu-Derived Fragment Impairs Glutamate Biosynthesis by Targeting Mitochondrial Translation-Dependent Cristae Organization. Cell Metab. 2024, 36, 1059–1075.e9. [Google Scholar] [CrossRef]
- Mahaman, Y.A.R.; Embaye, K.S.; Huang, F.; Li, L.; Zhu, F.; Wang, J.-Z.; Liu, R.; Feng, J.; Wang, X. Biomarkers Used in Alzheimer’s Disease Diagnosis, Treatment, and Prevention. Ageing Res. Rev. 2022, 74, 101544. [Google Scholar] [CrossRef]
- Oh, H.S.-H.; Urey, D.Y.; Karlsson, L.; Zhu, Z.; Shen, Y.; Farinas, A.; Timsina, J.; Duggan, M.R.; Chen, J.; Guldner, I.H.; et al. A Cerebrospinal Fluid Synaptic Protein Biomarker for Prediction of Cognitive Resilience versus Decline in Alzheimer’s Disease. Nat. Med. 2025, 31, 1592–1603. [Google Scholar] [CrossRef]
- Correia, S.C.; Santos, R.X.; Carvalho, C.; Cardoso, S.; Candeias, E.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Insulin Signaling, Glucose Metabolism and Mitochondria: Major Players in Alzheimer’s Disease and Diabetes Interrelation. Brain Res. 2012, 1441, 64–78. [Google Scholar] [CrossRef]
- Alzheimer’s Association Calcium Hypothesis Workgroup. Calcium Hypothesis of Alzheimer’s Disease and Brain Aging: A Framework for Integrating New Evidence into a Comprehensive Theory of Pathogenesis. Alzheimers Dement. J. Alzheimers Assoc. 2017, 13, 178–182.e17. [Google Scholar] [CrossRef]
- Adlimoghaddam, A.; Snow, W.M.; Stortz, G.; Perez, C.; Djordjevic, J.; Goertzen, A.L.; Ko, J.H.; Albensi, B.C. Regional Hypometabolism in the 3xTg Mouse Model of Alzheimer’s Disease. Neurobiol. Dis. 2019, 127, 264–277. [Google Scholar] [CrossRef]
- Ashleigh, T.; Swerdlow, R.H.; Beal, M.F. The Role of Mitochondrial Dysfunction in Alzheimer’s Disease Pathogenesis. Alzheimers Dement 2023, 19, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Schapira, A.H. Mitochondria and Degenerative Disorders. Am. J. Med. Genet. 2001, 106, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Mutisya, E.M.; Bowling, A.C.; Beal, M.F. Cortical Cytochrome Oxidase Activity Is Reduced in Alzheimer’s Disease. J. Neurochem. 1994, 63, 2179–2184. [Google Scholar] [CrossRef] [PubMed]
- Potenza, M.A.; Sgarra, L.; Desantis, V.; Nacci, C.; Montagnani, M. Diabetes and Alzheimer’s Disease: Might Mitochondrial Dysfunction Help Deciphering the Common Path? Antioxidants 2021, 10, 1257. [Google Scholar] [CrossRef]
- Orr, A.L.; Kim, C.; Jimenez-Morales, D.; Newton, B.W.; Johnson, J.R.; Krogan, N.J.; Swaney, D.L.; Mahley, R.W. Neuronal Apolipoprotein E4 Expression Results in Proteome-Wide Alterations and Compromises Bioenergetic Capacity by Disrupting Mitochondrial Function. J. Alzheimers Dis. 2019, 68, 991–1011. [Google Scholar] [CrossRef]
- Liu, G.; Yang, C.; Wang, X.; Chen, X.; Wang, Y.; Le, W. Oxygen Metabolism Abnormality and Alzheimer’s Disease: An Update. Redox Biol. 2023, 68, 102955. [Google Scholar] [CrossRef]
- Han, X. Neurolipidomics: Challenges and Developments. Front. Biosci. J. Virtual Libr. 2007, 12, 2601–2615. [Google Scholar] [CrossRef]
- Yin, F. Lipid Metabolism and Alzheimer’s Disease: Clinical Evidence, Mechanistic Link and Therapeutic Promise. FEBS J. 2023, 290, 1420–1453. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s Disease Mitochondrial Cascade Hypothesis: Progress and Perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, M.; Chang, W. Iron Dyshomeostasis and Ferroptosis in Alzheimer’s Disease: Molecular Mechanisms of Cell Death and Novel Therapeutic Drugs and Targets for AD. Front. Pharmacol. 2022, 13, 983623. [Google Scholar] [CrossRef]
- Cobb, C.A.; Cole, M.P. Oxidative and Nitrative Stress in Neurodegeneration. Neurobiol. Dis. 2015, 84, 4–21. [Google Scholar] [CrossRef]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and Oxidative Stress in Neurodegenerative Diseases. J. Alzheimers Dis. JAD 2014, 42 (Suppl. 3), S125–S152. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.W.K.; Tzvetkov, N.T.; Georgieva, M.G.; Ognyanov, I.V.; Kordos, K.; Jóźwik, A.; Kühl, T.; Perry, G.; Petralia, M.C.; Mazzon, E.; et al. Reactive Oxygen Species and Their Impact in Neurodegenerative Diseases: Literature Landscape Analysis. Antioxid. Redox Signal. 2021, 34, 402–420. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.-H.; Lu, C.-Y.; Lee, H.-C.; Pang, C.-Y.; Ma, Y.-S. Oxidative Damage and Mutation to Mitochondrial DNA and Age-Dependent Decline of Mitochondrial Respiratory Function. Ann. N. Y. Acad. Sci. 1998, 854, 155–170. [Google Scholar] [CrossRef]
- Selivanov, V.A.; Votyakova, T.V.; Pivtoraiko, V.N.; Zeak, J.; Sukhomlin, T.; Trucco, M.; Roca, J.; Cascante, M. Reactive Oxygen Species Production by Forward and Reverse Electron Fluxes in the Mitochondrial Respiratory Chain. PLoS Comput. Biol. 2011, 7, e1001115. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, Oxidative Stress and the Biology of Ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Kushwah, N.; Bora, K.; Maurya, M.; Pavlovich, M.C.; Chen, J. Oxidative Stress and Antioxidants in Age-Related Macular Degeneration. Antioxid. Basel Switz. 2023, 12, 1379. [Google Scholar] [CrossRef]
- Belarbi, K.; Cuvelier, E.; Bonte, M.-A.; Desplanque, M.; Gressier, B.; Devos, D.; Chartier-Harlin, M.-C. Glycosphingolipids and Neuroinflammation in Parkinson’s Disease. Mol. Neurodegener. 2020, 15, 59. [Google Scholar] [CrossRef] [PubMed]
- Chvilicek, M.M.; Titos, I.; Merrill, C.B.; Cummins-Beebee, P.N.; Chen, J.D.; Rodan, A.R.; Rothenfluh, A. Alcohol Induces Long-Lasting Sleep Deficits in Drosophila via Subsets of Cholinergic Neurons. Curr. Biol. 2025, 35, 1033–1046.e3. [Google Scholar] [CrossRef]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial Dynamics and Mitochondrial Quality Control. Redox Biol. 2014, 4, 6–13. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 Stabilized by Mitochondrial Depolarization Recruits Parkin to Damaged Mitochondria and Activates Latent Parkin for Mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-Mediated Mitophagy in Mammalian Cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Rubinsztein, D.C. Mechanisms of Autophagy–Lysosome Dysfunction in Neurodegenerative Diseases. Nat. Rev. Mol. Cell Biol. 2024, 25, 926–946. [Google Scholar] [CrossRef]
- Coleman, P.D.; Delvaux, E.; Kordower, J.H.; Boehringer, A.; Huseby, C.J. Massive Changes in Gene Expression and Their Cause(s) Can Be a Unifying Principle in the Pathobiology of Alzheimer’s Disease. Alzheimers Dement. 2025, 21, e14555. [Google Scholar] [CrossRef] [PubMed]
- Anchelin, M.; Alcaraz-Pérez, F.; Martínez, C.M.; Bernabé-García, M.; Mulero, V.; Cayuela, M.L. Premature Aging in Telomerase-Deficient Zebrafish. Dis. Model. Mech. 2013, 6, 1101–1112. [Google Scholar] [CrossRef]
- Seldeen, K.L.; Lasky, G.; Leiker, M.M.; Pang, M.; Personius, K.E.; Troen, B.R. High Intensity Interval Training Improves Physical Performance and Frailty in Aged Mice. J. Gerontol. Ser. A 2018, 73, 429–437. [Google Scholar] [CrossRef]
- Shen, Y.; Xu, S.; Wei, W.; Sun, X.; Yang, J.; Liu, L.; Dong, C. Melatonin Reduces Memory Changes and Neural Oxidative Damage in Mice Treated with D -galactose. J. Pineal Res. 2002, 32, 173–178. [Google Scholar] [CrossRef]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F. Alzheimer-Type Neuropathology in Transgenic Mice Overexpressing V717F Beta-Amyloid Precursor Protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Arendash, G.W.; King, D.L. Intra- and Intertask Relationships in a Behavioral Test Battery given to Tg2576 Transgenic Mice and Controls. Physiol. Behav. 2002, 75, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Westerman, M.A.; Cooper-Blacketer, D.; Mariash, A.; Kotilinek, L.; Kawarabayashi, T.; Younkin, L.H.; Carlson, G.A.; Younkin, S.G.; Ashe, K.H. The Relationship between Abeta and Memory in the Tg2576 Mouse Model of Alzheimer’s Disease. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 1858–1867. [Google Scholar] [CrossRef]
- Lalonde, R.; Dumont, M.; Staufenbiel, M.; Sturchler-Pierrat, C.; Strazielle, C. Spatial Learning, Exploration, Anxiety, and Motor Coordination in Female APP23 Transgenic Mice with the Swedish Mutation. Brain Res. 2002, 956, 36–44. [Google Scholar] [CrossRef]
- Chishti, M.A.; Yang, D.-S.; Janus, C.; Phinney, A.L.; Horne, P.; Pearson, J.; Strome, R.; Zuker, N.; Loukides, J.; French, J.; et al. Early-Onset Amyloid Deposition and Cognitive Deficits in Transgenic Mice Expressing a Double Mutant Form of Amyloid Precursor Protein 695. J. Biol. Chem. 2001, 276, 21562–21570. [Google Scholar] [CrossRef]
- Hyde, L.A.; Kazdoba, T.M.; Grilli, M.; Lozza, G.; Brussa, R.; Zhang, Q.; Wong, G.T.; McCool, M.F.; Zhang, L.; Parker, E.M.; et al. Age-Progressing Cognitive Impairments and Neuropathology in Transgenic CRND8 Mice. Behav. Brain Res. 2005, 160, 344–355. [Google Scholar] [CrossRef]
- McGowan, E.; Pickford, F.; Kim, J.; Onstead, L.; Eriksen, J.; Yu, C.; Skipper, L.; Murphy, M.P.; Beard, J.; Das, P.; et al. Aβ42 Is Essential for Parenchymal and Vascular Amyloid Deposition in Mice. Neuron 2005, 47, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn-Hardy, K.; Murphy, M.P.; Baker, M.; Yu, X.; et al. Neurofibrillary Tangles, Amyotrophy and Progressive Motor Disturbance in Mice Expressing Mutant (P301L) Tau Protein. Nat. Genet. 2000, 26, 127. [Google Scholar] [CrossRef]
- Oddo, S. Amyloid Deposition Precedes Tangle Formation in a Triple Transgenic Model of Alzheimer’s Disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef]
- Lewis, J.; Dickson, D.W.; Lin, W.-L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.-H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced Neurofibrillary Degeneration in Transgenic Mice Expressing Mutant Tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef]
- Takeda, T.; Hosokawa, M.; Higuchi, K. Senescence-Accelerated Mouse (SAM): A Novel Murine Model of Senescence. Exp. Gerontol. 1997, 32, 105–109. [Google Scholar] [CrossRef]
- Tissenbaum, H.A.; Guarente, L. Model Organisms as a Guide to Mammalian Aging. Dev. Cell 2002, 2, 9–19. [Google Scholar] [CrossRef]
- Piper, M.D.W.; Partridge, L. Drosophila as a Model for Ageing. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2018, 1864, 2707–2717. [Google Scholar] [CrossRef] [PubMed]
- Kishi, S.; Uchiyama, J.; Baughman, A.M.; Goto, T.; Lin, M.C.; Tsai, S.B. The Zebrafish as a Vertebrate Model of Functional Aging and Very Gradual Senescence. Exp. Gerontol. 2003, 38, 777–786. [Google Scholar] [CrossRef]
- Guo, Y.; Niu, K.; Okazaki, T.; Wu, H.; Yoshikawa, T.; Ohrui, T.; Furukawa, K.; Ichinose, M.; Yanai, K.; Arai, H.; et al. Coffee Treatment Prevents the Progression of Sarcopenia in Aged Mice in Vivo and in Vitro. Exp. Gerontol. 2014, 50, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Camporez, J.-P.G.; Petersen, M.C.; Abudukadier, A.; Moreira, G.V.; Jurczak, M.J.; Friedman, G.; Haqq, C.M.; Petersen, K.F.; Shulman, G.I. Anti-Myostatin Antibody Increases Muscle Mass and Strength and Improves Insulin Sensitivity in Old Mice. Proc. Natl. Acad. Sci. USA 2016, 113, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-K.; Tsai, Y.-L.; Shibu, M.A.; Shen, C.-Y.; Chang-Lee, S.N.; Chen, R.-J.; Yao, C.-H.; Ban, B.; Kuo, W.-W.; Huang, C.-Y. Exercise Training Augments Sirt1-Signaling and Attenuates Cardiac Inflammation in D-Galactose Induced-Aging Rats. Aging 2018, 10, 4166–4174. [Google Scholar] [CrossRef]
- Fan, J.; Yang, X.; Li, J.; Shu, Z.; Dai, J.; Liu, X.; Li, B.; Jia, S.; Kou, X.; Yang, Y.; et al. Spermidine Coupled with Exercise Rescues Skeletal Muscle Atrophy from D-Gal-Induced Aging Rats through Enhanced Autophagy and Reduced Apoptosis via AMPK-FOXO3a Signal Pathway. Oncotarget 2017, 8, 17475–17490. [Google Scholar] [CrossRef]
- Harel, I.; Benayoun, B.A.; Machado, B.; Singh, P.P.; Hu, C.-K.; Pech, M.F.; Valenzano, D.R.; Zhang, E.; Sharp, S.C.; Artandi, S.E.; et al. A Platform for Rapid Exploration of Aging and Diseases in a Naturally Short-Lived Vertebrate. Cell 2015, 160, 1013–1026. [Google Scholar] [CrossRef]
- Shcherbakov, D.; Nigri, M.; Akbergenov, R.; Brilkova, M.; Mantovani, M.; Petit, P.I.; Grimm, A.; Karol, A.A.; Teo, Y.; Sanchón, A.C.; et al. Premature Aging in Mice with Error-Prone Protein Synthesis. Sci. Adv. 2022, 8, eabl9051. [Google Scholar] [CrossRef]
- Varela, I.; Cadiñanos, J.; Pendás, A.M.; Gutiérrez-Fernández, A.; Folgueras, A.R.; Sánchez, L.M.; Zhou, Z.; Rodríguez, F.J.; Stewart, C.L.; Vega, J.A.; et al. Accelerated Ageing in Mice Deficient in Zmpste24 Protease Is Linked to P53 Signalling Activation. Nature 2005, 437, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-Positive Senescent Cells Delays Ageing-Associated Disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly-Y, M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature Ageing in Mice Expressing Defective Mitochondrial DNA Polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef]
- Dineley, K.T.; Xia, X.; Bui, D.; Sweatt, J.D.; Zheng, H. Accelerated Plaque Accumulation, Associative Learning Deficits, and Up-Regulation of A7 Nicotinic Receptor Protein in Transgenic Mice Co-Expressing Mutant Human Presenilin 1 and Amyloid Precursor Proteins. J. Biol. Chem. 2002, 277, 22768–22780. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Slunt, H.H.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Borchelt, D.R. APP Processing and Amyloid Deposition in Mice Haplo-Insufficient for Presenilin 1. Neurobiol. Aging 2004, 25, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Savonenko, A.; Xu, G.M.; Melnikova, T.; Morton, J.L.; Gonzales, V.; Wong, M.P.F.; Price, D.L.; Tang, F.; Markowska, A.L.; Borchelt, D.R. Episodic-like Memory Deficits in the APPswe/PS1dE9 Mouse Model of Alzheimer’s Disease: Relationships to β-Amyloid Deposition and Neurotransmitter Abnormalities. Neurobiol. Dis. 2005, 18, 602–617. [Google Scholar] [CrossRef]
- Kimura, R.; Ohno, M. Impairments in Remote Memory Stabilization Precede Hippocampal Synaptic and Cognitive Failures in 5XFAD Alzheimer Mouse Model. Neurobiol. Dis. 2009, 33, 229–235. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Zhang, C.; McNeil, E.; Dressler, L.; Siman, R. Long-Lasting Impairment in Hippocampal Neurogenesis Associated with Amyloid Deposition in a Knock-in Mouse Model of Familial Alzheimer’s Disease. Exp. Neurol. 2007, 204, 77–87. [Google Scholar] [CrossRef]
- Chang, E.H.; Savage, M.J.; Flood, D.G.; Thomas, J.M.; Levy, R.B.; Mahadomrongkul, V.; Shirao, T.; Aoki, C.; Huerta, P.T. AMPA Receptor Downscaling at the Onset of Alzheimer’s Disease Pathology in Double Knockin Mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3410–3415. [Google Scholar] [CrossRef]
- Thapar, N.; Eid, M.A.F.; Raj, N.; Kantas, T.; Billing, H.S.; Sadhu, D. Application of CRISPR/Cas9 in the Management of Alzheimer’s Disease and Parkinson’s Disease: A Review. Ann. Med. Surg. 2012 2024, 86, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Sharma, Y.; Rane, R.; Kumar, D. CRISPR/Cas9 Gene Editing: A Novel Approach Towards Alzheimer’s Disease Treatment. CNS Neurol. Disord. Drug Targets 2024, 23, 1405–1424. [Google Scholar] [CrossRef]
- Bhardwaj, S.; Kesari, K.K.; Rachamalla, M.; Mani, S.; Ashraf, G.M.; Jha, S.K.; Kumar, P.; Ambasta, R.K.; Dureja, H.; Devkota, H.P.; et al. CRISPR/Cas9 Gene Editing: New Hope for Alzheimer’s Disease Therapeutics. J. Adv. Res. 2022, 40, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Oh, J.; Shim, G.; Cho, B.; Chang, Y.; Kim, S.; Baek, S.; Kim, H.; Shin, J.; Choi, H.; et al. In Vivo Neuronal Gene Editing via CRISPR–Cas9 Amphiphilic Nanocomplexes Alleviates Deficits in Mouse Models of Alzheimer’s Disease. Nat. Neurosci. 2019, 22, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Saurat, N.; Minotti, A.P.; Rahman, M.T.; Sikder, T.; Zhang, C.; Cornacchia, D.; Jungverdorben, J.; Ciceri, G.; Betel, D.; Studer, L. Genome-Wide CRISPR Screen Identifies Neddylation as a Regulator of Neuronal Aging and AD Neurodegeneration. Cell Stem Cell 2024, 31, 1162–1174.e8. [Google Scholar] [CrossRef]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A Single-Cell Atlas of Entorhinal Cortex from Individuals with Alzheimer’s Disease Reveals Cell-Type-Specific Gene Expression Regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef]
- Kim, S.; Lim, K.; Yang, S.; Joo, J. Boosting of Tau Protein Aggregation by CD40 and CD48 Gene Expression in Alzheimer’s Disease. FASEB J. 2023, 37, e22702. [Google Scholar] [CrossRef]
- Chang, J.; Li, Y.; Shan, X.; Chen, X.; Yan, X.; Liu, J.; Zhao, L. Neural Stem Cells Promote Neuroplasticity: A Promising Therapeutic Strategy for the Treatment of Alzheimer’s Disease. Neural Regen. Res. 2024, 19, 619–628. [Google Scholar] [CrossRef]
- Zhang, H.-A.; Yuan, C.-X.; Liu, K.-F.; Yang, Q.-F.; Zhao, J.; Li, H.; Yang, Q.-H.; Song, D.; Quan, Z.-Z.; Qing, H. Neural Stem Cell Transplantation Alleviates Functional Cognitive Deficits in a Mouse Model of Tauopathy. Neural Regen. Res. 2022, 17, 152–162. [Google Scholar] [CrossRef]
- Abdi, S.; Javanmehr, N.; Ghasemi-Kasman, M.; Bali, H.Y.; Pirzadeh, M. Stem Cell-Based Therapeutic and Diagnostic Approaches in Alzheimer’s Disease. Curr. Neuropharmacol. 2022, 20, 1093–1115. [Google Scholar] [CrossRef]
- Chen, K.S.; Noureldein, M.H.; McGinley, L.M.; Hayes, J.M.; Rigan, D.M.; Kwentus, J.F.; Mason, S.N.; Mendelson, F.E.; Savelieff, M.G.; Feldman, E.L. Human Neural Stem Cells Restore Spatial Memory in a Transgenic Alzheimer’s Disease Mouse Model by an Immunomodulating Mechanism. Front. Aging Neurosci. 2023, 15, 1306004. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Lee, Y.-K.; Tsai, J.-K.; Su, Y.-T.; Ho, Y.-C.; Chu, T.-H.; Chen, K.-T.; Chang, C.-L.; Chen, J.-S. Cholinesterase Inhibitor Reveals Synergistic Potential for Neural Stem Cell-Based Therapy in the 5xFAD Mouse Model of Alzheimer’s Disease. Biol. Targets Ther. 2024, 18, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Zhan, L.; Li, Q.; Li, Y.; Wu, G.; Wei, H.; Dong, X. LncRNA FER1L4 Promotes Differentiation and Inhibits Proliferation of NSCs via miR-874-3p/Ascl2. Am. J. Transl. Res. 2022, 14, 2256–2266. [Google Scholar]
- Shigematsu, K.; Takeda, T.; Komori, N.; Tahara, K.; Yamagishi, H. Hypothesis: Intravenous Administration of Mesenchymal Stem Cells Is Effective in the Treatment of Alzheimer’s Disease. Med. Hypotheses 2021, 150, 110572. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Lu, Y.; Wang, K.; Bai, L.; Shi, G.; Huang, Y.; Li, Y. Transplantation of Bone Marrow Mesenchymal Stem Cells Improves Cognitive Deficits and Alleviates Neuropathology in Animal Models of Alzheimer’s Disease: A Meta-Analytic Review on Potential Mechanisms. Transl. Neurodegener. 2020, 9, 20. [Google Scholar] [CrossRef]
- Rash, B.G.; Ramdas, K.N.; Agafonova, N.; Naioti, E.; McClain-Moss, L.; Zainul, Z.; Varnado, B.; Peterson, K.; Brown, M.; Leal, T.; et al. Allogeneic Mesenchymal Stem Cell Therapy with Laromestrocel in Mild Alzheimer’s Disease: A Randomized Controlled Phase 2a Trial. Nat. Med. 2025, 31, 1257–1266. [Google Scholar] [CrossRef]
- Brody, M.; Agronin, M.; Herskowitz, B.J.; Bookheimer, S.Y.; Small, G.W.; Hitchinson, B.; Ramdas, K.; Wishard, T.; McInerney, K.F.; Vellas, B.; et al. Results and Insights from a Phase I Clinical Trial of Lomecel-B for Alzheimer’s Disease. Alzheimers Dement. 2023, 19, 261–273. [Google Scholar] [CrossRef]
- Hu, W.T.; Ozturk, T.; Kollhoff, A.; Wharton, W.; Christina Howell, J.; Weiner, M.; Aisen, P.; Petersen, R.; Jack, C.R.; Jagust, W.; et al. Higher CSF sTNFR1-Related Proteins Associate with Better Prognosis in Very Early Alzheimer’s Disease. Nat. Commun. 2021, 12, 4001. [Google Scholar] [CrossRef]
- Yang, H.-S.; Yau, W.-Y.W.; Carlyle, B.C.; Trombetta, B.A.; Zhang, C.; Shirzadi, Z.; Schultz, A.P.; Pruzin, J.J.; Fitzpatrick, C.D.; Kirn, D.R.; et al. Plasma VEGFA and PGF Impact Longitudinal Tau and Cognition in Preclinical Alzheimer’s Disease. Brain 2024, 147, 2158–2168. [Google Scholar] [CrossRef]
- Barry, J.-L.; Baufeld, C.; Guneykaya, D.; Zhang, X.; Litvinchuk, A.; Jiang, H.; Rosenzweig, N.; Pitts, K.M.; Aronchik, M.; Yahya, T.; et al. Inhaled Xenon Modulates Microglia and Ameliorates Disease in Mouse Models of Amyloidosis and Tauopathy. Sci. Transl. Med. 2025, 17, eadk3690. [Google Scholar] [CrossRef]
- Shao, L.; Dong, C.; Geng, D.; He, Q.; Shi, Y. Ginkgolide B Inactivates the NLRP3 Inflammasome by Promoting Autophagic Degradation to Improve Learning and Memory Impairment in Alzheimer’s Disease. Metab. Brain Dis. 2022, 37, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; Zoia, C.P.; La Rosa, F.; Bazzini, C.; Sala, G.; Grassenis, E.; Marventano, I.; Hernis, A.; Piancone, F.; Conti, E.; et al. Glibenclamide-Loaded Engineered Nanovectors (GNVs) Modulate Autophagy and NLRP3-Inflammasome Activation. Pharmaceuticals 2023, 16, 1725. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Jiang, Z.; Song, Y.; Huang, S.; Du, Y.; Yang, X.; Xiao, Y.; Ma, Z.; Xu, D.; Li, J. 3,4-Methylenedioxy-β-Nitrostyrene Alleviates Dextran Sulfate Sodium–Induced Mouse Colitis by Inhibiting the NLRP3 Inflammasome. Front. Pharmacol. 2022, 13, 866228. [Google Scholar] [CrossRef]
- Liu, B.; Li, X.; Yu, H.; Shi, X.; Zhou, Y.; Alvarez, S.; Naldrett, M.J.; Kachman, S.D.; Ro, S.-H.; Sun, X.; et al. Therapeutic Potential of Garlic Chive-Derived Vesicle-like Nanoparticles in NLRP3 Inflammasome-Mediated Inflammatory Diseases. Theranostics 2021, 11, 9311–9330. [Google Scholar] [CrossRef]
- Yu, Y.; Martins, L.M. Mitochondrial One-Carbon Metabolism and Alzheimer’s Disease. Int. J. Mol. Sci. 2024, 25, 6302. [Google Scholar] [CrossRef]
- Schneeberger, S.; Kim, S.J.; Geesdorf, M.N.; Friebel, E.; Eede, P.; Jendrach, M.; Boltengagen, A.; Braeuning, C.; Ruhwedel, T.; Hülsmeier, A.J.; et al. Interleukin-12 Signaling Drives Alzheimer’s Disease Pathology through Disrupting Neuronal and Oligodendrocyte Homeostasis. Nat. Aging 2025, 5, 622–641. [Google Scholar] [CrossRef]
- Samir, M.; Abdelkader, R.M.; Boushehri, M.S.; Mansour, S.; Lamprecht, A.; Tammam, S.N. Enhancement of Mitochondrial Function Using NO Releasing Nanoparticles; a Potential Approach for Therapy of Alzheimer’s Disease. Eur. J. Pharm. Biopharm. 2023, 184, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Phogat, A.; Kumar, V.; Malik, V. N-Acetylcysteine Ameliorates Monocrotophos Exposure-Induced Mitochondrial Dysfunctions in Rat Liver. Toxicol. Mech. Methods 2022, 32, 686–694. [Google Scholar] [CrossRef]
- Shi, H.-Z.; Wang, Y.-J.; Wang, Y.-X.; Xu, L.-F.; Pan, W.; Shi, L.; Wang, J. The Potential Benefits of PGC-1α in Treating Alzheimer’s Disease Are Dependent on the Integrity of the LLKYL L3 Motif: Effect of Regulating Mitochondrial Axonal Transportation. Exp. Gerontol. 2024, 194, 112514. [Google Scholar] [CrossRef]
- Pal, C. Mitochondria-Targeting by Small Molecules against Alzheimer’s Disease: A Mechanistic Perspective. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2025, 1871, 167617. [Google Scholar] [CrossRef]
- Chen, B.; Wu, J.; Hu, S.; Liu, Q.; Yang, H.; You, Y. Apelin-13 Improves Cognitive Impairment and Repairs Hippocampal Neuronal Damage by Activating PGC-1α/PPARγ Signaling. Neurochem. Res. 2023, 48, 1504–1515. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhou, P.; Zhao, Z.; Li, J.; Fan, Z.; Li, X.; Cui, Z.; Fu, A. Improvement Effect of Mitotherapy on the Cognitive Ability of Alzheimer’s Disease through NAD+/SIRT1-Mediated Autophagy. Antioxidants 2023, 12, 2006. [Google Scholar] [CrossRef]
- Ji, L.L.; Yeo, D.; Kang, C. Muscle Disuse Atrophy Caused by Discord of Intracellular Signaling. Antioxid. Redox Signal. 2020, 33, 727–744. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhang, H.; Yan, C.; Shen, B.; Zhang, Y.; Guo, X.; Sun, S.; Yu, F.; Yan, J.; Liu, R.; et al. Small Molecule Agonist of Mitochondrial Fusion Repairs Mitochondrial Dysfunction. Nat. Chem. Biol. 2023, 19, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Malar, D.S.; Thitilertdecha, P.; Ruckvongacheep, K.S.; Brimson, S.; Tencomnao, T.; Brimson, J.M. Targeting Sigma Receptors for the Treatment of Neurodegenerative and Neurodevelopmental Disorders. CNS Drugs 2023, 37, 399–440. [Google Scholar] [CrossRef]
- Hampel, H.; Williams, C.; Etcheto, A.; Goodsaid, F.; Parmentier, F.; Sallantin, J.; Kaufmann, W.E.; Missling, C.U.; Afshar, M. A Precision Medicine Framework Using Artificial Intelligence for the Identification and Confirmation of Genomic Biomarkers of Response to an Alzheimer’s Disease Therapy: Analysis of the Blarcamesine (ANAVEX2-73) Phase 2a Clinical Study. Alzheimers Dement. Transl. Res. Clin. Interv. 2020, 6, e12013. [Google Scholar] [CrossRef]
- Macfarlane, S.; Grimmer, T.; Teo, K.; O’Brien, T.J.; Woodward, M.; Grunfeld, J.; Mander, A.; Brodtmann, A.; Brew, B.J.; Morris, P.; et al. Blarcamesine for the Treatment of Early Alzheimer’s Disease: Results from the ANAVEX2-73-AD-004 Phase IIB/III Trial. J. Prev. Alzheimers Dis. 2025, 12, 100016. [Google Scholar] [CrossRef]
- Li, X.; Shi, Q.; Xu, H.; Xiong, Y.; Wang, C.; Le, L.; Lian, J.; Wu, G.; Peng, F.; Liu, Q.; et al. Ebselen Interferes with Alzheimer’s Disease by Regulating Mitochondrial Function. Antioxidants 2022, 11, 1350. [Google Scholar] [CrossRef]
- Pahlavani, H.A. Exercise Therapy to Prevent and Treat Alzheimer’s Disease. Front. Aging Neurosci. 2023, 15, 1243869. [Google Scholar] [CrossRef]
- Lin, T.-W.; Tsai, S.-F.; Kuo, Y.-M. Physical Exercise Enhances Neuroplasticity and Delays Alzheimer’s Disease. Brain Plast. 2018, 4, 95–110. [Google Scholar] [CrossRef]
- Lu, X.; Chen, Y.; Shi, Y.; Shi, Y.; Su, X.; Chen, P.; Wu, D.; Shi, H. Exercise and Exerkines: Mechanisms and Roles in Anti-Aging and Disease Prevention. Exp. Gerontol. 2025, 200, 112685. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, M.; Merchant, R.A.; Morley, J.E.; Anker, S.D.; Aprahamian, I.; Arai, H.; Aubertin-Leheudre, M.; Bernabei, R.; Cadore, E.L.; Cesari, M.; et al. International Exercise Recommendations in Older Adults (ICFSR): Expert Consensus Guidelines. J. Nutr. Health Aging 2021, 25, 824–853. [Google Scholar] [CrossRef]
- Hoscheidt, S.; Sanderlin, A.H.; Baker, L.D.; Jung, Y.; Lockhart, S.; Kellar, D.; Whitlow, C.T.; Hanson, A.J.; Friedman, S.; Register, T.; et al. Mediterranean and Western Diet Effects on Alzheimer’s Disease Biomarkers, Cerebral Perfusion, and Cognition in Mid-life: A Randomized Trial. Alzheimers Dement. 2022, 18, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Judd, J.M.; Jasbi, P.; Winslow, W.; Serrano, G.E.; Beach, T.G.; Klein-Seetharaman, J.; Velazquez, R. Inflammation and the Pathological Progression of Alzheimer’s Disease Are Associated with Low Circulating Choline Levels. Acta Neuropathol. 2023, 146, 565–583. [Google Scholar] [CrossRef]
- Falony, G.; Vandeputte, D.; Caenepeel, C.; Vieira-Silva, S.; Daryoush, T.; Vermeire, S.; Raes, J. The Human Microbiome in Health and Disease: Hype or Hope. Acta Clin. Belg. 2019, 74, 53–64. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Bäckhed, F. Signals from the Gut Microbiota to Distant Organs in Physiology and Disease. Nat. Med. 2016, 22, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Biagi, E.; Nylund, L.; Candela, M.; Ostan, R.; Bucci, L.; Pini, E.; Nikkïla, J.; Monti, D.; Satokari, R.; Franceschi, C.; et al. Through Ageing, and Beyond: Gut Microbiota and Inflammatory Status in Seniors and Centenarians. PLoS ONE 2010, 5, e10667. [Google Scholar] [CrossRef]
- Claesson, M.J.; Cusack, S.; O’Sullivan, O.; Greene-Diniz, R.; De Weerd, H.; Flannery, E.; Marchesi, J.R.; Falush, D.; Dinan, T.; Fitzgerald, G.; et al. Composition, Variability, and Temporal Stability of the Intestinal Microbiota of the Elderly. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4586–4591. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’Riordan, K.J.; Sandhu, K.; Peterson, V.; Dinan, T.G. The Gut Microbiome in Neurological Disorders. Lancet Neurol. 2020, 19, 179–194. [Google Scholar] [CrossRef]
- Duff, E.P.; Zetterberg, H.; Heslegrave, A.; Dehghan, A.; Elliott, P.; Allen, N.; Runz, H.; Laban, R.; Veleva, E.; Whelan, C.D.; et al. Plasma Proteomic Evidence for Increased β-Amyloid Pathology after SARS-CoV-2 Infection. Nat. Med. 2025, 31, 797–806. [Google Scholar] [CrossRef]
- Feng, Y.; Cao, S.-Q.; Shi, Y.; Sun, A.; Flanagan, M.E.; Leverenz, J.B.; Pieper, A.A.; Jung, J.U.; Cummings, J.; Fang, E.F.; et al. Human Herpesvirus-Associated Transposable Element Activation in Human Aging Brains with Alzheimer’s Disease. Alzheimers Dement. 2025, 21, e14595. [Google Scholar] [CrossRef] [PubMed]
- Bigio, B.; Lima-Filho, R.A.S.; Barnhill, O.; Sudo, F.K.; Drummond, C.; Assunção, N.; Vanderborght, B.; Beasley, J.; Young, S.; Korman, A.; et al. Sex Differences in Mitochondrial Free-Carnitine Levels in Subjects at-Risk and with Alzheimer’s Disease in Two Independent Study Cohorts. Mol. Psychiatry 2025, 30, 2573–2583. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Tan, E.C.K.; Goudey, B.; Jin, L.; Pan, Y. Unraveling the Bidirectional Link between Cancer and Dementia and the Impact of Cancer Therapies on Dementia Risk: A Systematic Review and Meta-analysis. Alzheimers Dement. 2025, 21, e14540. [Google Scholar] [CrossRef] [PubMed]
- Nihart, A.J.; Garcia, M.A.; El Hayek, E.; Liu, R.; Olewine, M.; Kingston, J.D.; Castillo, E.F.; Gullapalli, R.R.; Howard, T.; Bleske, B.; et al. Bioaccumulation of Microplastics in Decedent Human Brains. Nat. Med. 2025, 31, 1114–1119. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |

{kind=link}
{kind=link}
| Model | Animals | Method | Refs. |
|---|---|---|---|
| Natural aging models | Nematode worm | Natural growth to senescence stage; typical lifespan: 1 month. | [153] |
| Fruit fly | Natural growth to senescence; typical lifespan: 3 months. | [154] | |
| Danio rerio | Natural growth to senescence; typical life span: 36–42 months. | [155] | |
| Mice | Natural growth to senescence; typical life span: 2–4 years. | [144,156,157] | |
| Accelerated-aging model | Mice | Rapidly aging (SAMP) mice; rapidly developing aging characteristics after 4–6 months of age. | [152] |
| Rats | D-galactose induction | [158,159] | |
| Gene editing models of aging | African killifish (Medaka) | Breeding with gene editing reduces lifespan to 9–26 weeks. | [160] |
| Danio rerio | Telomerase-deficient; median life expectancy reduced to 9 months. | [139] | |
| Mice | Rps9 D95N mutation | [161] | |
| Hyperactivation of the tumor suppressor P53. | [162] | ||
| INK-ATTAC type (knockout P16) | [163] | ||
| PolgA gene proofreading functionally defective phenotype. | [164] | ||
| hAPP transgenic AD model | Mice | PDAPP type (V717F mutation introduced in APP) | [142] |
| Tg2576. (Introduction of the K670N/M671L mutation in APP) | [143,144] | ||
| APP23 type. (K670N/M671L and V717I double mutation in the APP gene) | [145] | ||
| TgCRND8 type (K670N/M671L and V717I double mutation in APP gene) | [146,147] | ||
| Aβ transgenic AD model | Mice | BRI-Aβ42 Type A (BRI protein was used as a carrier to fuse Aβ42 to its C-terminus) | [148] |
| APP/precocene double-turn AD model | Mice | PSAPP type (Tg2576 × PSI) | [165] |
| APPswe/PS1 ΔE9 type. (synergistic expression of APP with the K670N/M671L double-site mutation and deletion of exon 9 of PSEN1 (ΔE9)) | [54,166,167] | ||
| 5 × FAD type (APP triple mutation and PSEN1 double mutation hybridization) | [168,169] | ||
| 2 × KI type (APP and PSEN1 double knock-in mutation) | [170,171] | ||
| hTau transgenic AD model | Mice | JNPL3 type (introduction of the tau gene carrying the P301L mutation) | [149] |
| 3 × Tg type (Integration of APP double mutation, single mutation in PSEN1 gene, triple mutation in Tau gene P301L) | [150] | ||
| TAPP type (Tg2576 × JNPL3) | [151] | ||
| Aging-AD hybrid model | Mice | D-galactose induction | [141] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Zhu, Z.; Xu, Y. Signs of Alzheimer’s Disease: Tied to Aging. Int. J. Mol. Sci. 2025, 26, 4974. https://doi.org/10.3390/ijms26114974
Chen J, Zhu Z, Xu Y. Signs of Alzheimer’s Disease: Tied to Aging. International Journal of Molecular Sciences. 2025; 26(11):4974. https://doi.org/10.3390/ijms26114974
Chicago/Turabian StyleChen, Jiahui, Zhongying Zhu, and Yuanyuan Xu. 2025. "Signs of Alzheimer’s Disease: Tied to Aging" International Journal of Molecular Sciences 26, no. 11: 4974. https://doi.org/10.3390/ijms26114974
APA StyleChen, J., Zhu, Z., & Xu, Y. (2025). Signs of Alzheimer’s Disease: Tied to Aging. International Journal of Molecular Sciences, 26(11), 4974. https://doi.org/10.3390/ijms26114974