
Effect of Corticosterone on Gene Expression in the Context of Global Hippocampal Transcription

 , , and
, , and
Abstract
1. Introduction
2. Results
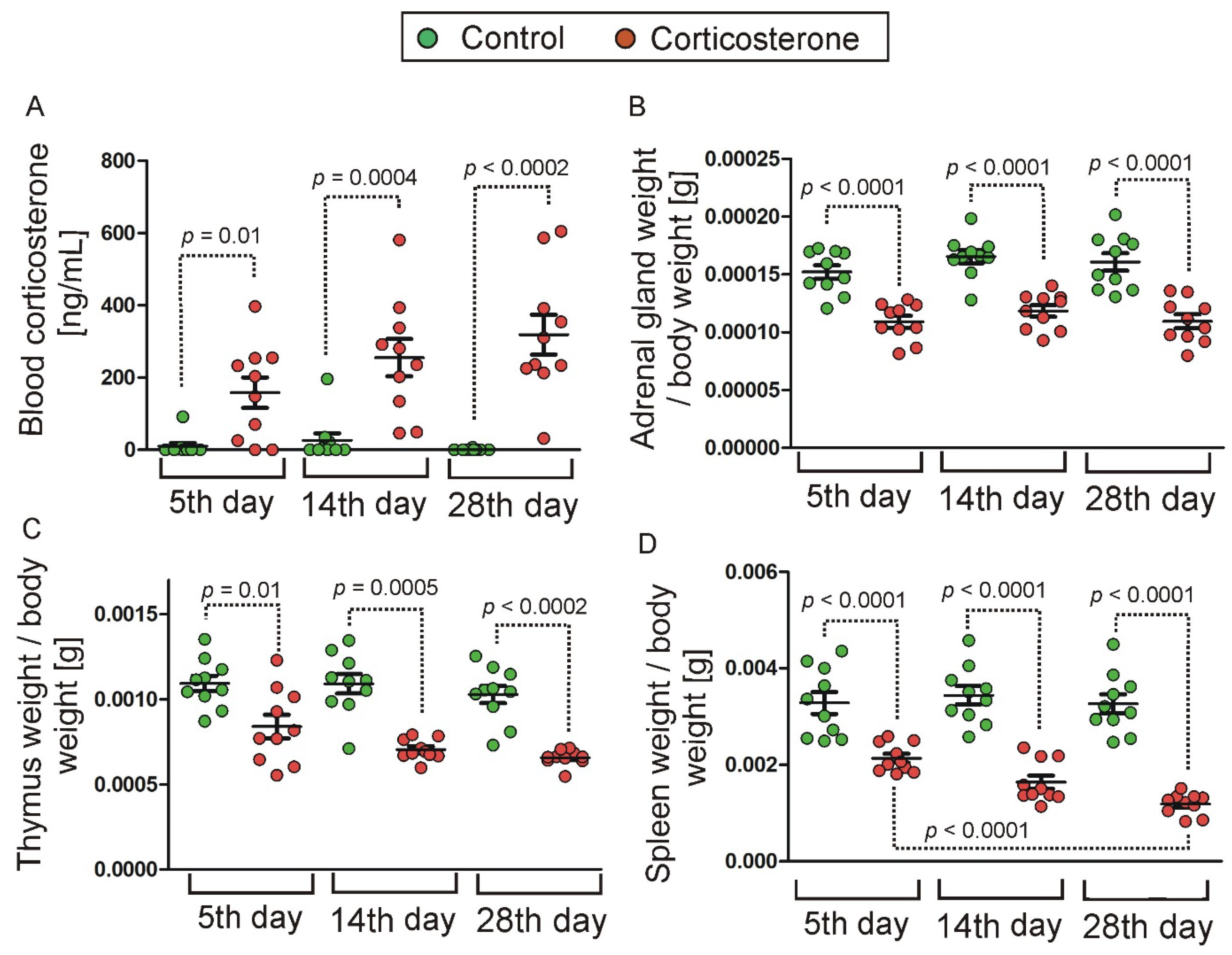
2.1. Effect of Treatment with Corticosterone on Blood Level of Corticosterone and Weight of Internal Organs
2.2. Global Assessment of Hippocampal Transcriptome
2.2.1. Validation of the Dataset
2.2.2. Summary of Hippocampal Transcriptome
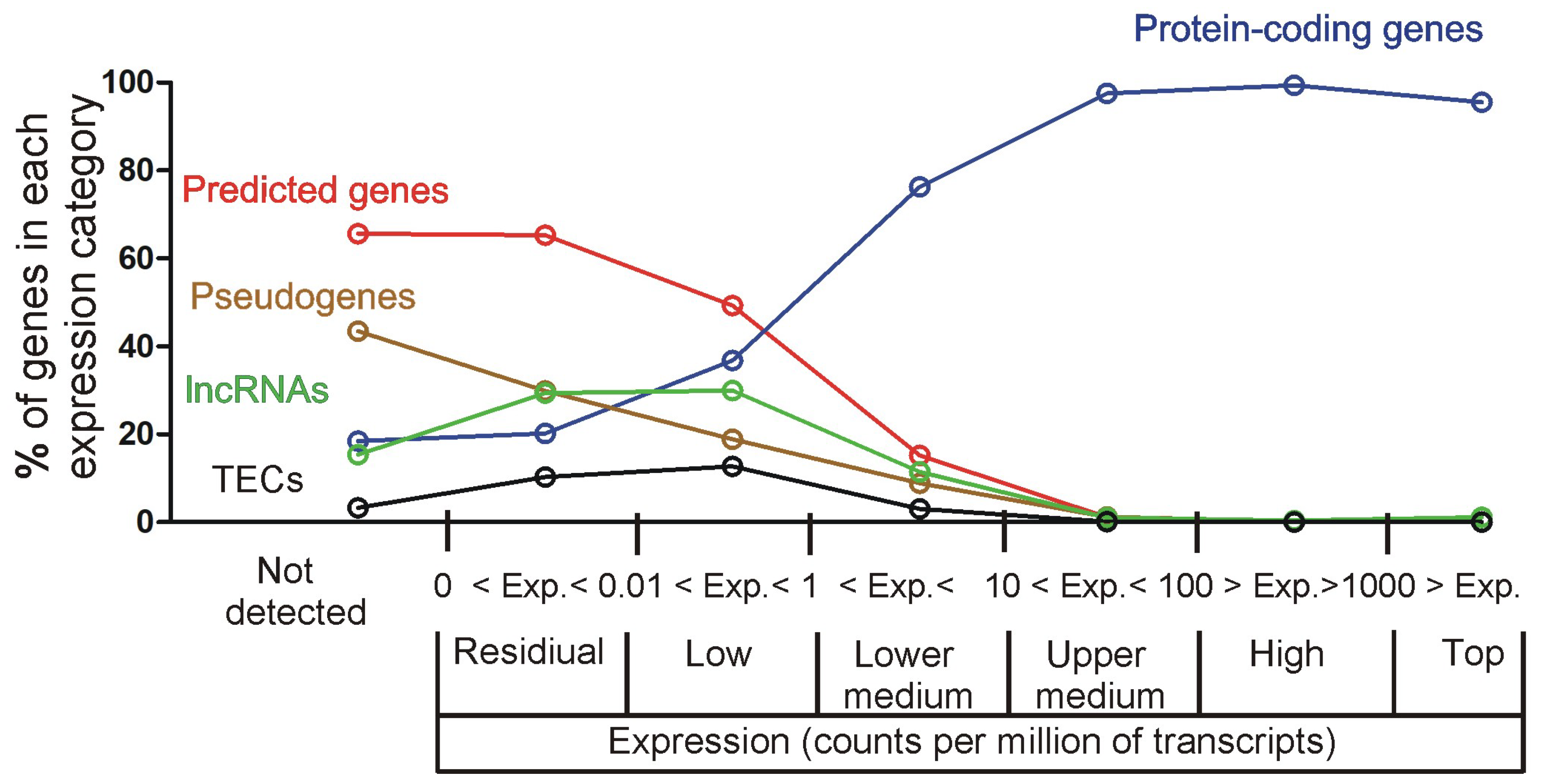
2.2.3. Determining the Lower Threshold of Biologically Meaningful Expression
2.2.4. Characteristics of Expression Categories
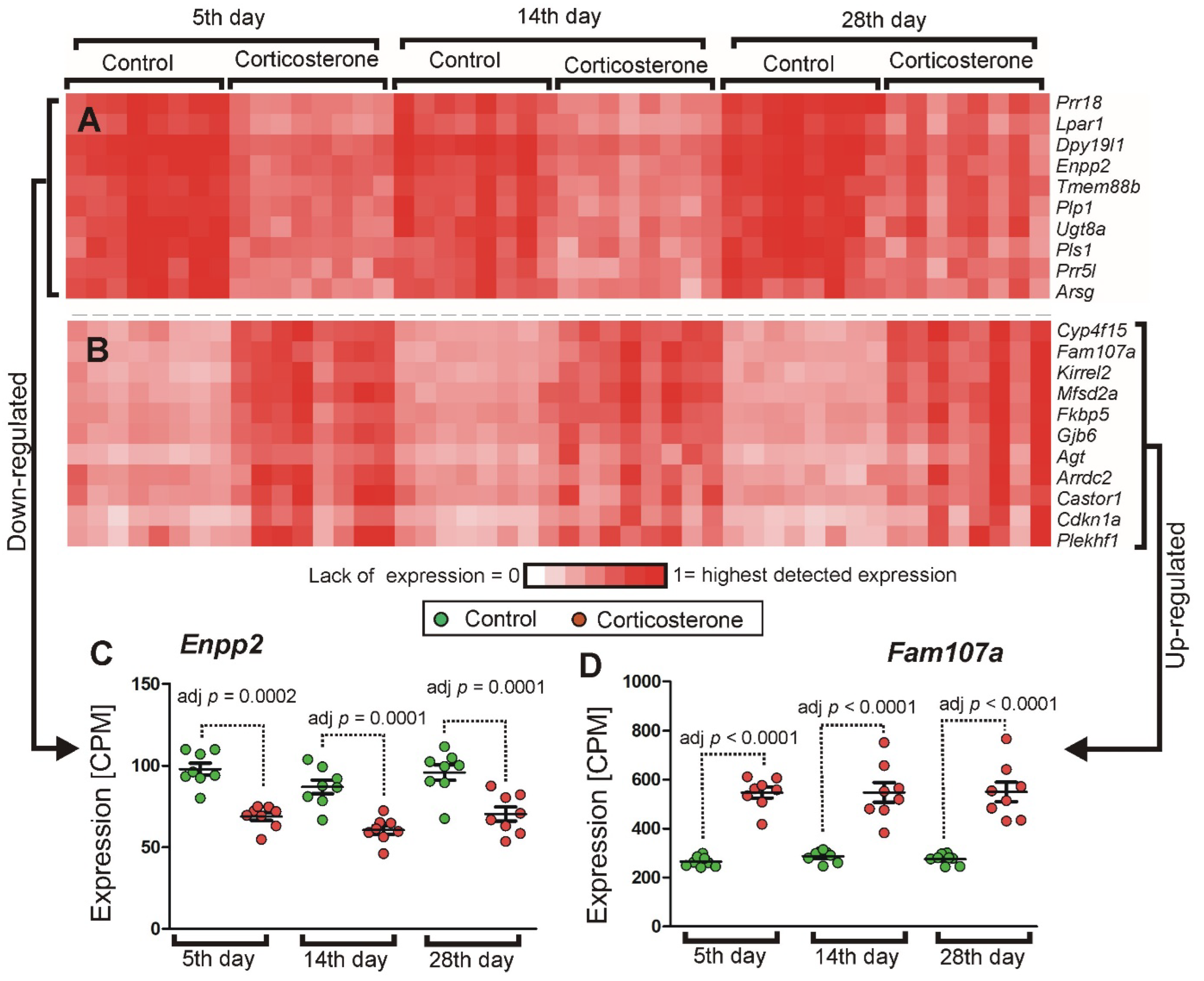
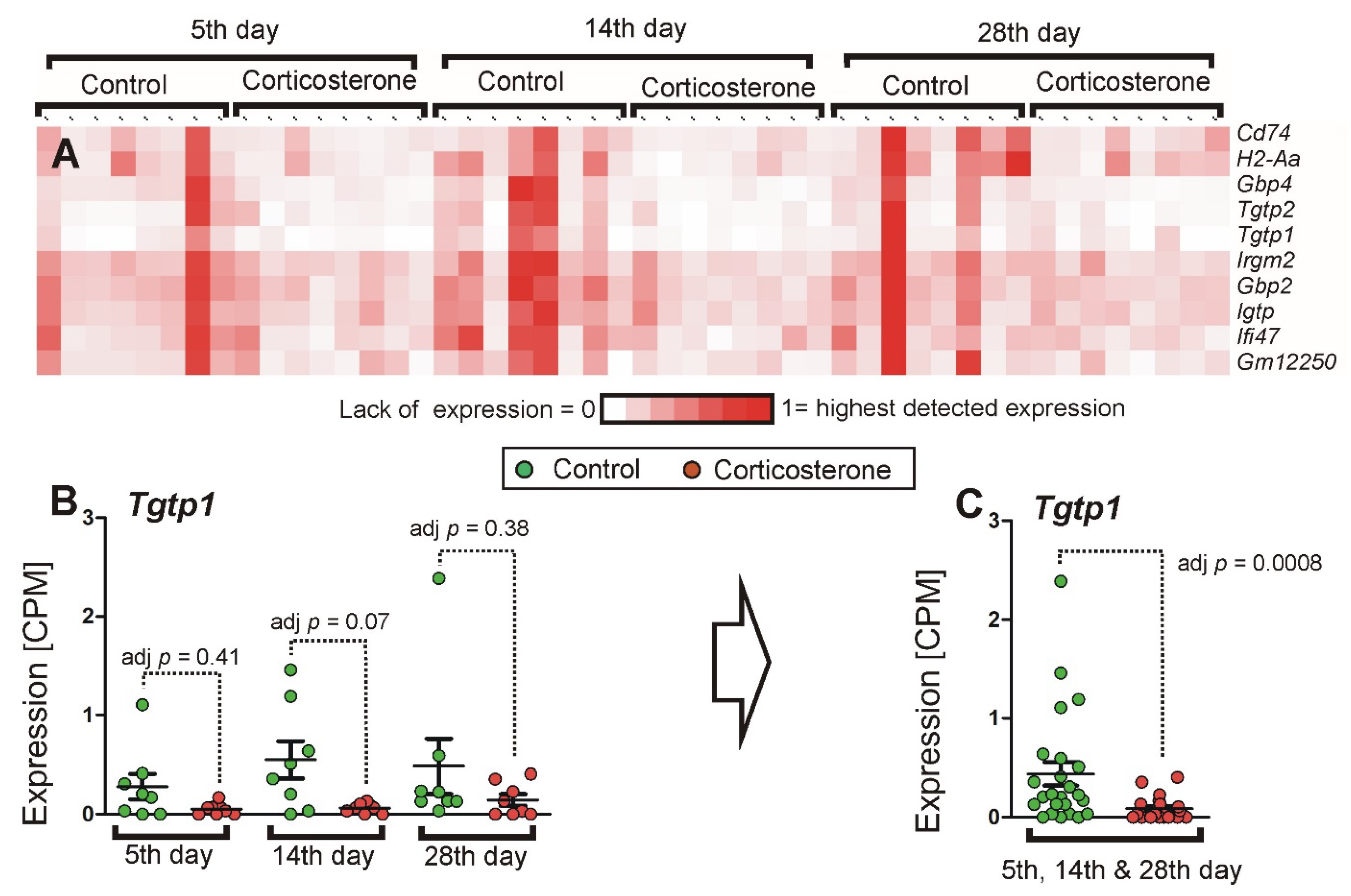
2.3. Effect of Corticosterone on Hippocampal Transcriptome
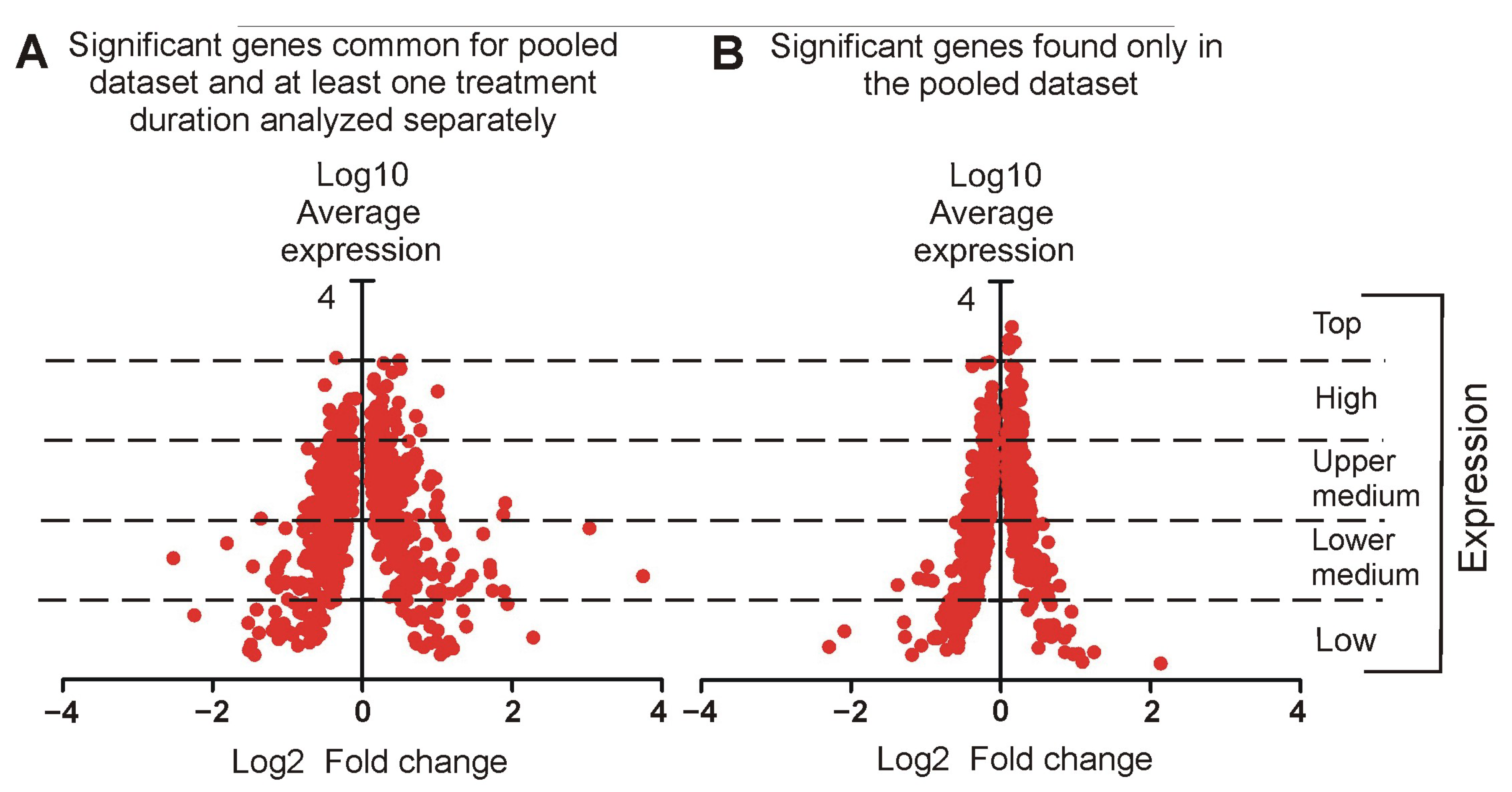
2.3.1. General Characteristics of Hippocampal Transcriptome
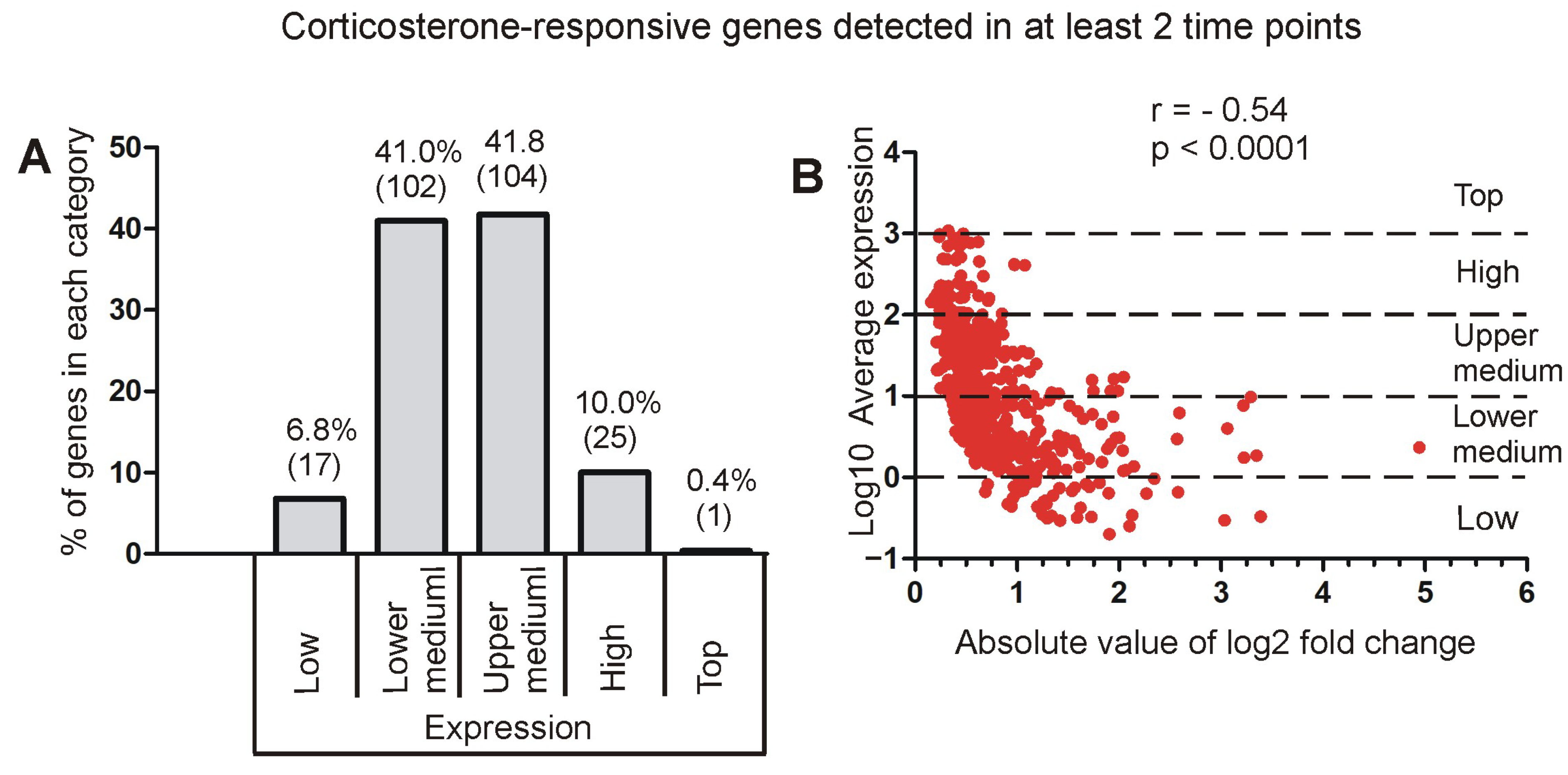
2.3.2. Relationship Between Fold Changes and Average Expression Level
2.3.3. Assessment of Cell Expression Specificity
2.3.4. Cluster Analysis
2.3.5. The Effect of Corticosterone on Total Transcriptomic Activity
2.3.6. Comparison with Referential Glucocorticoid Datasets
2.3.7. Comparison with Referential Stress Datasets
2.3.8. qPCR Validation
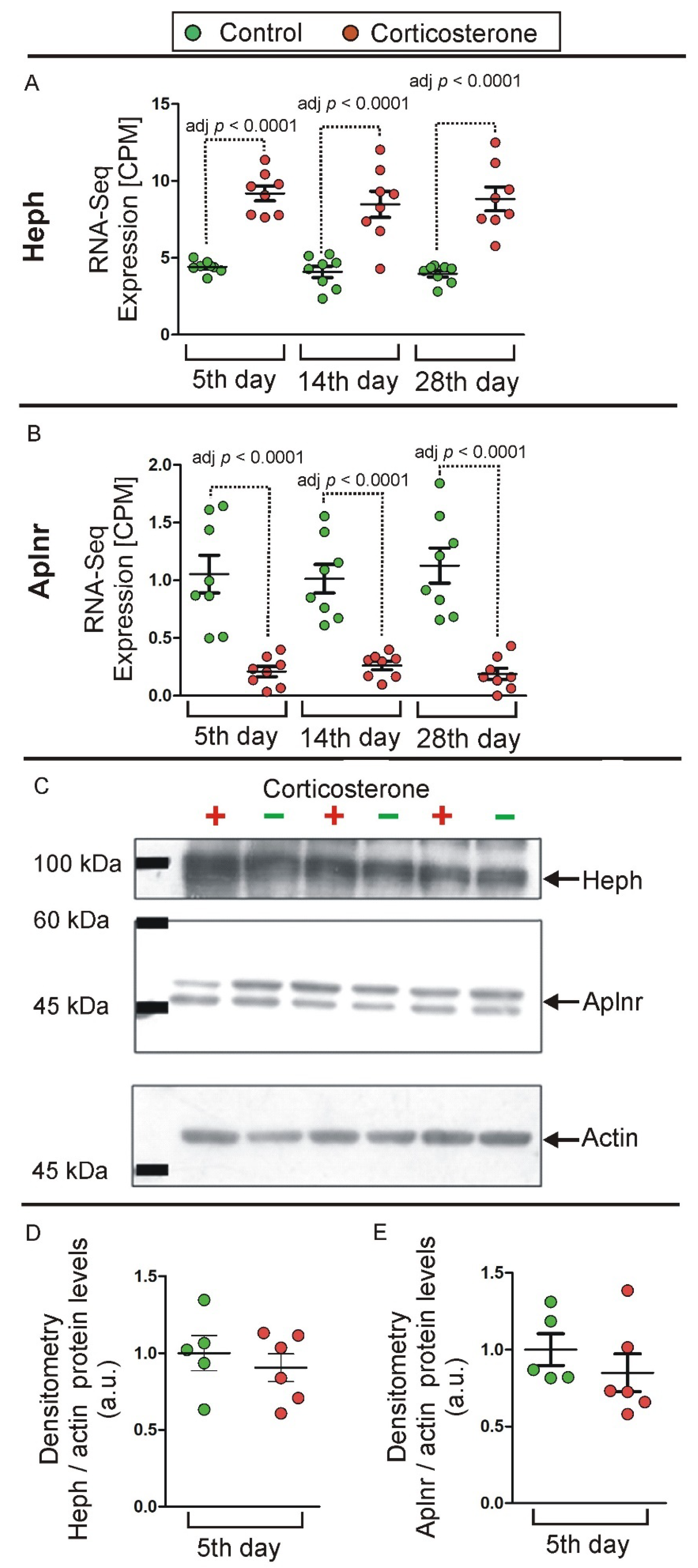
2.4. Western Blot
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Pharmacological RNA-Seq Experiment
4.3. Analysis of Blood Samples
4.4. Sample Preparation and RNA Sequencing (RNA-Seq)
4.5. Analysis of RNA-Seq Data
4.6. Assessment of Cell Expression Specificity
4.7. Cluster Analysis
4.8. Real-Time qPCR Validation of RNA-Seq Data
4.9. Comparison with Referential Datasets
4.10. Western Blot
4.11. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CPM | Counts Per Million |
| lncRNA | Long non-coding RNA |
| TEC | To be Experimentally Confirmed |
References
- Peterkofsky, B.; Tomkins, G.M. Effect of inhibitors of nucleic acid synthesis on steroid-mediated induction of tyrosine aminotransferase in hepatoma cell cultures. J. Mol. Biol. 1967, 30, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, G.R.; Stankiewicz, A.M. Glucocorticoids, genes and brain function. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 82, 136–168. [Google Scholar] [CrossRef]
- Stankiewicz, A.M.; Jaszczyk, A.; Goscik, J.; Juszczak, G.R. Stress and the brain transcriptome: Identifying commonalities and clusters in standardized data from published experiments. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2022, 119, 110558. [Google Scholar] [CrossRef]
- Jorstad, T.S.; Langaas, M.; Bones, A.M. Understanding sample size: What determines the required number of microarrays for an experiment? Trends Plant Sci. 2007, 12, 46–50. [Google Scholar] [CrossRef]
- Button, K.S.; Ioannidis, J.P.; Mokrysz, C.; Nosek, B.A.; Flint, J.; Robinson, E.S.; Munafò, M.R. Power failure: Why small sample size undermines the reliability of neuroscience. Nat. Rev. Neurosci. 2013, 14, 365–376. [Google Scholar] [CrossRef]
- Schurch, N.J.; Schofield, P.; Gierlinski, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T.; et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Smyth, G.K. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 2009, 25, 765–771. [Google Scholar] [CrossRef]
- Dalman, M.R.; Deeter, A.; Nimishakavi, G.; Duan, Z.H. Fold change and p-value cutoffs significantly alter microarray interpretations. BMC Bioinform. 2012, 13 (Suppl. 2), S11. [Google Scholar] [CrossRef]
- Walser, M.; Karlsson, L.; Motalleb, R.; Isgaard, J.; Kuhn, H.G.; Aberg, N.D. Brain tissue haemoglobin expression in saline-perfused vs non-perfused rodents. Heliyon 2024, 10, e23343. [Google Scholar] [CrossRef]
- Lee, R.S.; Tamashiro, K.L.; Yang, X.; Purcell, R.H.; Harvey, A.; Willour, V.L.; Huo, Y.; Rongione, M.; Wand, G.S.; Potash, J.B. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology 2010, 151, 4332–4343. [Google Scholar] [CrossRef] [PubMed]
- Claman, H.N. Corticosteroids and lymphoid cells. N. Engl. J. Med. 1972, 287, 388–397. [Google Scholar] [CrossRef]
- Stankiewicz, A.M.; Goscik, J.; Majewska, A.; Swiergiel, A.H.; Juszczak, G.R. The Effect of Acute and Chronic Social Stress on the Hippocampal Transcriptome in Mice. PLoS ONE 2015, 10, e0142195. [Google Scholar] [CrossRef]
- Jaszczyk, A.; Stankiewicz, A.M.; Juszczak, G.R. Dissection of Mouse Hippocampus with Its Dorsal, Intermediate and Ventral Subdivisions Combined with Molecular Validation. Brain Sci. 2022, 12, 799. [Google Scholar] [CrossRef]
- Zeisel, A.; Hochgerner, H.; Lonnerberg, P.; Johnsson, A.; Memic, F.; van der Zwan, J.; Häring, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular Architecture of the Mouse Nervous System. Cell 2018, 174, 999–1014.e22. [Google Scholar] [CrossRef]
- Jaszczyk, A.; Stankiewicz, A.M.; Goscik, J.; Majewska, A.; Jezierski, T.; Juszczak, G.R. Overnight Corticosterone and Gene Expression in Mouse Hippocampus: Time Course during Resting Period. Int. J. Mol. Sci. 2023, 24, 2828. [Google Scholar] [CrossRef]
- Chen, Y.; Lun, A.T.; Smyth, G.K. From reads to genes to pathways: Differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Research 2016, 5, 1438. [Google Scholar]
- Chen, Y.; McCarthy, D.; Baldoni, P.; Ritchie, M.; Robinson, M.; Smyth, G. edgeR: Differential Analysis of Sequence Read Count Data. User’s Guide 2023. Available online: https://bioconductor.org/packages/release/bioc/vignettes/edgeR/inst/doc/edgeRUsersGuide.pdf (accessed on 30 October 2023).
- Ruiz-Villalba, A.; Ruijter, J.M.; van den Hoff, M.J.B. Use and Misuse of C(q) in qPCR Data Analysis and Reporting. Life 2021, 11, 496. [Google Scholar] [CrossRef]
- Peccoud, J.; Jacob, C. Theoretical uncertainty of measurements using quantitative polymerase chain reaction. Biophys. J. 1996, 71, 101–108. [Google Scholar] [CrossRef]
- Wolff, C.A.; Gutierrez-Monreal, M.A.; Meng, L.; Zhang, X.; Douma, L.G.; Costello, H.M.; Douglas, C.M.; Ebrahimi, E.; Pham, A.; Oliveira, A.C.; et al. Defining the age-dependent and tissue-specific circadian transcriptome in male mice. Cell Rep. 2023, 42, 111982. [Google Scholar] [CrossRef]
- Renaud, J.; Dumont, F.; Khelfaoui, M.; Foisset, S.R.; Letourneur, F.; Bienvenu, T.; Khwaja, O.; Dorseuil, O.; Billuart, P. Identification of intellectual disability genes showing circadian clock-dependent expression in the mouse hippocampus. Neuroscience 2015, 308, 11–50. [Google Scholar] [CrossRef]
- van der Meulen, M.; Amaya, J.M.; Dekkers, O.M.; Meijer, O.C. Association between use of systemic and inhaled glucocorticoids and changes in brain volume and white matter microstructure: A cross-sectional study using data from the UK Biobank. BMJ Open 2022, 12, e062446. [Google Scholar] [CrossRef]
- Viho, E.M.G.; Buurstede, J.C.; Berkhout, J.B.; Mahfouz, A.; Meijer, O.C. Cell type specificity of glucocorticoid signaling in the adult mouse hippocampus. J. Neuroendocrinol. 2022, 34, e13072. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, A.M.; Goscik, J.; Swiergiel, A.H.; Majewska, A.; Wieczorek, M.; Juszczak, G.R.; Lisowski, P. Social stress increases expression of hemoglobin genes in mouse prefrontal cortex. BMC Neurosci. 2014, 15, 130. [Google Scholar] [CrossRef]
- Buccitelli, C.; Selbach, M. mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 2020, 21, 630–644. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]
- Price, J.C.; Guan, S.; Burlingame, A.; Prusiner, S.B.; Ghaemmaghami, S. Analysis of proteome dynamics in the mouse brain. Proc. Natl. Acad. Sci. USA 2010, 107, 14508–14513. [Google Scholar] [CrossRef]
- Jaszczyk, A.; Juszczak, G.R. Glucocorticoids, metabolism and brain activity. Neurosci. Biobehav. Rev. 2021, 126, 113–145. [Google Scholar] [CrossRef]
- Koning, A.; van der Meulen, M.; Schaap, D.; Satoer, D.D.; Vinkers, C.H.; Rossum, E.F.C.; van Furth, W.R.; Pereira, A.M.; Meijer, O.C.; Dekkers, O.M. Neuropsychiatric Adverse Effects of Synthetic Glucocorticoids: A Systematic Review and Meta-Analysis. J. Clin. Endocrinol. Metab. 2024, 109, e1442–e1451. [Google Scholar] [CrossRef]
- Meijer, O.C.; Buurstede, J.C.; Viho, E.M.G.; Amaya, J.M.; Koning, A.; van der Meulen, M.; van Weert, L.T.C.M.; Paul, S.N.; Kroon, J.; Koorneef, L.L. Transcriptional glucocorticoid effects in the brain: Finding the relevant target genes. J. Neuroendocrinol. 2023, 35, e13213. [Google Scholar] [CrossRef]
- Van Loo, P.L.; Mol, J.A.; Koolhaas, J.M.; Van Zutphen, B.F.; Baumans, V. Modulation of aggression in male mice: Influence of group size and cage size. Physiol. Behav. 2001, 72, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Mondragon, R.; Mayagoitia, L.; Lopez-Lujan, A.; Diaz, J.L. Social structure features in three inbred strains of mice, C57Bl/6J, Balb/cj, and NIH: A comparative study. Behav. Neural Biol. 1987, 47, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Hunt, C.; Hambly, C. Faecal corticosterone concentrations indicate that separately housed male mice are not more stressed than group housed males. Physiol. Behav. 2006, 87, 519–526. [Google Scholar] [CrossRef]
- Bowles, N.P.; Hill, M.N.; Bhagat, S.M.; Karatsoreos, I.N.; Hillard, C.J.; McEwen, B.S. Chronic, noninvasive glucocorticoid administration suppresses limbic endocannabinoid signaling in mice. Neuroscience 2012, 204, 83–89. [Google Scholar] [CrossRef]
- Bowles, N.P.; Karatsoreos, I.N.; Li, X.; Vemuri, V.K.; Wood, J.A.; Li, Z.; Tamashiro, K.L.K.; Schwartz, G.J.; Makriyannis, A.M.; Kunos, G.; et al. A peripheral endocannabinoid mechanism contributes to glucocorticoid-mediated metabolic syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, 285–290. [Google Scholar] [CrossRef]
- Solas, M.; Gerenu, G.; Gil-Bea, F.J.; Ramirez, M.J. Mineralocorticoid receptor activation induces insulin resistance through c-Jun N-terminal kinases in response to chronic corticosterone: Cognitive implications. J. Neuroendocrinol. 2013, 25, 350–356. [Google Scholar] [CrossRef]
- Karatsoreos, I.N.; Bhagat, S.M.; Bowles, N.P.; Weil ZMPfaff, D.W.; McEwen, B.S. Endocrine and physiological changes in response to chronic corticosterone: A potential model of the metabolic syndrome in mouse. Endocrinology 2010, 151, 2117–2127. [Google Scholar] [CrossRef]
- Jene, T.; Gassen, N.C.; Opitz, V.; Endres, K.; Muller, M.B.; van der Kooij, M.A. Temporal profiling of an acute stress-induced behavioral phenotype in mice and role of hippocampal DRR1. Psychoneuroendocrinology 2018, 91, 149–158. [Google Scholar] [CrossRef]
- Engler, H.; Bailey, M.T.; Engler, A.; Stiner-Jones, L.M.; Quan, N.; Sheridan, J.F. Interleukin-1 receptor type 1-deficient mice fail to develop social stress-associated glucocorticoid resistance in the spleen. Psychoneuroendocrinology 2008, 33, 108–117. [Google Scholar] [CrossRef]
- McGill, B.E.; Bundle, S.F.; Yaylaoglu, M.B.; Carson, J.P.; Thaller, C.; Zoghbi, H.Y. Enhanced anxiety and stress-induced corticosterone release are associated with increased Crh expression in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 18267–18272. [Google Scholar] [CrossRef]
- Norman, K.J.; Seiden, J.A.; Klickstein, J.A.; Han, X.; Hwa, L.S.; DeBold, J.F.; Miczek, K.A. Social stress and escalated drug self-administration in mice I. Alcohol and corticosterone. Psychopharmacology 2015, 232, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Okutsu, M.; Lira, V.A.; Higashida, K.; Peake, J.; Higuchi, M.; Suzuki, K. Corticosterone accelerates atherosclerosis in the apolipoprotein E-deficient mouse. Atherosclerosis 2014, 232, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jonassen, I. RASflow: An RNA-Seq analysis workflow with Snakemake. BMC Bioinform. 2020, 21, 110. [Google Scholar] [CrossRef]
- Kurtzer, G.M.; Sochat, V.; Bauer, M.W. Singularity: Scientific containers for mobility of compute. PLoS ONE 2017, 12, e0177459. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Chen, Y.; Lun, A.T.L.; McCarthy, D.J.; Chen, L.; Ritchie, M.E.; Phipson, B. Empirical Analysis of Digital Gene Expression Data in R. Reference Manual 2023. Available online: https://bioconductor.org/packages/release/bioc/manuals/edgeR/man/edgeR.pdf (accessed on 30 October 2023).
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Gibbons, F.D.; Roth, F.P. Judging the quality of gene expression-based clustering methods using gene annotation. Genome Res. 2002, 12, 1574–1581. [Google Scholar] [CrossRef]
- Eisen, M.; de Hoon, M. Cluster 3.0 Manual; University of Tokyo, Human Genome Center: Tokyo, Japan, 2002; Available online: http://bonsai.hgc.jp/~mdehoon/software/cluster/cluster3.pdf (accessed on 11 January 2024).
- Saldanha, A.J. Java Treeview--Extensible visualization of microarray data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Carter, B.S.; Meng, F.; Thompson, R.C. Glucocorticoid treatment of astrocytes results in temporally dynamic transcriptome regulation and astrocyte-enriched mRNA changes in vitro. Physiol. Genom. 2012, 44, 1188–1200. [Google Scholar] [CrossRef]
- Gray, J.D.; Rubin, T.G.; Hunter, R.G.; McEwen, B.S. Hippocampal gene expression changes underlying stress sensitization and recovery. Mol. Psychiatry 2014, 19, 1171–1178. [Google Scholar] [CrossRef]
- Sato, H.; Horikawa, Y.; Iizuka, K.; Sakurai, N.; Tanaka, T.; Shihara, N.; Oshima, A.; Takeda, J.; Mikuni, M. Large-scale analysis of glucocorticoid target genes in rat hypothalamus. J. Neurochem. 2008, 106, 805–814. [Google Scholar] [CrossRef]
- Datson, N.A.; van den Oever, J.M.; Korobko, O.B.; Magarinos, A.M.; de Kloet, E.R.; McEwen, B.S. Previous history of chronic stress changes the transcriptional response to glucocorticoid challenge in the dentate gyrus region of the male rat hippocampus. Endocrinology 2013, 154, 3261–3272. [Google Scholar] [CrossRef]
- Datson, N.A.; Polman, J.A.; de Jonge, R.T.; van Boheemen, P.T.; van Maanen, E.M.; Welten, J.; McEwen, B.S.; Meiland, H.C.; Meijer, O.C. Specific regulatory motifs predict glucocorticoid responsiveness of hippocampal gene expression. Endocrinology 2011, 152, 3749–3757. [Google Scholar] [CrossRef]
- Polman, J.A.; Hunter, R.G.; Speksnijder, N.; van den Oever, J.M.; Korobko, O.B.; McEwen, B.S.; de Kloet, E.R.; Datson, N.A. Glucocorticoids modulate the mTOR pathway in the hippocampus: Differential effects depending on stress history. Endocrinology 2012, 153, 4317–4327. [Google Scholar] [CrossRef]
- Datson, N.A.; van der Perk, J.; de Kloet, E.R.; Vreugdenhil, E. Identification of corticosteroid-responsive genes in rat hippocampus using serial analysis of gene expression. Eur. J. Neurosci. 2001, 14, 675–689. [Google Scholar] [CrossRef]
- Chen, K.C.; Blalock, E.M.; Curran-Rauhut, M.A.; Kadish, I.; Blalock, S.J.; Brewer, L.; Porter, N.M.; Landfield, P.W. Glucocorticoid-dependent hippocampal transcriptome in male rats: Pathway-specific alterations with aging. Endocrinology 2013, 154, 2807–2820. [Google Scholar] [CrossRef]
- Alfonso, J.; Aguero, F.; Sanchez, D.O.; Flugge, G.; Fuchs, E.; Frasch, A.C.; Pollevick, G.D. Gene expression analysis in the hippocampal formation of tree shrews chronically treated with cortisol. J. Neurosci. Res. 2004, 78, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Slezak, M.; Korostynski, M.; Gieryk, A.; Golda, S.; Dzbek, J.; Piechota, M.; Wlazlo, E.; Bilecki, W.; Przewlocki, R. Astrocytes are a neural target of morphine action via glucocorticoid receptor-dependent signaling. Glia 2013, 61, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Peffer, M.E.; Chandran, U.R.; Luthra, S.; Volonte, D.; Galbiati, F.; Garabedian, M.J.; Monaghan, A.P.; DeFranco, D.B. Caveolin-1 regulates genomic action of the glucocorticoid receptor in neural stem cells. Mol. Cell. Biol. 2014, 34, 2611–2623. [Google Scholar] [CrossRef] [PubMed]
- Morsink, M.C.; Steenbergen, P.J.; Vos, J.B.; Karst, H.; Joels, M.; De Kloet, E.R.; Datson, N.A. Acute activation of hippocampal glucocorticoid receptors results in different waves of gene expression throughout time. J. Neuroendocrinol. 2006, 18, 239–252. [Google Scholar] [CrossRef]
- Morsink, M.C.; Joels, M.; Sarabdjitsingh, R.A.; Meijer, O.C.; De Kloet, E.R.; Datson, N.A. The dynamic pattern of glucocorticoid receptor-mediated transcriptional responses in neuronal PC12 cells. J. Neurochem. 2006, 99, 1282–1298. [Google Scholar] [CrossRef]
- Jenkins, S.I.; Pickard, M.R.; Khong, M.; Smith, H.L.; Mann, C.L.; Emes, R.D.; Chari, D.M. Identifying the cellular targets of drug action in the central nervous system following corticosteroid therapy. ACS Chem. Neurosci. 2014, 5, 51–63. [Google Scholar] [CrossRef]
- Fukumoto, K.; Morita, T.; Mayanagi, T.; Tanokashira, D.; Yoshida, T.; Sakai, A.; Sobue, K. Detrimental effects of glucocorticoids on neuronal migration during brain development. Mol. Psychiatry 2009, 14, 1119–1131. [Google Scholar] [CrossRef]
- Anacker, C.; Cattaneo, A.; Musaelyan, K.; Zunszain, P.A.; Horowitz, M.; Molteni, R.; Luoni, A.; Calabrese, F.; Tansey, K.; Gennarelli, M.; et al. Role for the kinase SGK1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 8708–8713. [Google Scholar] [CrossRef]
- Salaria, S.; Chana, G.; Caldara, F.; Feltrin, E.; Altieri, M.; Faggioni, F.; Domenici, E.; Merlo-Pich, E.; Everall, I.P. Microarray analysis of cultured human brain aggregates following cortisol exposure: Implications for cellular functions relevant to mood disorders. Neurobiol. Dis. 2006, 23, 630–636. [Google Scholar] [CrossRef]
- Itoi, K.; Motoike, I.; Liu, Y.; Clokie, S.; Iwasaki, Y.; Uchida, K.; Sato, T.; Aguilera, G. Genome-Wide Analysis of Glucocorticoid-Responsive Transcripts in the Hypothalamic Paraventricular Region of Male Rats. Endocrinology 2019, 160, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Frahm, K.A.; Waldman, J.K.; Luthra, S.; Rudine, A.C.; Monaghan-Nichols, A.P.; Chandran, U.R.; DeFranco, D.B. A comparison of the sexually dimorphic dexamethasone transcriptome in mouse cerebral cortical and hypothalamic embryonic neural stem cells. Mol. Cell. Endocrinol. 2018, 471, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Caradonna, S.G.; Einhorn, N.R.; Saudagar, V.; Khalil, H.; Petty, G.H.; Lihagen, A.; LeFloch, C.; Lee, F.S.; Akil, H.; Guidotti, A.; et al. Corticosterone induces discrete epigenetic signatures in the dorsal and ventral hippocampus that depend upon sex and genotype: Focus on methylated Nr3c1 gene. Transl. Psychiatry 2022, 12, 109. [Google Scholar] [CrossRef]
- Caradonna, S.G.; Zhang, T.Y.; O’Toole, N.; Shen, M.J.; Khalil, H.; Einhorn, N.R.; Wen, X.; Parent, C.; Lee, F.S.; Akil, H.; et al. Genomic modules and intramodular network concordance in susceptible and resilient male mice across models of stress. Neuropsychopharmacology 2022, 47, 987–999. [Google Scholar] [CrossRef]
- Hancock, A.A.; Bush, E.N.; Stanisic, D.; Kyncl, J.J.; Lin, C.T. Data normalization before statistical analysis: Keeping the horse before the cart. Trends Pharmacol. Sci. 1988, 9, 29–32. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward or Reverse Primer Sequence | Annealing Temperature | Efficiency | |
|---|---|---|---|---|
| Agt | F | GCGGAGGCAAATCTGAACAAC | 60 °C | 100.6% |
| R | CTCGTAGATGGCGAACAGGA | |||
| Aplnr | F | TCGTGGTGCTTGTAGTGACC | 60 °C | 91.5% |
| R | ATGCAGGTGCAGTACGGAAA | |||
| Bfsp2 | F | TGCTGCCCTCAGTGTAGAGTTA | 60 °C | 115.8% |
| R | GGCGTCGTGCAAGCTGTTTT | |||
| Etnppl | F | TTGGTGAAGGACCGTGAGAAA | 60 °C | 94.9% |
| R | AACTTTGCATCGTCTTCCGTG | |||
| Fabp7 | F | GGTGGCAAAGTGGTGATCC | 60 °C | 92.6% |
| R | ATCCCCAAAGGTAAGAGTCACG | |||
| Fmo2 | F | AGGCTCCATCTTCCCAACCG | 60 °C | 94.3% |
| R | AAGGCGAGTTCATCCAGGTAGT | |||
| Lrg1 | F | GTCTCTTGGCAGCATCAAGG | 60 °C | 107.6% |
| R | GAATTCCACCGACAGATGGAC | |||
| Opalin | F | AGACTGTGGTCCCTCTATTGG | 60 °C | 86.6% |
| R | GGGTTCATTTCATGTGTGGGTG | |||
| Sult1a1 | F | GATGGGAAAGTGTCCTATGGGT | 60 °C | 92.9% |
| R | TGAAGGATGTGTGGTGAACAATTA | |||
| Tekt4 | F | GCCTTTACCAGCGGTCACA | 60 °C | 71.2% |
| R | CTGTTGGTCTTGACTGCGATG | |||
| Gapdh | F | TCAAGCTCATTTCCTGGTATGACAA | 60 °C | 99.8% |
| R | TCTCTTGCTCAGTGTCCTTGCT | |||
| Ywhaz | F | TTGAGCAGAAGACGGAAGGT | 60 °C | 96.9% |
| R | GAAGCATTGGGGATCAAGAA | |||
| Tbp | F | GCAGTGCCCAGCATCACTATT | 60 °C | 108.5% |
| R | AAGCCCTGAGCATAAGGTGG | |||
| Hmbs | F | TCCTGGCTTTACTATTGGAG | 60 °C | 87.4% |
| R | TGAATTCCAGGTGGGGGAAC | |||
| Target Protein | Primary Ab | Dilution | Secondary Ab | Dilution | |
|---|---|---|---|---|---|
| Apelin receptor (Invitrogen, Waltham, MA, USA, Catalog # PA5-114830, RRID:AB_2899466) | Affinity-purified rabbit anti-recombinant protein corresponding to the human APLNR (Accession P35414), amino acid residues A147-W197 https://www.thermofisher.com/order/genome-database/dataSheetPdf?producttype=antibody&productsubtype=antibody_primary&productId=PA5-114830&version=Local (accessed on 28 May 2024) | 1:1000 | Goat anti-rabbit IgG (whole molecule), polyclonal, peroxidase conjugate, affinity isolated antibody (Sigma-Aldrich Catalog # A6154, RRID: AB_258284) https://www.sigmaaldrich.com/PL/pl/product/sigma/a6154#product-documentation (accessed on 28 May 2024) | 1:20,000 | |
| Hephaestin (Affinity Biosciences, Cincinnati, OH, USA, Catalog # DF13057, RRID:AB_2846018) | Affinity-purified rabbit anti-human recombinant protein, corresponding to a region within the internal amino acids https://www.affbiotech.com/download/pdf/DF13057 (accessed on 28 May 2024) | 1:1000 | Goat anti-rabbit IgG (whole molecule), polyclonal, peroxidase conjugate, affinity isolated antibody (Sigma-Aldrich Catalog # A6154, RRID: AB_258284) https://www.sigmaaldrich.com/PL/pl/product/sigma/a6154#product-documentation (accessed on 28 May 2024) | 1:20,000 | |
| Actin (ACTN05 (C4) Thermo Fisher Scientific, Cat# MA5-11869, RRID:AB_11004139) | Rabbit monoclonal, anti-chicken gizzard actin https://www.thermofisher.com/order/genome-database/dataSheetPdf?producttype=antibody&productsubtype=antibody_primary&productId=MA5-11869&version=Local (accessed on 28 May 2024) | 1:1000 | Goat anti-rabbit IgG (whole molecule), polyclonal, peroxidase conjugate, affinity isolated antibody (Sigma-Aldrich Catalog # A6154, RRID: AB_258284) https://www.sigmaaldrich.com/PL/pl/product/sigma/a6154#product-documentation (accessed on 28 May 2024) | 1:20,000 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juszczak, G.R.; Stankiewicz, A.M.; Starzyński, R.R.; Ogłuszka, M.; Jaszczyk, A. Effect of Corticosterone on Gene Expression in the Context of Global Hippocampal Transcription. Int. J. Mol. Sci. 2025, 26, 4889. https://doi.org/10.3390/ijms26104889
Juszczak GR, Stankiewicz AM, Starzyński RR, Ogłuszka M, Jaszczyk A. Effect of Corticosterone on Gene Expression in the Context of Global Hippocampal Transcription. International Journal of Molecular Sciences. 2025; 26(10):4889. https://doi.org/10.3390/ijms26104889
Chicago/Turabian StyleJuszczak, Grzegorz R., Adrian M. Stankiewicz, Rafał R. Starzyński, Magdalena Ogłuszka, and Aneta Jaszczyk. 2025. "Effect of Corticosterone on Gene Expression in the Context of Global Hippocampal Transcription" International Journal of Molecular Sciences 26, no. 10: 4889. https://doi.org/10.3390/ijms26104889
APA StyleJuszczak, G. R., Stankiewicz, A. M., Starzyński, R. R., Ogłuszka, M., & Jaszczyk, A. (2025). Effect of Corticosterone on Gene Expression in the Context of Global Hippocampal Transcription. International Journal of Molecular Sciences, 26(10), 4889. https://doi.org/10.3390/ijms26104889