
Distribution of NECAB1-Positive Neurons in Normal and Epileptic Brain—Expression Changes in Temporal Lobe Epilepsy and Modulation by Levetiracetam and Brivaracetam
 , , , ,
, , , ,  , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Mapping of NECAB1 Expression and Its Colocalization with PV and CR in Physiological Conditions Along the Epileptic Circuitry
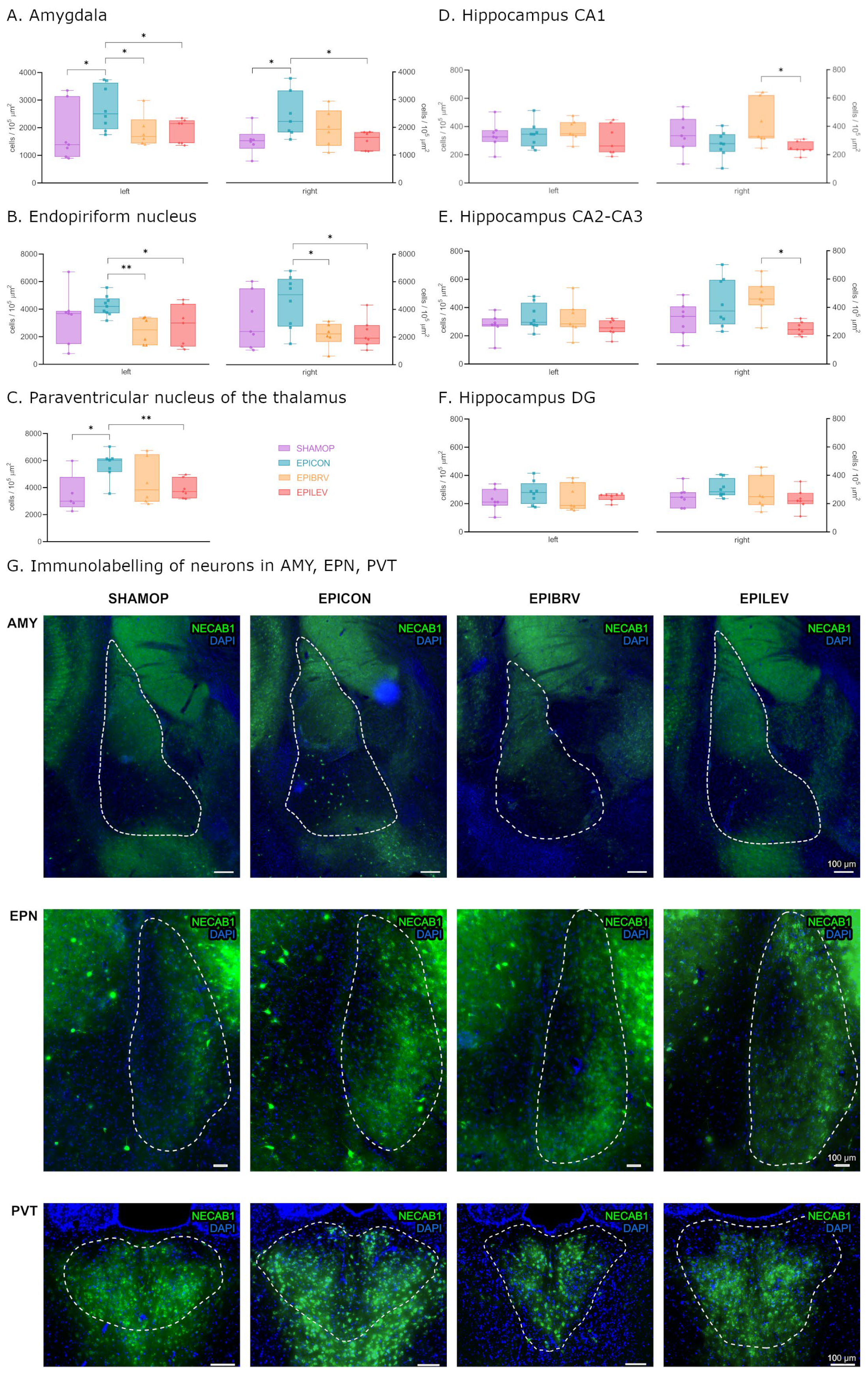
2.2. Epilepsy Increases the Density of NECAB1+ Cells in Several Brain Regions, Which Is Attenuated by the Antiseizure Medications Brivaracetam and Levetiracetam
2.3. Comparative Analysis of NECAB1 Expression Patterns in Brain Regions in the Context of Epilepsy and ASM Treatment
3. Discussion
4. Materials and Methods
4.1. Ethical Approval and Compliance
4.2. Animals
4.3. Epilepsy Induction
4.4. Pharmacological Interventions
4.5. Tissue Processing for Immunohistochemistry
4.6. Immunohistochemical Staining
4.7. Image Acquisition and Analysis
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMY | Amygdala |
| ASMs | Antiseizure medication |
| BRV | Brivaracetam |
| CaBPs | Calcium-binding proteins |
| CB1 | Cannabinoid receptor 1 |
| CCK | Cholecystokinin |
| CNS | Central nervous system |
| CR | Calretinin |
| DG | Dentate gyrus |
| EPIBRV | Brivaracetam-treated |
| EPICON | Epileptic control |
| EPILEV | Levetiracetam-treated |
| EPN | Endopiriform nucleus |
| GABA | Gamma-aminobutyric acid |
| KA | Kainic acid |
| LEV | Levetiracetam |
| mAChRs | Muscarinic acetylcholine receptors |
| N1 | NECAB1 |
| N1-CR | NECAB1–calretinin co-expressing cells |
| N1-PV | NECAB1–parvalbumin co-expressing cell |
| N1-PV-CR | NECAB1–parvalbumin–calretinin co-expressing cells |
| NECAB1 | N-Terminal EF-Hand Calcium-Binding Protein 1 |
| PV | Parvalbumin |
| PVT | Paraventricular nucleus of the thalamus |
| SHAMOP | Sham-operated group |
| SV2A | Synaptic vesicle glycoprotein 2A |
| Syt1 | Synaptotagmin-1 |
| TLE | Temporal lobe epilepsy |
References
- Trevelyan, A.J.; Muldoon, S.F.; Merricks, E.M.; Racca, C.; Staley, K.J. The Role of Inhibition in Epileptic Networks. J. Clin. Neurophysiol. 2015, 32, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Royer, J.; Rodriguez-Cruces, R.; Horwood, L.; Ngo, A.; Arafat, T.; Auer, H.; Sahlas, E.; Chen, J.; Zhou, Y.; et al. Temporal Lobe Epilepsy Perturbs the Brain-Wide Excitation-Inhibition Balance: Associations with Microcircuit Organization, Clinical Parameters, and Cognitive Dysfunction. Adv. Sci. 2025, 12, 2406835. [Google Scholar] [CrossRef]
- Engel, J. Mesial Temporal Lobe Epilepsy: What Have We Learned? Neuroscient 2001, 7, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat. Rev. Neurosci. 2005, 6, 591–602. [Google Scholar] [CrossRef]
- Engel, J.; Pedley, T.A.; Aicardi, J. (Eds.) Epilepsy: A Comprehensive Textbook, 2nd ed.; Lippincott Williams & Wilkins, A Wolters Kluwer Business: Philadelphia, PA, USA, 2007. [Google Scholar]
- Roux, L.; Buzsáki, G. Tasks for inhibitory interneurons in intact brain circuits. Neuropharmacology 2015, 88, 10–23. [Google Scholar] [CrossRef]
- Cossart, R.; Bernard, C.; Ben-Ari, Y. Multiple facets of GABAergic neurons and synapses: Multiple fates of GABA signalling in epilepsies. Trends Neurosci. 2005, 28, 108–115. [Google Scholar] [CrossRef]
- Baimbridge, K.G.; Celio, M.R.; Rogers, J.H. Calcium-binding proteins in the nervous system. Trends Neurosci. 1992, 15, 303–308. [Google Scholar] [CrossRef]
- Schwaller, B. The continuing disappearance of “pure” Ca2+ buffers. Cell. Mol. Life Sci. 2009, 66, 275–300. [Google Scholar] [CrossRef]
- Burgoyne, R.D.; Weiss, J.L. The neuronal calcium sensor family of Ca2+-binding proteins. Biochem. J. 2001, 353, 1–12. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Miczán, V.; Kelemen, K.; Glavinics, J.R.; László, Z.I.; Barti, B.; Kenesei, K.; Kisfali, M.; Katona, I. Necab1 and necab2 are prevalent calcium-binding proteins of cb1/cck-positive gabaergic interneurons. Cereb. Cortex 2021, 31, 1786–1806. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Wang, X.; Chen, Y.; Qiu, W.; Ge, W.; Ma, C. Proteomic and Transcriptomic Analyses Reveal Pathological Changes in the Entorhinal Cortex Region that Correlate Well with Dysregulation of Ion Transport in Patients with Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 4007–4027. [Google Scholar] [CrossRef] [PubMed]
- Underwood, R.; Gannon, M.; Pathak, A.; Kapa, N.; Chandra, S.; Klop, A.; Yacoubian, T.A. 14-3-3 mitigates alpha-synuclein aggregation and toxicity in the in vivo preformed fibril model. Acta Neuropathol. Commun. 2021, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Kornilov, S.A.; Rakhlin, N.; Koposov, R.; Lee, M.; Yrigollen, C.; Caglayan, A.O.; Magnuson, J.S.; Mane, S.; Chang, J.T.; Grigorenko, E.L. Genome-Wide Association and Exome Sequencing Study of Language Disorder in an Isolated Population. Pediatrics 2016, 137, e20152469. [Google Scholar] [CrossRef]
- Zhang, M.W.; Liang, X.Y.; Wang, J.; Gao, L.D.; Liao, H.J.; He, Y.H.; Yi, Y.-H.; He, N.; Liao, W.P. Epilepsy-Associated Genes: An Update. Seizure: Eur. J. Epilepsy 2024, 116, 4–13. [Google Scholar] [CrossRef]
- Sik, A.; Penttonen, M.; Buzsáki, G. Interneurons in the Hippocampal Dentate Gyrus: An In Vivo intracellular Study. Eur. J. Neurosci. 1997, 9, 573–588. [Google Scholar] [CrossRef]
- Löscher, W.; Brandt, C. Prevention or Modification of Epileptogenesis after Brain Insults: Experimental Approaches and Translational Research. Pharmacol. Rev. 2010, 62, 668–700. [Google Scholar] [CrossRef]
- Löscher, W.; Gillard, M.; Sands, Z.A.; Kaminski, R.M.; Klitgaard, H. Synaptic Vesicle Glycoprotein 2A Ligands in the Treatment of Epilepsy and Beyond. CNS Drugs 2016, 30, 1055–1077. [Google Scholar] [CrossRef]
- Itoh, K.; Inamine, M.; Oshima, W.; Kotani, M.; Chiba, Y.; Ueno, M.; Ishihara, Y. Prevention of status epilepticus-induced brain edema and neuronal cell loss by repeated treatment with high-dose levetiracetam. Brain Res. 2015, 1608, 225–234. [Google Scholar] [CrossRef]
- Bertram, E.H. Neuronal circuits in epilepsy: Do they matter? Exp. Neurol. 2013, 244, 67–74. [Google Scholar] [CrossRef]
- Kelemen, K.; Szilágyi, T. New approach for untangling the role of uncommon calcium-binding proteins in the central nervous system. Brain Sci. 2021, 11, 634. [Google Scholar] [CrossRef] [PubMed]
- Maliković, J.; Amrein, I.; Vinciguerra, L.; Wolfer, D.P.; Slomianka, L. NECAB1-3, parvalbumin, calbindin, and calretinin in the hippocampus of the European mole. Front. Neuroanat. 2024, 18, 1452722. [Google Scholar] [CrossRef] [PubMed]
- Butler, R.K.; Oliver, E.M.; Fadel, J.R.; Wilson, M.A. Hemispheric differences in the number of parvalbumin-positive neurons in subdivisions of the rat basolateral amygdala complex. Brain Res. 2018, 1678, 214–219. [Google Scholar] [CrossRef]
- Bakken, T.E.; van Velthoven, C.T.; Menon, V.; Hodge, R.D.; Yao, Z.; Nguyen, T.N.; Graybuck, L.T.; Horwitz, G.D.; Bertagnolli, D.; Goldy, J.; et al. Single-cell and single-nucleus RNA-seq uncovers shared and distinct axes of variation in dorsal LGN neurons in mice, non-human primates, and humans. Elife 2021, 10, e64875. [Google Scholar] [CrossRef]
- Viney, T.J.; Lasztoczi, B.; Katona, L.; Crump, M.G.; Tukker, J.J.; Klausberger, T.; Somogyi, P. Network state-dependent inhibition of identified hippocampal CA3 axo-axonic cells in vivo. Nat. Neurosci. 2013, 16, 1802–1811. [Google Scholar] [CrossRef]
- Roy, D.S.; Zhang, Y.; Halassa, M.M.; Feng, G. Thalamic Subnetworks as Units of Function. Nat. Neurosci. 2022, 25, 140–153. [Google Scholar] [CrossRef]
- Phillips, J.W.; Schulmann, A.; Hara, E.; Winnubst, J.; Liu, C.; Valakh, V.; Wang, L.; Shields, B.C.; Korff, W.; Chandrashekar, J.; et al. A repeated molecular architecture across thalamic pathways. Nat. Neurosci. 2019, 22, 1925–1935. [Google Scholar] [CrossRef]
- Kapustina, M.; Zhang, A.A.; Tsai, J.Y.J.; Bristow, B.N.; Kraus, L.; Sullivan, K.E.; Erwin, S.R.; Wang, L.; Stach, T.R.; Clements, J.; et al. The cell-type-specific spatial organization of the anterior thalamic nuclei of the mouse brain. Cell Rep. 2024, 43, 113842. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, Z.; Xiao, L.; Zhong, Y.; Liu, Y.; Wu, J.; Tao, H. Intracellular calcium homeostasis and its dysregulation underlying epileptic seizures. Seizure Eur. J. Epilepsy 2022, 103, 126–136. [Google Scholar] [CrossRef]
- Xu, J.-H.; Tang, F.-R. Voltage-Dependent Calcium Channels, Calcium Binding Proteins, and Their Interaction in the Pathological Process of Epilepsy. Int. J. Mol. Sci. 2018, 19, 2735. [Google Scholar] [CrossRef]
- Thomas, N.L.; Dart, C.; Helassa, N. Editorial: The role of calcium and calcium binding proteins in cell physiology and disease. Front. Physiol. 2023, 14, 1228885. [Google Scholar] [CrossRef] [PubMed]
- Chazin, W.J. Relating Form and Function of EF-Hand Calcium Binding Proteins. Acc. Chem. Res. 2011, 44, 171–179. [Google Scholar] [CrossRef]
- Schwaller, B. Cytosolic Ca2+ Buffers. Cold Spring Harb. Perspect. Biol. 2010, 2, a004051. [Google Scholar] [CrossRef]
- Kuchukhidze, G.; Wieselthaler-Hölzl, A.; Drexel, M.; Unterberger, I.; Luef, G.; Ortler, M.; Becker, A.J.; Trinka, E.; Sperk, G. Calcium-binding proteins in focal cortical dysplasia. Epilepsia 2015, 56, 1207–1216. [Google Scholar] [CrossRef]
- Mihály, I.; Molnár, T.; Berki, Á.-J.; Bod, R.-B.; Orbán-Kis, K.; Gáll, Z.; Szilágyi, T. Short-Term Amygdala Low-Frequency Stimulation Does not Influence Hippocampal Interneuron Changes Observed in the Pilocarpine Model of Epilepsy. Cells 2021, 10, 520. [Google Scholar] [CrossRef]
- Godoy, L.D.; Prizon, T.; Rossignoli, M.T.; Leite, J.P.; Liberato, J.L. Parvalbumin Role in Epilepsy and Psychiatric Comorbidities: From Mechanism to Intervention. Front. Integr. Neurosci. 2022, 16, 765324. [Google Scholar] [CrossRef]
- Maglóczky, Z.; Tóth, K.; Karlócai, R.; Nagy, S.; Erőss, L.; Czirják, S.; Vajda, J.; Rásonyi, G.; Kelemen, A.; Juhos, V.; et al. Dynamic changes of CB1-receptor expression in hippocampi of epileptic mice and humans. Epilepsia 2010, 51, 115–120. [Google Scholar] [CrossRef]
- Karlócai, M.R.; Tóth, K.; Watanabe, M.; Ledent, C.; Juhász, G.; Freund, T.F.; Maglóczky, Z. Redistribution of CB1 Cannabinoid Receptors in the Acute and Chronic Phases of Pilocarpine-Induced Epilepsy. PLoS ONE 2011, 6, e27196. [Google Scholar] [CrossRef]
- Lazarini-Lopes, W.; da Silva-Júnior, R.M.P.; Servilha-Menezes, G.; Do Val-da Silva, R.A.; Garcia-Cairasco, N. Cannabinoid Receptor Type 1 (CB1R) Expression in Limbic Brain Structures After Acute and Chronic Seizures in a Genetic Model of Epilepsy. Front. Behav. Neurosci. 2020, 14, 602258. [Google Scholar] [CrossRef]
- Soltesz, I.; Alger, B.E.; Kano, M.; Lee, S.-H.; Lovinger, D.M.; Ohno-Shosaku, T.; Watanabe, M. Weeding out bad waves: Towards selective cannabinoid circuit control in epilepsy. Nat. Rev. Neurosci. 2015, 16, 264–277. [Google Scholar] [CrossRef]
- Ludányi, A.; Eross, L.; Czirják, S. Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. J. Neurosci. 2008, 28, 2976–2990. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Cheng, H.; Wang, Y.; Chen, Z. Revealing the Precise Role of Calretinin Neurons in Epilepsy: We Are on the Way. Neurosci. Bull. 2022, 38, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Toth, K.; Eross, L.; Vajda, J.; Halasz, P.; Freund, T.F.; Magloczky, Z. Loss and reorganization of calretinin-containing interneurons in the epileptic human hippocampus. Brain 2010, 133, 2763–2777. [Google Scholar] [CrossRef] [PubMed]
- Navarro, D.; Alvarado, M.; Figueroa, A.; Gonzalez-Liencres, C.; Salas-Lucia, F.; Pacheco, P.; Sanzhez-Vives, M.V.; Berbel, P. Distribution of GABAergic Neurons and VGluT1 and VGAT Immunoreactive Boutons in the Ferret (Mustela putorius) Piriform Cortex and Endopiriform Nucleus. Comparison With Visual Areas 17, 18 and 19. Front. Neuroanat. 2019, 13, 54. [Google Scholar] [CrossRef]
- Crepeau, A.Z.; Treiman, D.M. Levetiracetam: A comprehensive review. Expert. Rev. Neurother. 2010, 10, 159–171. [Google Scholar] [CrossRef]
- Klein, P.; Diaz, A.; Gasalla, T.; Whitesides, J. A review of the pharmacology and clinical efficacy of brivaracetam. Clin. Pharmacol. 2018, 10, 1–22. [Google Scholar] [CrossRef]
- Kelemen, K.; Sárosi, M.; Csüdör, Á.; Orbán-Kis, K.; Kelemen, H.; Bába, L.; Gáll, Z.; Horváth, E.; Katona, I.; Szilágyi, T. Marked differences in the effects of levetiracetam and its analogue brivaracetam on microglial, astrocytic, and neuronal density in the rat model of kainic acid-induced temporal lobe epilepsy. Front. Pharmacol. 2025, 16, 888784. [Google Scholar] [CrossRef]
- Hogg, J.A.; Cousin, M.A. Control of Synaptotagmin-1 Trafficking by SV2A—Mechanism and Consequences for Presynaptic Function and Dysfunction. J. Neurochem. 2025, 169, e16308. [Google Scholar] [CrossRef]
- Schurmans, S.; Schiffmann, S.N.; Gurden, H.; Lemaire, M.; Lipp, H.-P.; Schwam, V.; Pochet, R.; Imperato, A.; Böhme, G.A.; Parmentier, M. Impaired long-term potentiation induction in dentate gyrus of calretinin-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 10415–10420. [Google Scholar] [CrossRef]
- Maglóczky Zs Halász, P.; Vajda, J.; Czirják, S.; Freund, T.F. Loss of Calbindin-D28K immunoreactivity from dentate granule cells in human temporal lobe epilepsy. Neuroscience 1997, 76, 377–385. [Google Scholar] [CrossRef]
- Sloviter, R.S.; Zappone, C.A.; Harvey, B.D.; Bumanglag, A.V.; Bender, R.A.; Frotscher, M. “Dormant basket cell” hypothesis revisited: Relative vulnerabilities of dentate gyrus mossy cells and inhibitory interneurons after hippocampal status epilepticus in the rat. J. Comp. Neurol. 2003, 459, 44–76. [Google Scholar] [CrossRef] [PubMed]
- Wittner, L.; Maglóczky, Z.; Borhegyi, Z.; Halász, P.; Tóth, S.; Erőss, L.; Szabó, Z.; Freund, T.F. Preservation of perisomatic inhibitory input of granule cells in the epileptic human dentate gyrus. Neuroscience 2001, 108, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Hay, Y.A.; Jarzebowski, P.; Zhang, Y.; Digby, R.; Brendel, V.; Paulsen, O.; Paulsen, O.; Magloire, V. Cholinergic modulation of Up–Down states in the mouse medial entorhinal cortex in vitro. Eur. J. Neurosci. 2021, 53, 1378–1393. [Google Scholar] [CrossRef] [PubMed]
- Unal, G.; Joshi, A.; Viney, T.J.; Kis, V.; Somogyi, P. Synaptic Targets of Medial Septal Projections in the Hippocampus and Extrahippocampal Cortices of the Mouse. J. Neurosci. 2015, 35, 15812–15826. [Google Scholar] [CrossRef]
- Rusina, E.; Bernard, C.; Williamson, A. The kainic acid models of temporal lobe epilepsy. ENeuro 2021, 8, ENEURO.0337-20.2021. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C.; Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates: Hard Cover Edition, 6th ed.; Academic Press: London, UK, 2007. [Google Scholar]
- Lüttjohann, A.; Fabene, P.F.; van Luijtelaar, G. A revised Racine’s scale for PTZ-induced seizures in rats. Physiol. Behav. 2009, 98, 579–586. [Google Scholar] [CrossRef]
- Bába, L.I.; Gáll, Z.; Kolcsár, M.; Pap, Z.; Varga, Z.V.; Kovács, B.; Hack, B.; Kun, I.-Z. Effect on body weight and adipose tissue by cariprazine: A head-to-head comparison study to olanzapine and aripiprazole in rats. Sci. Pharm. 2020, 88, 50. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % | Left | Right | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CA1 | CA2-3 | DG | EPN | AMY | PVT | AMY | EPN | DG | CA2-3 | CA1 | |
| N1 | 54.25 | 62.59 | 80.62 | 98.69 | 95.64 | 41.00 | 94.13 | 98.91 | 76.23 | 64.03 | 53.84 |
| N1-PV | 7.67 | 1.53 | 1.28 | 0.92 | 0.94 | 0.00 | 1.58 | 1.09 | 0.00 | 4.87 | 10.64 |
| N1-CR | 31.69 | 33.44 | 18.10 | 0.39 | 3.42 | 59.00 | 4.29 | 0.00 | 23.77 | 29.11 | 29.78 |
| N1-PV-CR | 6.40 | 2.44 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 1.98 | 5.74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kelemen, K.; Orbán-Kis, K.; Szentes, Á.; Nagy, Z.A.; Kelemen, H.; Fehér, A.; Bába, L.-I.; Gáll, Z.; Horváth, E.; Katona, I.; et al. Distribution of NECAB1-Positive Neurons in Normal and Epileptic Brain—Expression Changes in Temporal Lobe Epilepsy and Modulation by Levetiracetam and Brivaracetam. Int. J. Mol. Sci. 2025, 26, 4906. https://doi.org/10.3390/ijms26104906
Kelemen K, Orbán-Kis K, Szentes Á, Nagy ZA, Kelemen H, Fehér A, Bába L-I, Gáll Z, Horváth E, Katona I, et al. Distribution of NECAB1-Positive Neurons in Normal and Epileptic Brain—Expression Changes in Temporal Lobe Epilepsy and Modulation by Levetiracetam and Brivaracetam. International Journal of Molecular Sciences. 2025; 26(10):4906. https://doi.org/10.3390/ijms26104906
Chicago/Turabian StyleKelemen, Krisztina, Károly Orbán-Kis, Ádám Szentes, Zsolt András Nagy, Hanga Kelemen, Anna Fehér, László-István Bába, Zsolt Gáll, Eszter Horváth, István Katona, and et al. 2025. "Distribution of NECAB1-Positive Neurons in Normal and Epileptic Brain—Expression Changes in Temporal Lobe Epilepsy and Modulation by Levetiracetam and Brivaracetam" International Journal of Molecular Sciences 26, no. 10: 4906. https://doi.org/10.3390/ijms26104906
APA StyleKelemen, K., Orbán-Kis, K., Szentes, Á., Nagy, Z. A., Kelemen, H., Fehér, A., Bába, L.-I., Gáll, Z., Horváth, E., Katona, I., Szatmári, S., Szász, J. A., & Szilágyi, T. (2025). Distribution of NECAB1-Positive Neurons in Normal and Epileptic Brain—Expression Changes in Temporal Lobe Epilepsy and Modulation by Levetiracetam and Brivaracetam. International Journal of Molecular Sciences, 26(10), 4906. https://doi.org/10.3390/ijms26104906