1. Introduction
According to the latest data from the World Health Organization in 2019, asthma has a global incidence rate of 3%, affecting over 600 million individuals with symptoms, resulting in 461,000 deaths annually [
1]. In China, the incidence rate stands at 4.2% [
2]; it is characterized by a large patient base, high incidence, and high deterioration rates. However, challenges such as insufficient diagnosis and treatment persist, leading to reduced quality of life and posing a significant financial burden, particularly for those with poorly controlled asthma and in low-income settings [
3].
Marked by airway inflammation, hyperresponsiveness, and airway remodeling, asthma is a condition closely associated with exposure to allergens or diverse physical and chemical stimuli. Its clinical presentation includes wheezing, coughing, shortness of breath, chest tightness, and breathing difficulties, significantly disrupting patients’ daily activities and overall quality of life [
4]. In more severe cases, asthma can damage airways and impair oxygen flow to the alveoli, potentially leading to life-threatening complications [
3]. Influenced by both genetic and environmental factors, including infections, allergens, and irritants, asthma exhibits heterogeneity in its inflammatory and remodeling processes [
5]. Given its complexity, asthma treatment aims to alleviate symptoms, preserve normal activity levels, and prevent the deterioration of lung function, rather than achieving a complete cure or absolute prevention [
6].
Asthma therapy is guided by a stepwise approach tailored to disease severity and control, as recommended by global guidelines [
7]. All patients require immediate access to a rapid-onset bronchodilator (e.g., short-acting β
2-agonists [SABAs]) for symptom relief. For mild intermittent asthma, as-needed SABA use may suffice, while patients with persistent symptoms or exacerbations require controller medications. First-line controllers include low-dose inhaled glucocorticoids (IGCs), which reduce airway inflammation and are the cornerstone of long-term management. For moderate disease, IGCs are often combined with long-acting β
2-agonists (LABAs) in a single inhaler (fixed-dose combination) to improve adherence and efficacy [
8]. Alternative options for mild-to-moderate asthma include leukotriene receptor antagonists (LTRAs) or theophylline.
Severe asthma warrants high-dose IGCs/LABA combinations, and patients should be evaluated for add-on therapies such as long-acting muscarinic antagonists (LAMAs) or biologic agents. Systemic glucocorticoids should be avoided whenever possible; instead, patients with eosinophilic or type 2 inflammation (e.g., elevated blood eosinophils or FeNO) may benefit from targeted biologics such as anti-IgE (omalizumab), anti-IL-5/IL-5R (mepolizumab, reslizumab, benralizumab), or anti-IL-4/IL-13 (dupilumab) [
9]. These therapies address specific inflammatory pathways and reduce the risk of exacerbation, aligning with the principles of personalized medicine [
10].
However, these treatments have limitations, such as the requirement for lifelong use, ineffectiveness in some patients, and potential side effects including suppression of the hypothalamic–pituitary–adrenal axis, growth retardation, osteoporosis, diabetes, and respiratory infections [
11]. Therefore, there is a significant unmet clinical demand for asthma therapies that are both highly effective and safe. This has prompted a shift towards exploring natural medicines, which offer a plethora of potential drug candidates with minimal adverse effects. These natural compounds often exhibit multiple therapeutic advantages, despite their complex mechanisms of action and potentially unidentified primary targets [
12,
13].
In pursuit of more effective and safer asthma therapies, our research has focused on
Hyssopus cuspidatus Boriss, a perennial herb of the Labiatae family with traditional uses in treating colds, fever, phlegm, and cough. Its anti-allergic inflammation properties have been reported in mouse asthma and anaphylaxis models, with Isochlorogenic acid C (ICGAC) identified as a primary bioactive component [
14,
15]. This dicaffeoylquinic acid, belonging to the caffeoylquinic class of acids, exhibits a wide range of pharmacological effects such as antioxidant, anti-inflammatory, and antimicrobial activities [
14]. Preclinical studies have shown that ICGAC can inhibit eosinophil activation and migration, suppress histamine release from mast cells, and restore the Th1/Th2 immune balance, thereby effectively alleviating asthma symptoms [
16]. Although the therapeutic potential of ICGAC in asthma is evident, its precise mechanisms of action remain to be fully elucidated, particularly given its low bioavailability, which ranges from 14.4% to 16.9%. This suggests a potential role for the gut microbiota in mediating some of these effects, given its critical involvement in immune and inflammatory regulation, as well as its emerging implications in respiratory diseases [
14].
Lipidomics, which involves the comprehensive analysis of thousands of lipid species and their functions, has revealed that lipids, as crucial signaling molecules, modulate various cellular processes in asthma patients’ airways, influencing disease onset and progression [
17]. Notably, asthma has been associated with alterations in glycerophospholipid and sphingomyelin metabolism, leading to the discovery of potential lipid biomarkers such as PC(18:1/18:2), PC(16:0/18:1), and PC(18:0/22:5), serving as valuable references in the research and development of asthma treatments [
18].
The aim of this study is to elucidate the anti-asthma mechanism of ICGAC by adopting an integrative gut–lung axis approach. Employing an Ovalbumin (OVA)-induced asthma mouse model, we place special emphasis on deciphering how ICGAC interacts with the gastrointestinal system when exerting its anti-asthmatic effects, particularly focusing on the gut microbiota and intestinal epithelial cells. Through the application of lipid metabolomics and network pharmacology, we aim to uncover the multifaceted mechanisms of ICGAC in asthma, including its regulation of the lipid metabolism, modulation of inflammatory mediators, immune cell function, and airway responsiveness. Ultimately, this research aims to lay a scientific foundation for the potential clinical application of ICGAC in asthma management.
3. Material and Method
3.1. Material
ELISA kits for the determination of tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, IL-4, IL-5, IL-6, and immunoglobulin E (IgE) were purchased from Cusabio Biotech Co., Ltd. (Wuhan, China). Glutathione (GSH) assay kits were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Mass spectrometry (MS)-grade water, acetonitrile, formic acid, acetic acid, ethanol, isopropanol, methyl tert-butyl ether, and methanol were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Glycerol, OVA, and butylated hydroxytoluene (BHT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Deuterated lipid mediator (LM) standards (IS) were purchased from Cayman (Ann Arbor, MI, USA). Deuterated phosphatidyl choline (PC) standards (IS) were purchased from Avanti (Alabaster, AL, USA). Deuterated propionic acid was purchased from Shanghai Zzbio Co., Ltd. (Shanghai, China). All other reagents were of analytical or high-performance liquid chromatography (HPLC) grade.
3.2. Establishment of the Asthma Mouse Model
All animal experiments were conducted in accordance with the guidelines established by the Animal Care and Use Committee (IACUC) of Xinjiang Medical University and the Ethics Committee of the same institution (XJIMM-20230704). After purchase, the animals were kept in an animal house at a constant temperature (22 °C) and 50% relative humidity; they were fed ad libitum and acclimatized for 1 week during the rearing period.
BALB/C female mice (18–22 g) were divided into five groups: a naïve control group (A), a model group (B), a high-dose ICGAC group (C) (2 mg/mL, 20 mg/kg), a low-dose ICGAC group (D) (1 mg/mL, 10 mg/kg), and a dexamethasone (DEX) group (E) (0.16 mg/mL, 1.6 mg/kg). Group A consists of 6 mice, while Groups B, C, D, E, and F each comprise 8 mice. Mice in groups B, C, D, and E were intraperitoneally injected with 0.2 mL of OVA sensitization solution (aluminum hydroxide gel saline dilution 0.5 mg/mL), and mice in group A were injected with an equal amount of saline from days 1–8. On days 15–21, mice in group A were given saline stimulation, and mice in group B, C, D, E were given atomized OVA stimulation solution (OVA 10 mg/mL) for 30 min once a day for 7 days. Groups C, D, E were administered ICGAC and dexamethasone intragastrically. The dose was determined according to the previous study. After 1 h, atomized OVA was used for stimulation, once a day for 30 min per dose for 7 days. After gavage on the first day, three mice were randomly selected as single-dosage groups from the control and high-dose ICGAC groups. During the experiment, the mice were allowed to eat and drink freely.
3.3. Collection of Bio-Samples
Serum: Blood serum was collected from mouse eye samples and transferred into Eppendorf tubes. The tubes were then left to sit at room temperature for 2 h, followed by centrifugation at 3000 rpm for 15 min. The serum was subsequently stored at −20 °C for IgE detection and a serum components assay.
Bronchoalveolar lavage fluid (BALF): Mice were euthanized and skinned, followed by immediate isolation of the peritracheal tissues. A transverse incision was made in the upper trachea (between the third and fourth tracheal rings), and a 1 mL syringe needle was inserted into the trachea (approximately 2 mm) and secured. The right main bronchus was ligated, and the left lung was flushed three times with saline, 0.5 mL each time (with a recovery rate of 80–90%). The collected BALF was centrifuged twice at 2000 rpm for 10 min each, and the supernatant was aliquoted into 200 μL portions and stored at −80 °C for a subsequent cytokine assay.
Lung tissues: After serum collection, the mice were dislocated and executed. Lung tissues were immediately removed and rinsed clean with saline. The left lung was placed in a 5 mL Eppendorf tube containing 4% paraformaldehyde solution for morphological analysis. The right lung was immediately frozen in liquid nitrogen for 30 min and then transferred to a −80 °C refrigerator for the later analysis of phosphatidyl cholines (PCs), polyunsaturated fatty acids (PUFAs), and lipid mediators (LMs).
Fecal samples: Fecal samples were collected from the colon and immediately snap-frozen in liquid nitrogen. Subsequently, they were stored at −80 °C for later biochemical analysis, including the determination of short-chain fatty acids (SCFAs).
3.4. Biological Sample Preparation for PUFA, LM, and PC Assays
Preparation for the LM Assay: The preparation of lipid mediators begins with homogenizing 5 mg of finely ground lung tissue in 175 μL of acetonitrile through vigorous vortexing for 10 min. After centrifugation at 13,300 rpm for 10 min, the clarified supernatant is carefully collected and combined with stabilizing reagents: 35 μL of 10% glycerol, 1 μL of 10 mg/mL BHT to prevent oxidation, and 10 μL of 50 ng/mL internal standard. The mixture is diluted with 359 μL of 25% acetonitrile and vortexed again to ensure homogeneity. For purification, the sample is passed through a Waters MAX solid phase extraction (SPE) column (Milford, MA, USA) preactivated with acetonitrile and equilibrated with 25% acetonitrile. Sequential washes with 25% acetonitrile and pure acetonitrile eliminate interfering substances, while the target analytes are eluted using 1% formic acid in acetonitrile. The eluate is concentrated under vacuum and stored at −20 °C until analysis. Prior to ultra-high-performance liquid chromatography—multiple reaction monitoring (UHPLC-MRM) detection, the samples are reconstituted in a methanol/acetonitrile solution (50:50, v/v) to optimize chromatographic compatibility.
Preparation for the PC Assay: Building on the residual material from the LMs assay, phosphatidylcholine extraction involves supplementing the remaining supernatant with 50 μL of methanol and 1 mL of methyl tert-butyl ether (MTBE). The biphasic system is vortexed for 10 min to enhance lipid partitioning, followed by centrifugation at 13,300 rpm to isolate the organic phase. A 200 μL aliquot of the upper layer is transferred and dried under vacuum to remove solvents, with the resulting residue stored at −20 °C for stability. For downstream ultra-high-performance liquid chromatography—high-resolution mass spectrometry (UHPLC-HRMS) analysis, dried PC extracts are resolubilized in acetonitrile containing a 200 ng/mL internal standard.
This sequential methodology not only maximizes sample utilization but also minimizes cross-contamination risks between lipid classes, ensuring analytical specificity for both LMs and PCs within a unified workflow.
3.5. Biochemical Assays in Bronchoalveolar Lavage Fluid
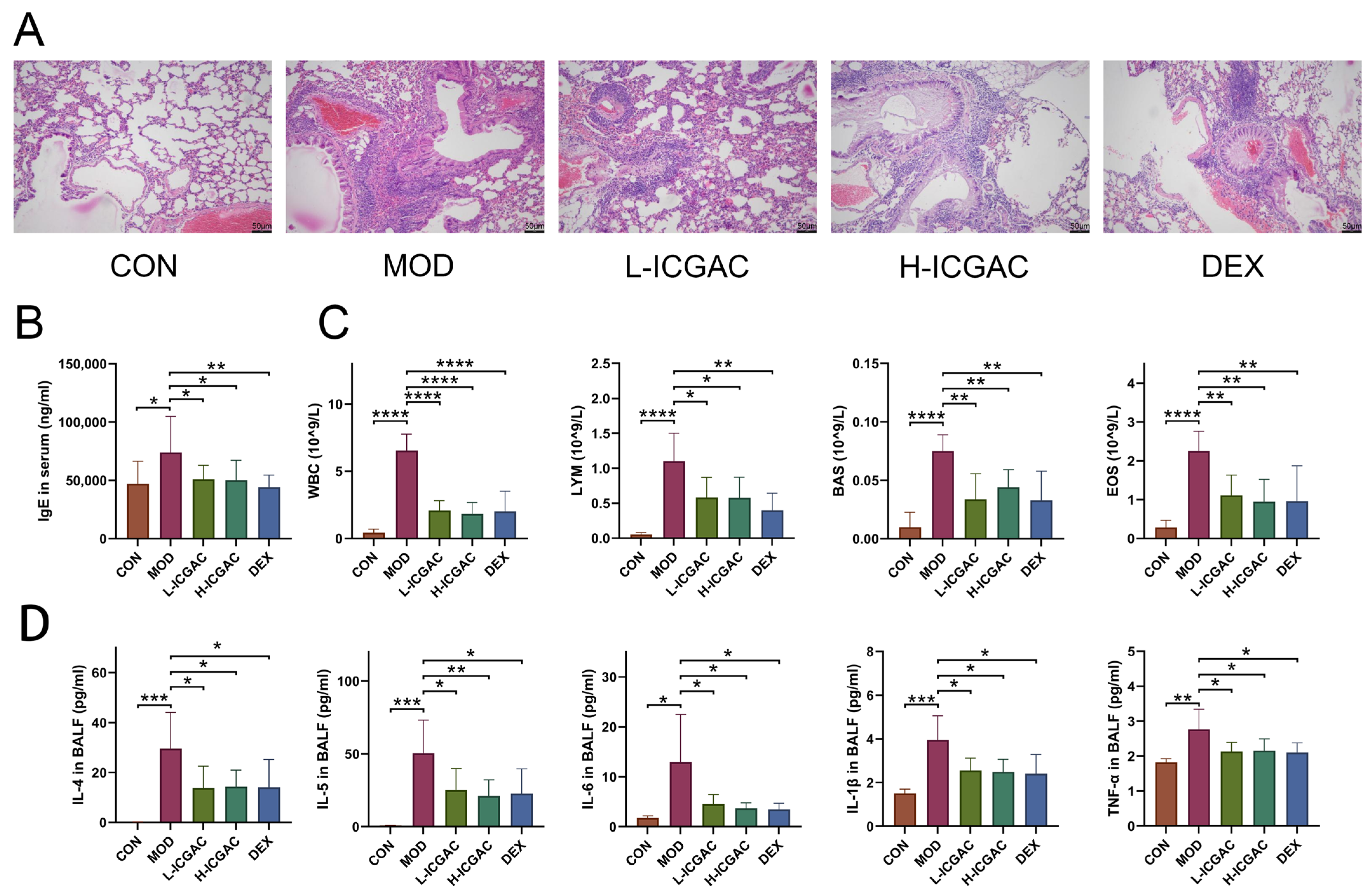
The collected BALF was centrifuged at 3500 rpm for 15 min. The cell pellet was suspended in 1 mL phosphate-buffered saline (PBS) for inflammatory cell counting and classification. Levels of basophils (BAS), eosinophils (EOS) white blood cells (WBC), and lymphocytes (LYM) in BALF were measured using an automatic cell counter (Nihon Kohden, Shinjuku, Japan) according to the manufacturer’s protocol. Levels of IL-4, IL-5, IL-6, IL-1β, TNF-α in BALF and the total IgE level in serum were measured using corresponding kits.
3.6. Biological Sample Preparation for Active Components Analysis
We mixed 10 µL of serum and 40 µL of methanol containing 0.1% formic acid. After centrifugation at 13,300 rpm for 10 min, 30 µL of the supernatant was taken and then subjected to mass spectrometry analysis.
3.7. Biological Sample Preparation for Fecal SCFAs Analysis
In brief, 10 mg of feces was mixed with 600 μL of water containing 0.5% concentrated sulfuric acid. The mixture was vortexed for 5 min and sonicated for 30 min and then centrifuged for 10 min at 13,300 rpm. Then, 500 μL of the supernatant was collected and extracted with 800 μL of MTBE. The mixture was vortexed for 5 min and centrifuged for 10 min at 13,300 rpm. Subsequently, 700 μL of the supernatant was collected and mixed with 0.15 g of anhydrous sodium sulfate. From this, another 500 μL aliquot was taken and combined with 500 μL of Propionic Acid-d3 (200 μM). After vortexing for another 5 min, the mixture was transferred to a chromatographic vial for analysis.
3.8. Real-Time Quantitative PCR Analysis
Total RNA was extracted from lung tissues using the RNAeasy™ Animal RNA Isolation Kit with Spin Column (Beyotime, Shanghai, China). The concentration of total RNA was quantified using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Then, 1 μg of purified RNA from each sample was reverse-transcribed into complementary DNA (cDNA) using the HiFi-Script cDNA Synthesis Kit (CWBIO, Taizhou, Jiangsu, China). Quantitative reverse transcription PCR (qRT-PCR) was performed using an ABI 7500 Fast PCR system (Applied Biosystems, San Francisco, CA, USA) using UltraSYBR Mixture (Low ROX) (CWBIO, Taizhou, China). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was employed as an internal control for normalization. The relative expression levels were calculated as fold changes in comparison to GAPDH, using the 2−ΔΔCT method.
3.9. The Determination of Glutathione (GSH)
First, we accurately weighed the tissue (approximately 0.005 g) and proceeded to homogenize it in physiological saline using a weight-to-volume ratio of 1:9 (g:mL). Subsequently, we centrifuged the homogenate at 2500 rpm for 10 min and then collected 0.1 mL of the supernatant. Following the manufacturer’s instructions from the Reduced Glutathione (GSH) Assay Kit (Njjcbio, Nanjing, China), we added 0.1 mL of the reagent and mixed it thoroughly. We then centrifuged the mixture at 3500 rpm for 10 min and collected the supernatant for colorimetric analysis.
3.10. Histology and Immunofluorescence Staining
Lung tissues were fixed with 4% paraformaldehyde for 48 h, routinely dehydrated, paraffin embedded, cut into 4 μm thick sections, stained with hematoxylin-eosin (H&E), and observed under light microscope for inflammatory cell infiltration in lung tissues. The expression of glutathione peroxidase 4 (GPX4) in the lung tissue was detected via an immunofluorescence assay using anti-GPX4 antibody. After washing, the slides were incubated with Alexa Fluor 488-conjugated goat anti-mouse (Lot: A10680) and CY3-conjugated goat anti-rabbit secondary antibody (Lot: A10520, Thermo Fisher Scientific). Slides were washed with PBS and counterstained with 4′,6-Diamidino-2-Phenylindole (DAPI). Fluorescence images were obtained using a whole-slide imaging system.
As described in our previous study [
14]. Microbiota analysis was conducted using the Majorbio Cloud Platform (
https://cloud.majorbio.com/; access date: 7 October 2024). α- and β-diversity, along with bacterial community composition, were assessed using the Quantitative Insights Into Microbial Ecology (QIIME) pipeline. Additionally, the linear discriminant analysis (LDA) effect size (LEfSe) was employed with a threshold LDA score >3.0 to identify differentially abundant taxa.
3.11. UPLC-MRM Analysis
The analysis of active components in serum samples was carried out using Waters Synapt G2-Si High-Definition Mass Spectrometry (HDMS) with H-class ultra-performance liquid chromatography (UPLC) and an Acquity Bridged Ethylene Hybrid (BEH) C18 column. Mobile phase A consisted of water containing 0.1% acetic acid, while mobile phase B comprised acetonitrile with 0.1% acetic acid. The solvent procedure is shown in
Table 1. The injection volume was 2 μL, and the column temperature was 30 °C. MS was operated in negative mode under MRM. Data acquisition was performed using MassLynx 4.2.
PUFAs and LMs analyses were performed on a SCIEX 5500plus mass spectrometer equipped with an Electrospray Ionization (ESI) source (Framingham, MA, USA) and a Thermo Scientific Dionex ultimate 3000 HPLC (Waltham, MA, USA). The Waters ACQUITY UPLC BEH C18 (1.7 μm, 2.1 × 50 mm) column was used. Mobile phase A consisted of water with 0.1% acetic acid, whereas mobile phase B was a mixture of acetonitrile and isopropanol in a 9:1 volume ratio. The solvent procedure is shown in
Table 2. The flow rate was set at 0.4 mL/min, with the column temperature maintained at 40 °C. An injection volume of 5 μL was used, and the MS was operated in negative mode under MRM. All data were acquired and processed using SCIEX Analyst software (version 1.7.1).
3.12. UPLC-MS Analysis
PCs analysis was performed on a SCIEX 5500plus mass spectrometer equipped with an ESI source (MA, USA) and a Thermo Scientific Dionex ultimate 3000 HPLC (MA, USA). The Waters ACQUITY UPLC C8 (1.7 μm, 2.1 × 100 mm) column was used. Mobile phase A consisted of a mixture of acetonitrile and water (3:2
v/
v) containing 0.1% acetic acid and 10 mM ammonium acetate. Mobile phase B was a blend of acetonitrile and isopropanol (1:9
v/
v) with 0.1% acetic acid and 10 mM ammonium acetate. The solvent procedure is shown in
Table 3. The flow rate was set at 0.3 mL/min and the column temperature was maintained at 55 °C. The injection volume was 2 μL. MS was operated in positive mode under full scans. All data were acquired and processed using SCIEX Analyst software (version 1.7.1).
3.13. GC-MS Analysis
The SCFA analysis was conducted on an Agilent 7890B (Santa Clara, CA, USA) equipped with an automatic sampler (7693A) and coupled to an Agilent 5973 mass selective detector. The column employed was a fused-silica capillary column featuring a free fatty acid phase (DB-FFAP 122-3232) with dimensions of 0.25 mm internal diameter, 30 m length, and 0.25 μm film thickness. The helium carrier gas flow rate was set at 1 mL/min. The oven temperature was initially set at 50 °C and held for 1.5 min. It was then ramped up to 90 °C at a rate of 20 °C/min and held for 2.1 min. Subsequently, it was increased to 120 °C at a rate of 5 °C/min and held for 1 min. Finally, it was raised to 180 °C at a rate of 25 °C/min and held for an additional 1.5 min. The injected sample volume for the GC analysis was 0.5 μL, and the run time for each analysis was 16.5 min. The detector was operated in the selection ionization mode (SIM). Ion selection of the SCFAs was based on the retention time of standard compounds. All data were acquired and processed using Agilent MassHunter (version B.07.01).
3.14. Data Processing and Statistical Analysis
The open-source tool MetaboAnalyst 6.0 (available at
http://www.metaboanalyst.ca/; access date: 15 January 2025) was utilized for the statistical analysis. The threshold for selecting important predictive variables was based on the variable importance in projection (VIP) score. Multivariate statistical differences were identified using an independent sample
t-test. Variables with a VIP score greater than 1 and a
p-value less than 0.05 were considered significant and selected as differential variables.
4. Discussion
In this study, ICGAC, identified as the key active ingredient in SXCF (a traditional folk remedy historically used for asthma relief in China), was assessed in an allergic asthma mouse model. Both dexamethasone and ICGAC significantly reduced typical lung morphological changes in asthma, such as pronounced inflammatory cell infiltration, bronchial epithelial degeneration, and interstitial inflammation. Remarkably, ICGAC exhibited therapeutic efficacy comparable to or exceeding that of dexamethasone, while demonstrating no experimentally detectable adverse effects commonly associated with steroid use. This highlights ICGAC’s potential as a safe and effective natural therapeutic option for managing allergic asthma.
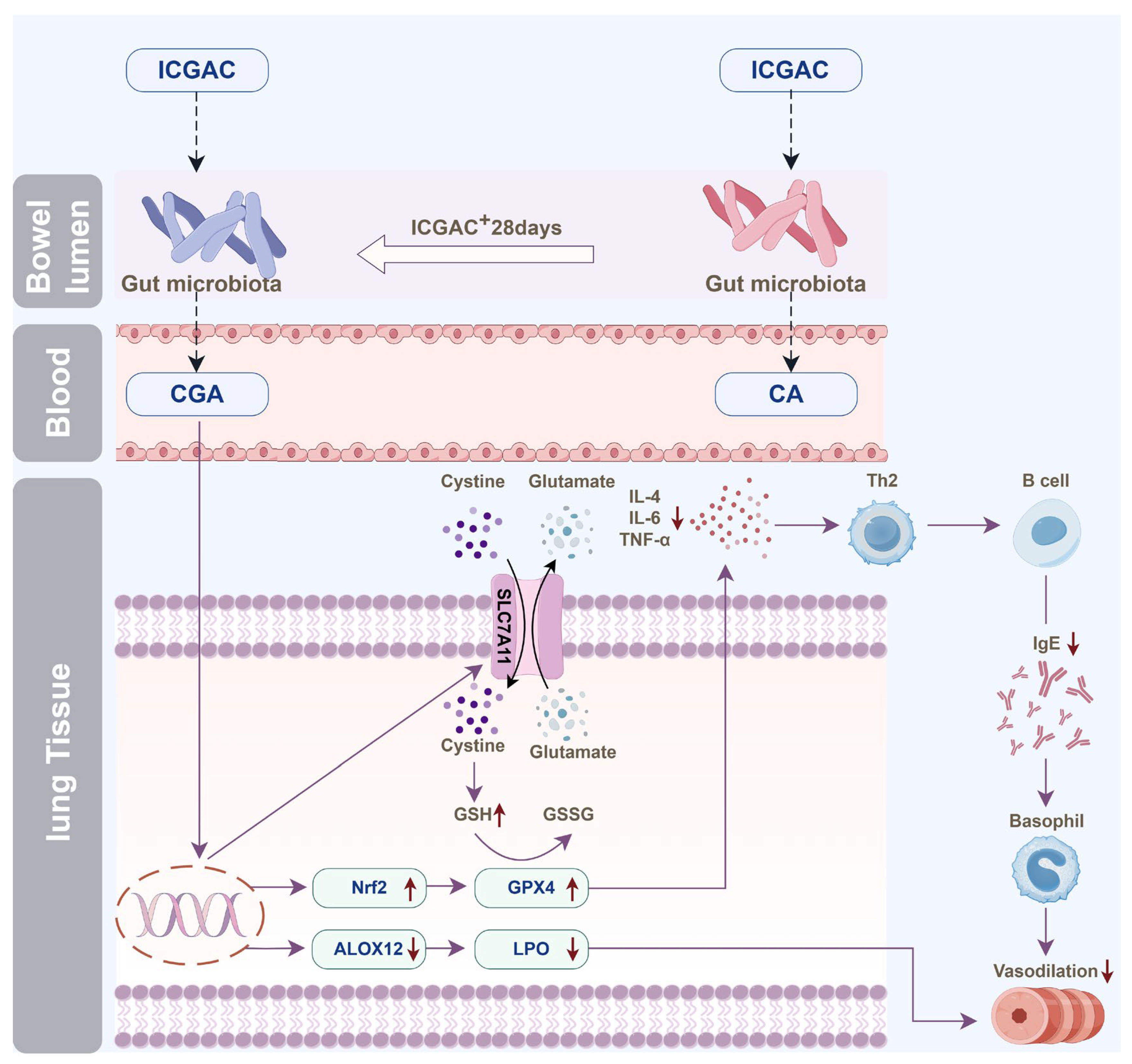
4.1. Oral ICGAC Boosts Gut-Microbiota-Derived CGA and SCFAs as Bioactive Metabolites
We discovered that orally administered ICGAC is not the direct effective component; instead, it undergoes transformation into Chlorogenic acid. This transformation process is mediated by the gut microbiota, a complex community of microorganisms that play crucial physiological roles in food digestion, nutrient absorption, drug metabolism, and maintenance of gut barrier function. Upon entering the intestine, ICGAC may interact with these microorganisms, leading to its conversion into CGA.
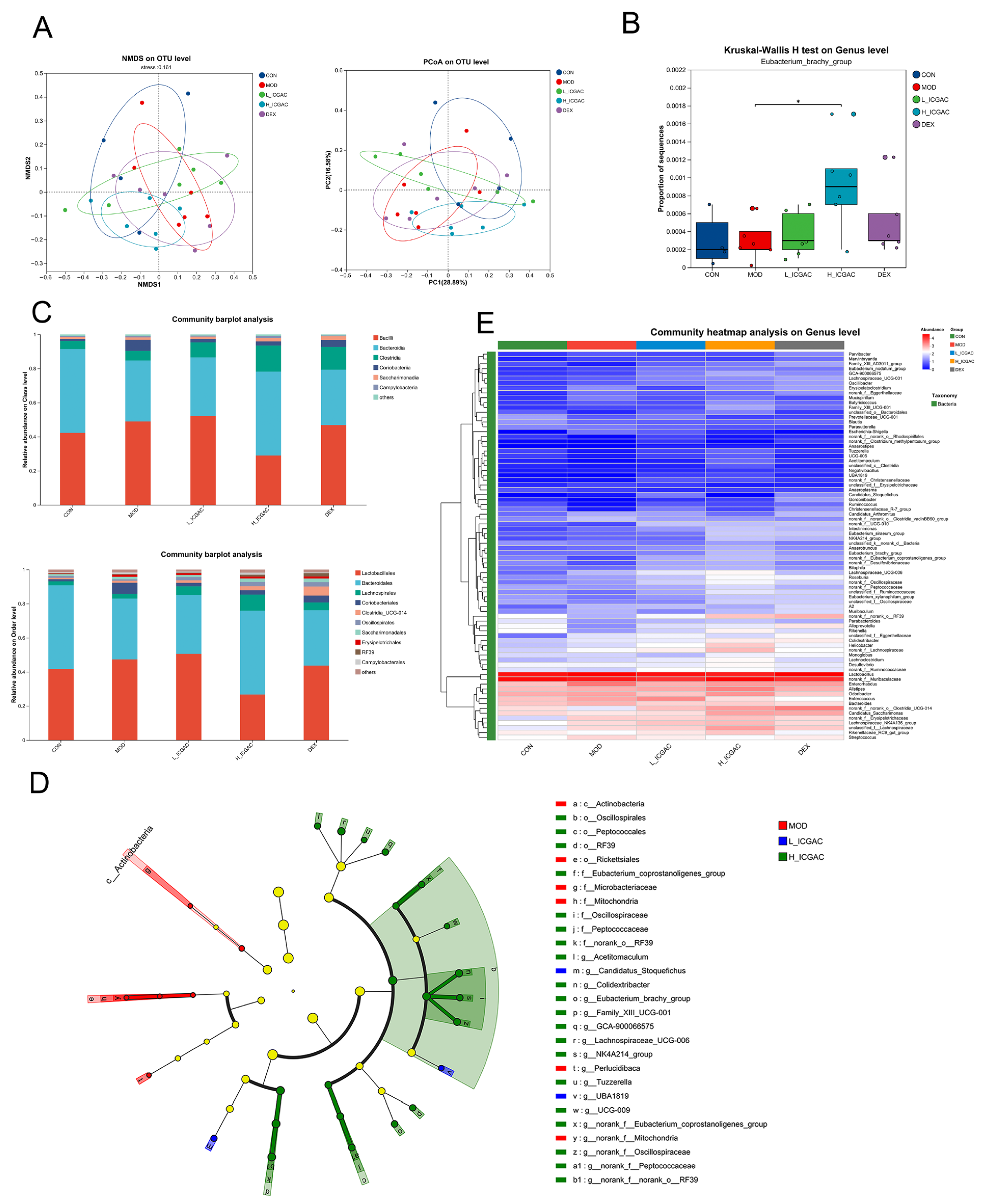
Although the involvement of the gut microbiota in such transformation processes is well recognized, the specific bacterial species or strains responsible for converting ICGAC to CGA remain unclear. However, several studies have emphasized the significant role of the gut microbiota in the biotransformation of bioactive compounds [
21]. For example, certain gut bacteria are capable of hydrolyzing, reducing, or methylating compounds, thereby altering their chemical structures and biological activities [
22]. Our study detected a significant increase in the
Eubacterium_brachy_group, suggesting its potential role in the conversion of ICGAC to CGA through analogous microbial metabolic pathways. To date, several studies have demonstrated that chlorogenic acid exhibits anti-inflammatory effects via the modulation of the intestinal microbiota [
23].
Natural compounds frequently undergo structural modifications during absorption. Moreover, the effective form and pharmacological mechanisms of natural drugs can vary between single-dose and prolonged administrations [
24]. Further research is essential to fully elucidate the intricate interactions between natural products, gut microbiota, and therapeutic efficacy.
Using UPLC-tandem mass spectrometry, we measured serum ICGAC and chlorogenic acid levels. Both H-ICGAC and L-ICGAC groups showed similar chlorogenic acid concentrations in the bloodstream (
Figure 2A). This similarity can be attributed to the enhanced expression of P-glycoprotein (PGP) in the gut endothelial cells of the H-ICGAC group, facilitated by ICGAC.
PGP, an ATPase efflux pump that is a member of the ATP-binding cassette transporter family, is recognized for its ability to extrude a variety of lipophilic drugs. Contemporary studies have underscored a significant association between gut microbiota and PGP expression within the colon [
25]. Specifically,
Clostridium and
Bacillus have been identified as key in stimulating PGP expression in the gut epithelium of mouse models, contributing to the observed correlation [
25]. This suggests that ICGAC may upregulate PGP, leading to stable chlorogenic acid serum levels irrespective of the initial dose administered.
ICGAC also enhances the synthesis of SCFAs—notably, acetic acid and butyric acid—upon administration. SCFAs play a crucial role in modulating the immune system and reducing inflammation [
26]. They also regulate various immune cells and influence T and B cell differentiation, impacting both innate and adaptive responses [
27]. Furthermore, SCFAs reduce airway inflammation, a hallmark feature of allergic asthma [
14].
Indeed, multiple studies emphasize the connection between SCFAs and allergic asthma. Changes in the gut microbiota, which produce SCFAs, can influence the risk of developing allergic asthma [
14]. While, early-life antibiotic exposure disrupts the gut microbiota, increasing the risk of asthma and other allergic conditions [
28]. Moreover, SCFAs modulate immune-related genes, potentially contributing to allergic asthma [
29]. Intriguingly, our findings further show a positive correlation between butyrate production and PGP expression, highlighting its importance. A detailed analysis revealed that
Eubacterium_brachy_group plays a key role in SCFA production following ICGAC administration (
Figure 3).
4.2. ICGAC Modulates ALOX Activity and LMs to Reduce Asthmatic Inflammation
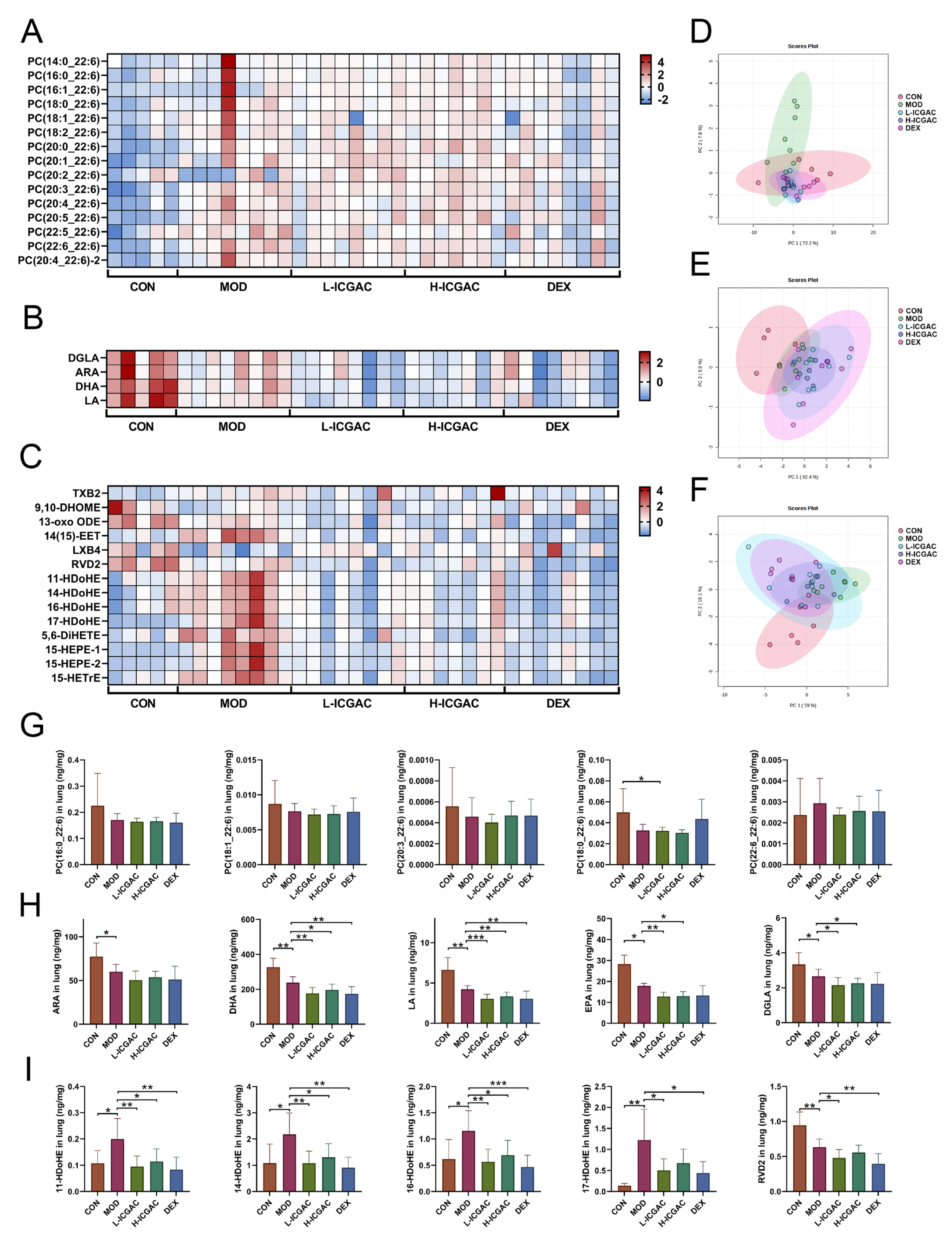
We explored the regulatory role of ICGAC in LMs metabolism and its anti-inflammatory potential through Lipidomics analysis. Our investigation focused on the activities of arachidonate lipoxygenases (ALOX-isoforms), particularly ALOX12 and ALOX15, which catalyze the oxidation of polyunsaturated fatty acids (PUFAs) and play crucial roles in inflammatory responses [
30].We found that ICGAC treatment inhibited ALOX12 and ALOX15 activities, specifically reducing the enzyme-catalyzed oxidation of DHA (
Figure 6A). DHA, an omega-3 PUFA, serves as a precursor for anti-inflammatory metabolites such as 11-HDoHE, 14-HDoHE, 16-HDoHE, 17-HDoHE, and RVD2, which are crucial in resolving inflammation in allergic asthma [
31]. Disruption of their balance can result in heightened inflammatory reactions and asthma symptoms.
Consistently with this, chlorogenic acid, the active compound in ICGAC, has also been identified as a competitive inhibitor of ALOX, thereby inhibiting lipid peroxidation [
32]. Taken together, our findings indicate that ICGAC modulates the metabolic networks of LMs and PUFAs, potentially exerting anti-inflammatory effects by regulating ALOX activity and LMs levels.
4.3. ICGAC Enhances GPX4, SLC7A11, and Nrf2 to Strengthen Antioxidant Defense
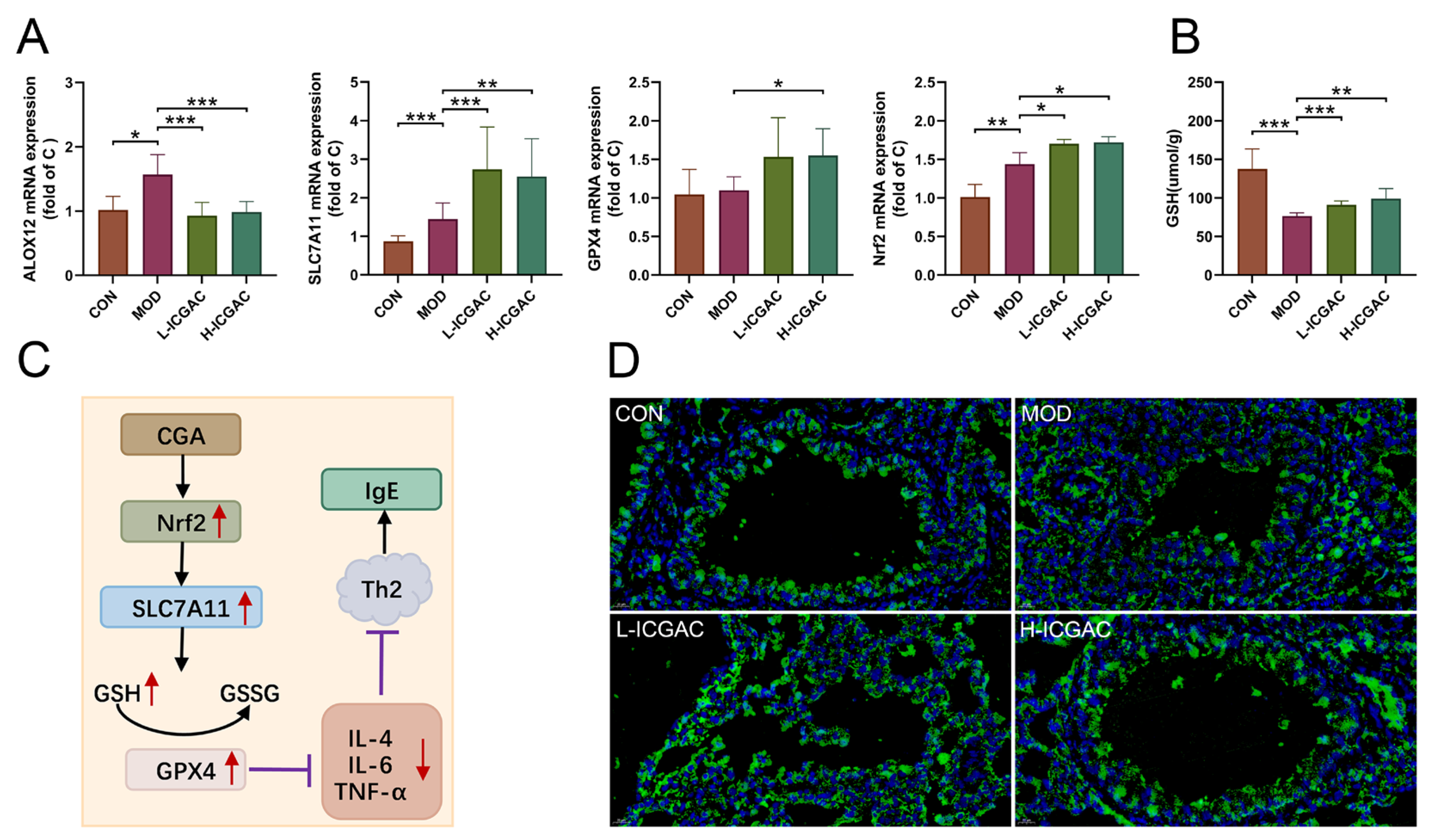
In this study, we explored how ICGAC suppresses asthma-related inflammation and oxidative stress, specifically by upregulating GPX4, SLC7A11, and Nrf2 in lung tissues.
Firstly, GPX4, an enzyme found on the outer surface of the inner mitochondrial membrane and within the mitochondrial matrix, is unique in its ability to directly reduce phospholipid peroxides (PL-OOH) in the mitochondrial membrane [
33]. By using GSH as a co-substrate, GPX4 plays a vital role in regulating cellular lipid peroxides. Our findings revealed that the ICGAC treatment markedly increased GPX4 expression (
Figure 6A), thereby enhancing the uptake and reduction in the peroxide load in PL-OOH, converting it into nontoxic lipid alcohol while oxidizing glutathione (reduced, GSH) to glutathione (oxidized, GSSG) [
34].
Secondly, ICGAC significantly upregulated SLC7A11 expression (
Figure 6A), which aids in the uptake of extracellular cystine for conversion to cysteine, a crucial precursor for glutathione synthesis [
35]. Consistently with this upregulation, we observed a notable increase in GSH content in the lung tissue of ICGAC-treated mice compared to the model group.
Additionally, Nrf2, a member of the basic leucine zipper transcription factor family, is a central regulator of the antioxidant response. GPX4, a well-known transcriptional target of Nrf2, is upregulated by Nrf2 activation, creating a positive feedback loop that enhances cellular antioxidant defenses [
36]. Our results indicated that ICGAC also increased Nrf2 and GPX4 expression (
Figure 6A), further strengthening the antioxidant response.
Moreover, dendritic cells (DCs) facilitate the differentiation of naive CD4
+ T cells to Th2 cells, which further stimulate B cells to produce IgE [
37]. IL-4 amplifies Th2 activity, but elevated levels of Nrf2 inhibit its release [
38]. ICGAC suppressed IL-4 and other asthma-related cytokines, thus reducing IgE production by B cells and alleviating asthma symptoms by lowering airway responsiveness (
Figure 7).


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}