
NDF/GLYR1 Promotes RNA Polymerase II Processivity via Pol II Binding and Nucleosome Destabilization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
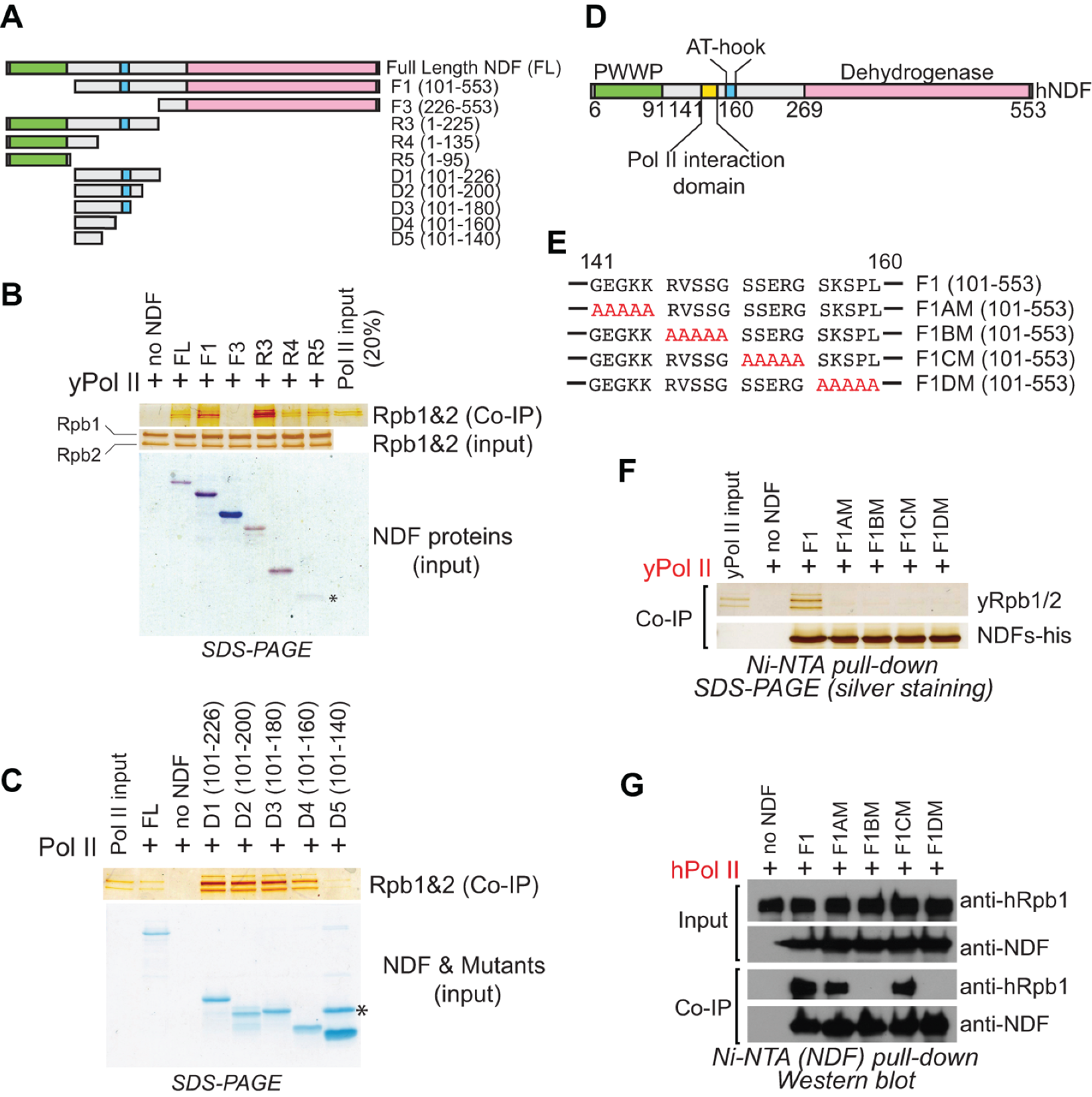
2.1. Identification of the Domain in NDF Required for Interaction with Pol II
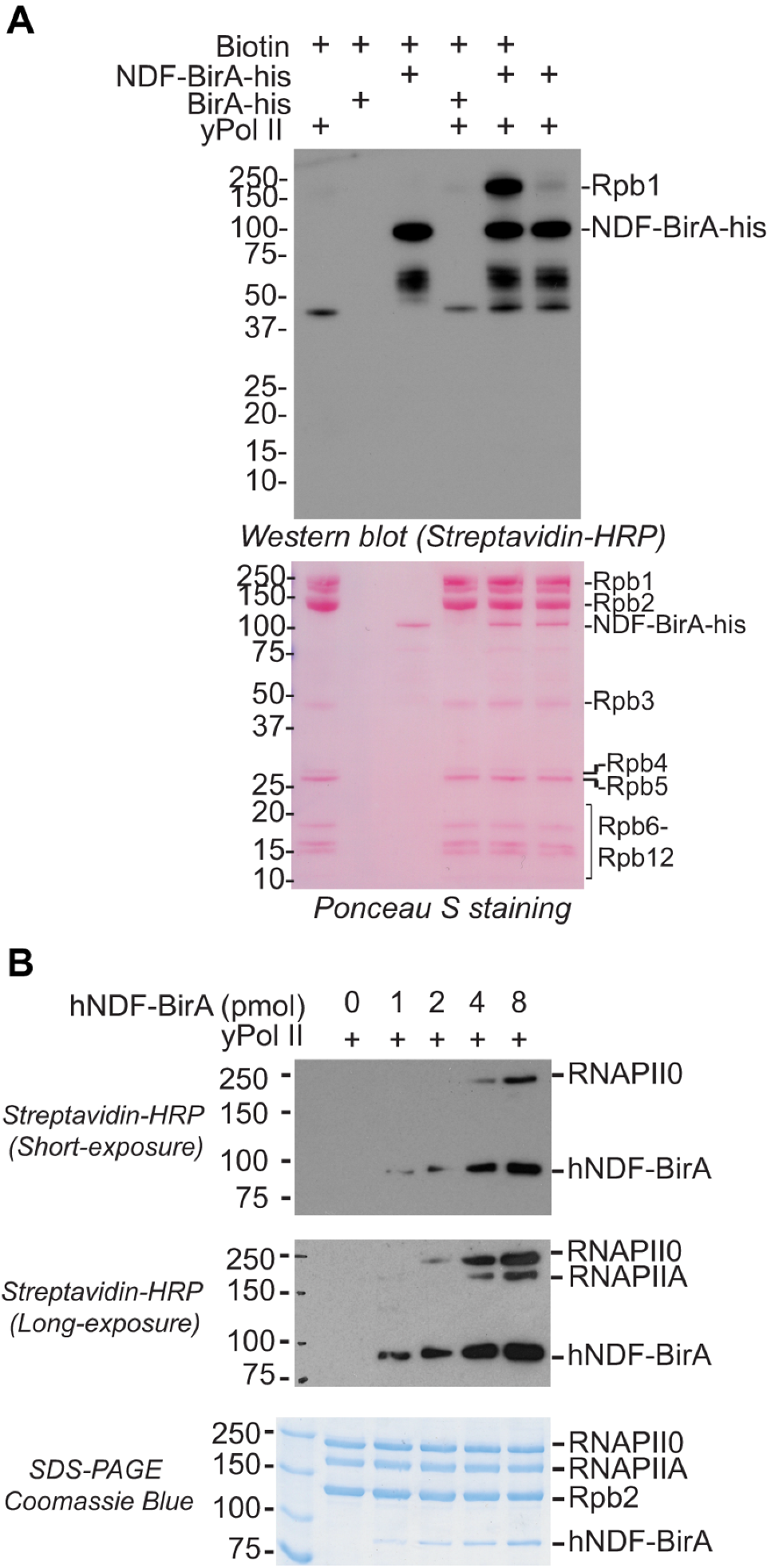
2.2. NDF Preferentially Interacts with Hyperphosphorylated Rpb1
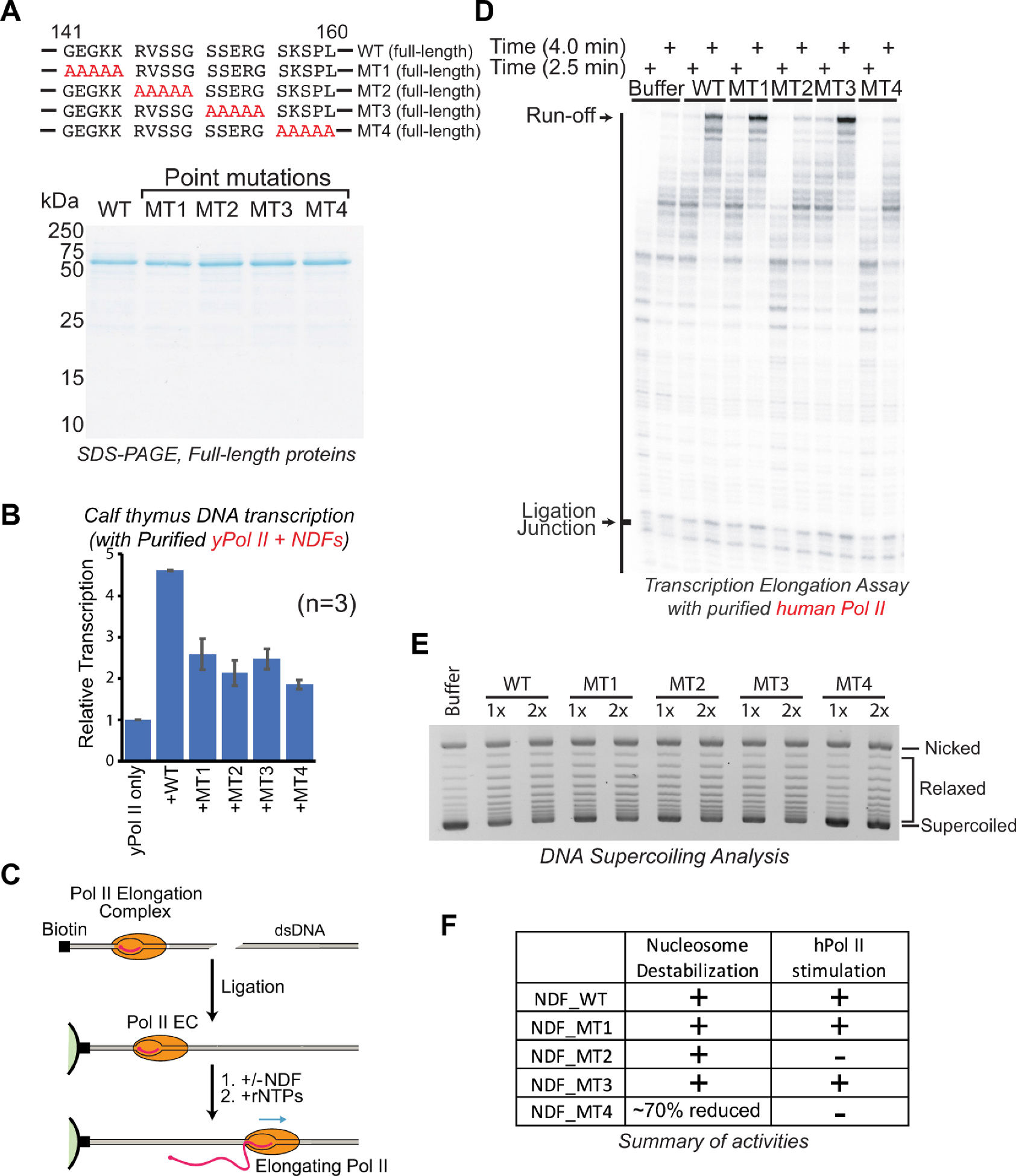
2.3. Specific Interaction Domain of NDF Is Required for Stimulating Transcription by Pol II In Vitro
2.4. Specific Interaction Domain of NDF Is Required for Stimulating Transcription Elongation by Pol II
2.5. Distinct Domains of NDF Separately Mediate Nucleosome Destabilization and Pol II Stimulation
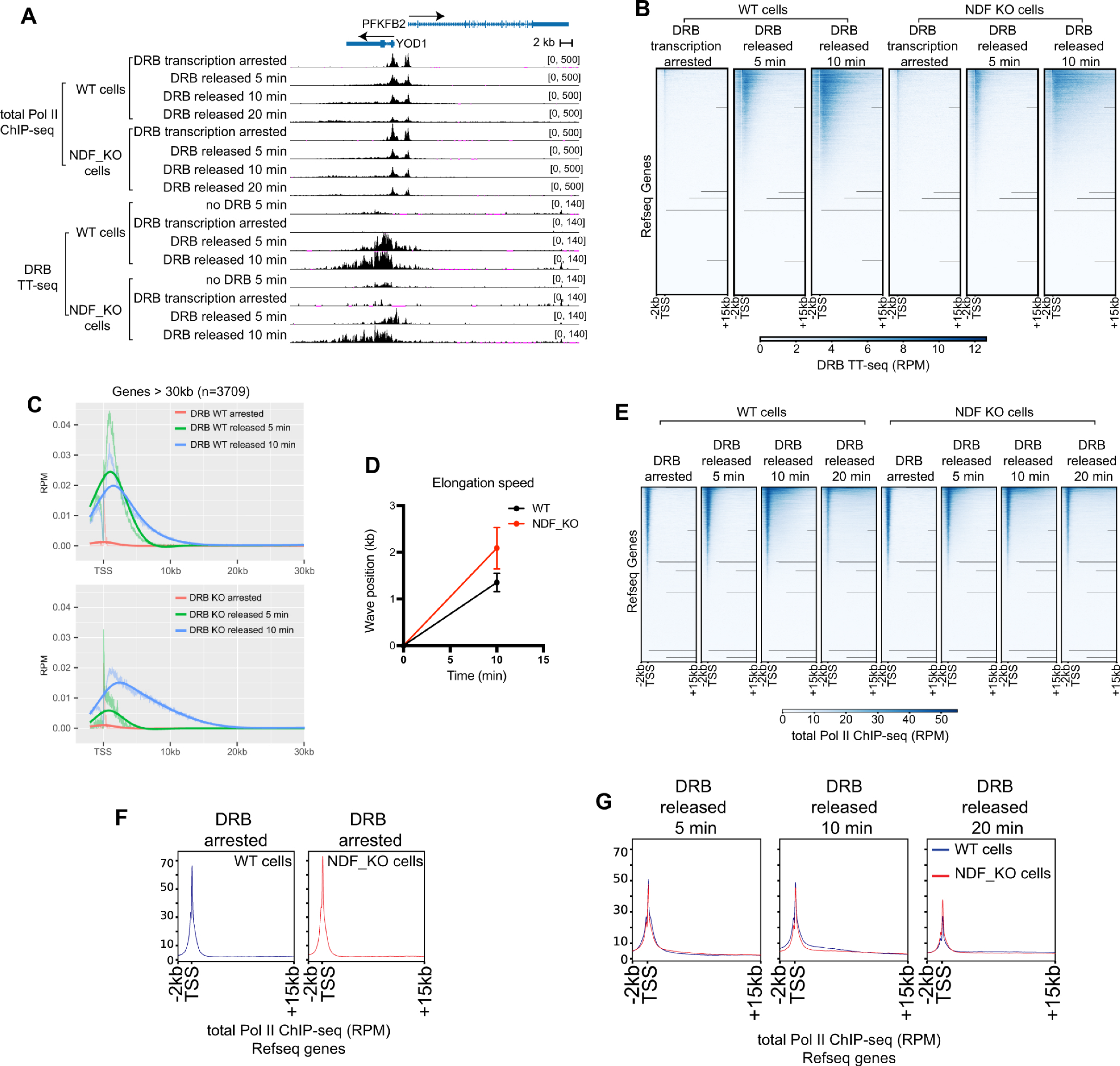
2.6. NDF Regulates Transcription Through Processivity Rather than Elongation Rate
2.7. NDF Mutants Affect Gene Expression in Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Antibodies
4.3. Plasmid Construction
4.4. Protein Purification
4.5. Pull-Down Assay
4.6. Ndf Knockout and Knockout Rescue in Sw480 Cells
4.7. In Vitro Biotinylation Assay
4.8. Western Blot
4.9. In Vitro Transcription Assays with Calf Thymus DNA
4.10. Transcription Elongation Assays
4.11. DNA Supercoiling Assays
4.12. ChIP-Seq Analysis
4.13. TT-Seq and BRU-Seq
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jonkers, I.; Lis, J.T. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Core, L.; Adelman, K. Promoter-proximal pausing of RNA polymerase II: A nexus of gene regulation. Genes Dev. 2019, 33, 960–982. [Google Scholar] [CrossRef]
- Lis, J.T. A 50 year history of technologies that drove discovery in eukaryotic transcription regulation. Nat. Struct. Mol. Biol. 2019, 26, 777–782. [Google Scholar] [CrossRef]
- Gilmour, D.S.; Lis, J.T. RNA polymerase II interacts with the promoter region of the noninduced hsp70 gene in Drosophila melanogaster cells. Mol. Cell. Biol. 1986, 6, 3984–3989. [Google Scholar] [CrossRef]
- Wada, T.; Takagi, T.; Yamaguchi, Y.; Ferdous, A.; Imai, T.; Hirose, S.; Sugimoto, S.; Yano, K.; Hartzog, G.A.; Winston, F.; et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998, 12, 343–356. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Takagi, T.; Wada, T.; Yano, K.; Furuya, A.; Sugimoto, S.; Hasegawa, J.; Handa, H. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 1999, 97, 41–51. [Google Scholar] [CrossRef]
- Vos, S.M.; Farnung, L.; Urlaub, H.; Cramer, P. Structure of paused transcription complex Pol II-DSIF-NELF. Nature 2018, 560, 601–606. [Google Scholar] [CrossRef]
- Bernecky, C.; Plitzko, J.M.; Cramer, P. Structure of a transcribing RNA polymerase II-DSIF complex reveals a multidentate DNA-RNA clamp. Nat. Struct. Mol. Biol. 2017, 24, 809–815. [Google Scholar] [CrossRef]
- Schier, A.C.; Taatjes, D.J. Structure and mechanism of the RNA polymerase II transcription machinery. Genes Dev. 2020, 34, 465–488. [Google Scholar] [CrossRef]
- Kireeva, M.L.; Hancock, B.; Cremona, G.H.; Walter, W.; Studitsky, V.M.; Kashlev, M. Nature of the nucleosomal barrier to RNA polymerase II. Mol. Cell 2005, 18, 97–108. [Google Scholar] [CrossRef]
- Weber, C.M.; Ramachandran, S.; Henikoff, S. Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol. Cell 2014, 53, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Studitsky, V.M.; Clark, D.J.; Felsenfeld, G. A histone octamer can step around a transcribing polymerase without leaving the template. Cell 1994, 76, 371–382. [Google Scholar] [CrossRef]
- Gilchrist, D.A.; Dos Santos, G.; Fargo, D.C.; Xie, B.; Gao, Y.; Li, L.; Adelman, K. Pausing of RNA polymerase II disrupts DNA-specified nucleosome organization to enable precise gene regulation. Cell 2010, 143, 540–551. [Google Scholar] [CrossRef]
- Kwak, H.; Fuda, N.J.; Core, L.J.; Lis, J.T. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 2013, 339, 950–953. [Google Scholar] [CrossRef]
- Li, J.; Gilmour, D.S. Distinct mechanisms of transcriptional pausing orchestrated by GAGA factor and M1BP, a novel transcription factor. EMBO J. 2013, 32, 1829–1841. [Google Scholar] [CrossRef]
- Sims, R.J., 3rd; Belotserkovskaya, R.; Reinberg, D. Elongation by RNA polymerase II: The short and long of it. Genes Dev. 2004, 18, 2437–2468. [Google Scholar] [CrossRef]
- Chen, F.X.; Smith, E.R.; Shilatifard, A. Born to run: Control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2018, 19, 464–478. [Google Scholar] [CrossRef]
- Belotserkovskaya, R.; Oh, S.; Bondarenko, V.A.; Orphanides, G.; Studitsky, V.M.; Reinberg, D. FACT facilitates transcription-dependent nucleosome alteration. Science 2003, 301, 1090–1093. [Google Scholar] [CrossRef]
- Formosa, T.; Winston, F. The role of FACT in managing chromatin: Disruption, assembly, or repair? Nucleic Acids Res. 2020, 48, 11929–11941. [Google Scholar] [CrossRef]
- Orphanides, G.; LeRoy, G.; Chang, C.H.; Luse, D.S.; Reinberg, D. FACT, a factor that facilitates transcript elongation through nucleosomes. Cell 1998, 92, 105–116. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, K.; Zhang, N.; Wei, H.; Tan, Y.Z.; Zhang, Z.; Carragher, B.; Potter, C.S.; D’Arcy, S.; Luger, K. FACT caught in the act of manipulating the nucleosome. Nature 2020, 577, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Ehara, H.; Kujirai, T.; Shirouzu, M.; Kurumizaka, H.; Sekine, S.I. Structural basis of nucleosome disassembly and reassembly by RNAPII elongation complex with FACT. Science 2022, 377, eabp9466. [Google Scholar] [CrossRef] [PubMed]
- Kireeva, M.L.; Walter, W.; Tchernajenko, V.; Bondarenko, V.; Kashlev, M.; Studitsky, V.M. Nucleosome remodeling induced by RNA polymerase II: Loss of the H2A/H2B dimer during transcription. Mol. Cell 2002, 9, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Zumer, K.; Ochmann, M.; Aljahani, A.; Zheenbekova, A.; Devadas, A.; Maier, K.C.; Rus, P.; Neef, U.; Oudelaar, A.M.; Cramer, P. FACT maintains chromatin architecture and thereby stimulates RNA polymerase II pausing during transcription in vivo. Mol. Cell 2024, 84, 2053–2069.E9. [Google Scholar] [CrossRef]
- Klein, D.C.; Lardo, S.M.; McCannell, K.N.; Hainer, S.J. FACT regulates pluripotency through proximal and distal regulation of gene expression in murine embryonic stem cells. BMC Biol. 2023, 21, 167. [Google Scholar] [CrossRef]
- Hsieh, F.K.; Kulaeva, O.I.; Patel, S.S.; Dyer, P.N.; Luger, K.; Reinberg, D.; Studitsky, V.M. Histone chaperone FACT action during transcription through chromatin by RNA polymerase II. Proc. Natl. Acad. Sci. USA 2013, 110, 7654–7659. [Google Scholar] [CrossRef]
- Farnung, L.; Ochmann, M.; Engeholm, M.; Cramer, P. Structural basis of nucleosome transcription mediated by Chd1 and FACT. Nat. Struct. Mol. Biol. 2021, 28, 382–387. [Google Scholar] [CrossRef]
- Kaplan, C.D.; Laprade, L.; Winston, F. Transcription elongation factors repress transcription initiation from cryptic sites. Science 2003, 301, 1096–1099. [Google Scholar] [CrossRef]
- Ahmad, K.; Brahma, S.; Henikoff, S. Epigenetic pioneering by SWI/SNF family remodelers. Mol. Cell 2024, 84, 194–201. [Google Scholar] [CrossRef]
- Narain, A.; Bhandare, P.; Adhikari, B.; Backes, S.; Eilers, M.; Dolken, L.; Schlosser, A.; Erhard, F.; Baluapuri, A.; Wolf, E. Targeted protein degradation reveals a direct role of SPT6 in RNAPII elongation and termination. Mol. Cell 2021, 81, 3110–3127.E14. [Google Scholar] [CrossRef]
- Simic, R.; Lindstrom, D.L.; Tran, H.G.; Roinick, K.L.; Costa, P.J.; Johnson, A.D.; Hartzog, G.A.; Arndt, K.M. Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. EMBO J. 2003, 22, 1846–1856. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, W.; Xu, L.; Chen, J.Y.; Chong, J.; Oh, J.; Leschziner, A.E.; Fu, X.D.; Wang, D. Cockayne syndrome B protein acts as an ATP-dependent processivity factor that helps RNA polymerase II overcome nucleosome barriers. Proc. Natl. Acad. Sci. USA 2020, 117, 25486–25493. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Price, D.H. RNA polymerase II transcription elongation control. Chem. Rev. 2013, 113, 8583–8603. [Google Scholar] [CrossRef]
- Conaway, R.C.; Conaway, J.W. The hunt for RNA polymerase II elongation factors: A historical perspective. Nat. Struct. Mol. Biol. 2019, 26, 771–776. [Google Scholar] [CrossRef]
- Cramer, P. Organization and regulation of gene transcription. Nature 2019, 573, 45–54. [Google Scholar] [CrossRef]
- Roeder, R.G. 50+ years of eukaryotic transcription: An expanding universe of factors and mechanisms. Nat. Struct. Mol. Biol. 2019, 26, 783–791. [Google Scholar] [CrossRef]
- Fang, R.; Chen, F.; Dong, Z.; Hu, D.; Barbera, A.J.; Clark, E.A.; Fang, J.; Yang, Y.; Mei, P.; Rutenberg, M.; et al. LSD2/KDM1B and its cofactor NPAC/GLYR1 endow a structural and molecular model for regulation of H3K4 demethylation. Mol. Cell 2013, 49, 558–570. [Google Scholar] [CrossRef]
- Vermeulen, M.; Eberl, H.C.; Matarese, F.; Marks, H.; Denissov, S.; Butter, F.; Lee, K.K.; Olsen, J.V.; Hyman, A.A.; Stunnenberg, H.G.; et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 2010, 142, 967–980. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef]
- Wang, C.I.; Alekseyenko, A.A.; LeRoy, G.; Elia, A.E.; Gorchakov, A.A.; Britton, L.M.; Elledge, S.J.; Kharchenko, P.V.; Garcia, B.A.; Kuroda, M.I. Chromatin proteins captured by ChIP-mass spectrometry are linked to dosage compensation in Drosophila. Nat. Struct. Mol. Biol. 2013, 20, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Fei, J.; Xu, J.; Li, Z.; Xu, K.; Wang, D.; Kassavetis, G.A.; Kadonaga, J.T. NDF is a transcription factor that stimulates elongation by RNA polymerase II. Genes Dev. 2022, 36, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Marabelli, C.; Marrocco, B.; Pilotto, S.; Chittori, S.; Picaud, S.; Marchese, S.; Ciossani, G.; Forneris, F.; Filippakopoulos, P.; Schoehn, G.; et al. A Tail-Based Mechanism Drives Nucleosome Demethylation by the LSD2/NPAC Multimeric Complex. Cell Rep. 2019, 27, 387–399.E7. [Google Scholar] [CrossRef]
- Fei, J.; Ishii, H.; Hoeksema, M.A.; Meitinger, F.; Kassavetis, G.A.; Glass, C.K.; Ren, B.; Kadonaga, J.T. NDF, a nucleosome-destabilizing factor that facilitates transcription through nucleosomes. Genes Dev. 2018, 32, 682–694. [Google Scholar] [CrossRef]
- Qin, S.; Min, J. Structure and function of the nucleosome-binding PWWP domain. Trends Biochem. Sci. 2014, 39, 536–547. [Google Scholar] [CrossRef]
- Huang, Y.J.; Acton, T.B.; Montelione, G.T. DisMeta: A meta server for construct design and optimization. Methods Mol. Biol. 2014, 1091, 3–16. [Google Scholar] [CrossRef]
- Tian, W.; Yan, P.; Xu, N.; Chakravorty, A.; Liefke, R.; Xi, Q.; Wang, Z. The HRP3 PWWP domain recognizes the minor groove of double-stranded DNA and recruits HRP3 to chromatin. Nucleic Acids Res. 2019, 47, 5436–5448. [Google Scholar] [CrossRef]
- Wang, H.; Farnung, L.; Dienemann, C.; Cramer, P. Structure of H3K36-methylated nucleosome-PWWP complex reveals multivalent cross-gyre binding. Nat. Struct. Mol. Biol. 2020, 27, 8–13. [Google Scholar] [CrossRef]
- Qiu, C.; Sawada, K.; Zhang, X.; Cheng, X. The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat. Struct. Biol. 2002, 9, 217–224. [Google Scholar] [CrossRef]
- Cramer, P.; Bushnell, D.A.; Kornberg, R.D. Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science 2001, 292, 1863–1876. [Google Scholar] [CrossRef]
- Larkin, R.M.; Guilfoyle, T.J. Two small subunits in Arabidopsis RNA polymerase II are related to yeast RPB4 and RPB7 and interact with one another. J. Biol. Chem. 1998, 273, 5631–5637. [Google Scholar] [CrossRef]
- Oostdyk, L.T.; Shank, L.; Jividen, K.; Dworak, N.; Sherman, N.E.; Paschal, B.M. Towards improving proximity labeling by the biotin ligase BirA. Methods 2019, 157, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.I.; Birendra, K.C.; Zhu, W.; Motamedchaboki, K.; Doye, V.; Roux, K.J. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. USA 2014, 111, E2453–E2461. [Google Scholar] [CrossRef] [PubMed]
- Cadena, D.L.; Dahmus, M.E. Messenger RNA synthesis in mammalian cells is catalyzed by the phosphorylated form of RNA polymerase II. J. Biol. Chem. 1987, 262, 12468–12474. [Google Scholar] [CrossRef]
- Payne, J.M.; Laybourn, P.J.; Dahmus, M.E. The transition of RNA polymerase II from initiation to elongation is associated with phosphorylation of the carboxyl-terminal domain of subunit IIa. J. Biol. Chem. 1989, 264, 19621–19629. [Google Scholar] [CrossRef]
- Reinberg, D.; Roeder, R.G. Factors involved in specific transcription by mammalian RNA polymerase II. Transcription factor IIS stimulates elongation of RNA chains. J. Biol. Chem. 1987, 262, 3331–3337. [Google Scholar] [CrossRef]
- Sekimizu, K.; Kobayashi, N.; Mizuno, D.; Natori, S. Purification of a factor from Ehrlich ascites tumor cells specifically stimulating RNA polymerase II. Biochemistry 1976, 15, 5064–5070. [Google Scholar] [CrossRef]
- Komissarova, N.; Kireeva, M.L.; Becker, J.; Sidorenkov, I.; Kashlev, M. Engineering of elongation complexes of bacterial and yeast RNA polymerases. Methods Enzym. 2003, 371, 233–251. [Google Scholar] [CrossRef]
- Sidorenkov, I.; Komissarova, N.; Kashlev, M. Crucial role of the RNA:DNA hybrid in the processivity of transcription. Mol. Cell 1998, 2, 55–64. [Google Scholar] [CrossRef]
- Kireeva, M.L.; Komissarova, N.; Waugh, D.S.; Kashlev, M. The 8-nucleotide-long RNA:DNA hybrid is a primary stability determinant of the RNA polymerase II elongation complex. J. Biol. Chem. 2000, 275, 6530–6536. [Google Scholar] [CrossRef]
- Mason, P.B.; Struhl, K. Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol. Cell 2005, 17, 831–840. [Google Scholar] [CrossRef]
- Paulsen, M.T.; Veloso, A.; Prasad, J.; Bedi, K.; Ljungman, E.A.; Tsan, Y.C.; Chang, C.W.; Tarrier, B.; Washburn, J.G.; Lyons, R.; et al. Coordinated regulation of synthesis and stability of RNA during the acute TNF-induced proinflammatory response. Proc. Natl. Acad. Sci. USA 2013, 110, 2240–2245. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Teran, B.; Pittman, M.; Felix, F.; Thomas, R.; Richmond-Buccola, D.; Huttenhain, R.; Choudhary, K.; Moroni, E.; Costa, M.W.; Huang, Y.; et al. Transcription factor protein interactomes reveal genetic determinants in heart disease. Cell 2022, 185, 794–814.E30. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, Y.; Edwards, G.; Krzizike, D.; Scherman, H.; Luger, K. The histone chaperone FACT modulates nucleosome structure by tethering its components. Life Sci. Alliance 2018, 1, e201800107. [Google Scholar] [CrossRef]
- Tsunaka, Y.; Fujiwara, Y.; Oyama, T.; Hirose, S.; Morikawa, K. Integrated molecular mechanism directing nucleosome reorganization by human FACT. Genes Dev. 2016, 30, 673–686. [Google Scholar] [CrossRef]
- Gurova, K.; Chang, H.W.; Valieva, M.E.; Sandlesh, P.; Studitsky, V.M. Structure and function of the histone chaperone FACT - Resolving FACTual issues. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 892–904. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Galaxy, C. The Galaxy platform for accessible, reproducible, and collaborative data analyses: 2024 update. Nucleic Acids Res. 2024, 52, W83–W94. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- F.K. Trim Galore (RRID: SCR_011847). Available online: https://github.com/FelixKrueger/TrimGalore (accessed on 5 October 2023).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, F.; Dundar, F.; Diehl, S.; Gruning, B.A.; Manke, T. deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef]
- Fuchs, G.; Voichek, Y.; Benjamin, S.; Gilad, S.; Amit, I.; Oren, M. 4sUDRB-seq: Measuring genomewide transcriptional elongation rates and initiation frequencies within cells. Genome Biol. 2014, 15, R69. [Google Scholar] [CrossRef]
- Fuchs, G.; Voichek, Y.; Rabani, M.; Benjamin, S.; Gilad, S.; Amit, I.; Oren, M. Simultaneous measurement of genome-wide transcription elongation speeds and rates of RNA polymerase II transition into active elongation with 4sUDRB-seq. Nat. Protoc. 2015, 10, 605–618. [Google Scholar] [CrossRef]
- Gregersen, L.H.; Mitter, R.; Svejstrup, J.Q. Using TT(chem)-seq for profiling nascent transcription and measuring transcript elongation. Nat. Protoc. 2020, 15, 604–627. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Fei, J. NDF/GLYR1 Promotes RNA Polymerase II Processivity via Pol II Binding and Nucleosome Destabilization. Int. J. Mol. Sci. 2025, 26, 4874. https://doi.org/10.3390/ijms26104874
Li Z, Fei J. NDF/GLYR1 Promotes RNA Polymerase II Processivity via Pol II Binding and Nucleosome Destabilization. International Journal of Molecular Sciences. 2025; 26(10):4874. https://doi.org/10.3390/ijms26104874
Chicago/Turabian StyleLi, Ziwei, and Jia Fei. 2025. "NDF/GLYR1 Promotes RNA Polymerase II Processivity via Pol II Binding and Nucleosome Destabilization" International Journal of Molecular Sciences 26, no. 10: 4874. https://doi.org/10.3390/ijms26104874
APA StyleLi, Z., & Fei, J. (2025). NDF/GLYR1 Promotes RNA Polymerase II Processivity via Pol II Binding and Nucleosome Destabilization. International Journal of Molecular Sciences, 26(10), 4874. https://doi.org/10.3390/ijms26104874