
Microbiome Markers in Gastrointestinal Disorders: Inflammatory Bowel Disease, Colorectal Cancer, and Celiac Disease

 ,
,  ,
,  , and
, and
Abstract
1. Introduction
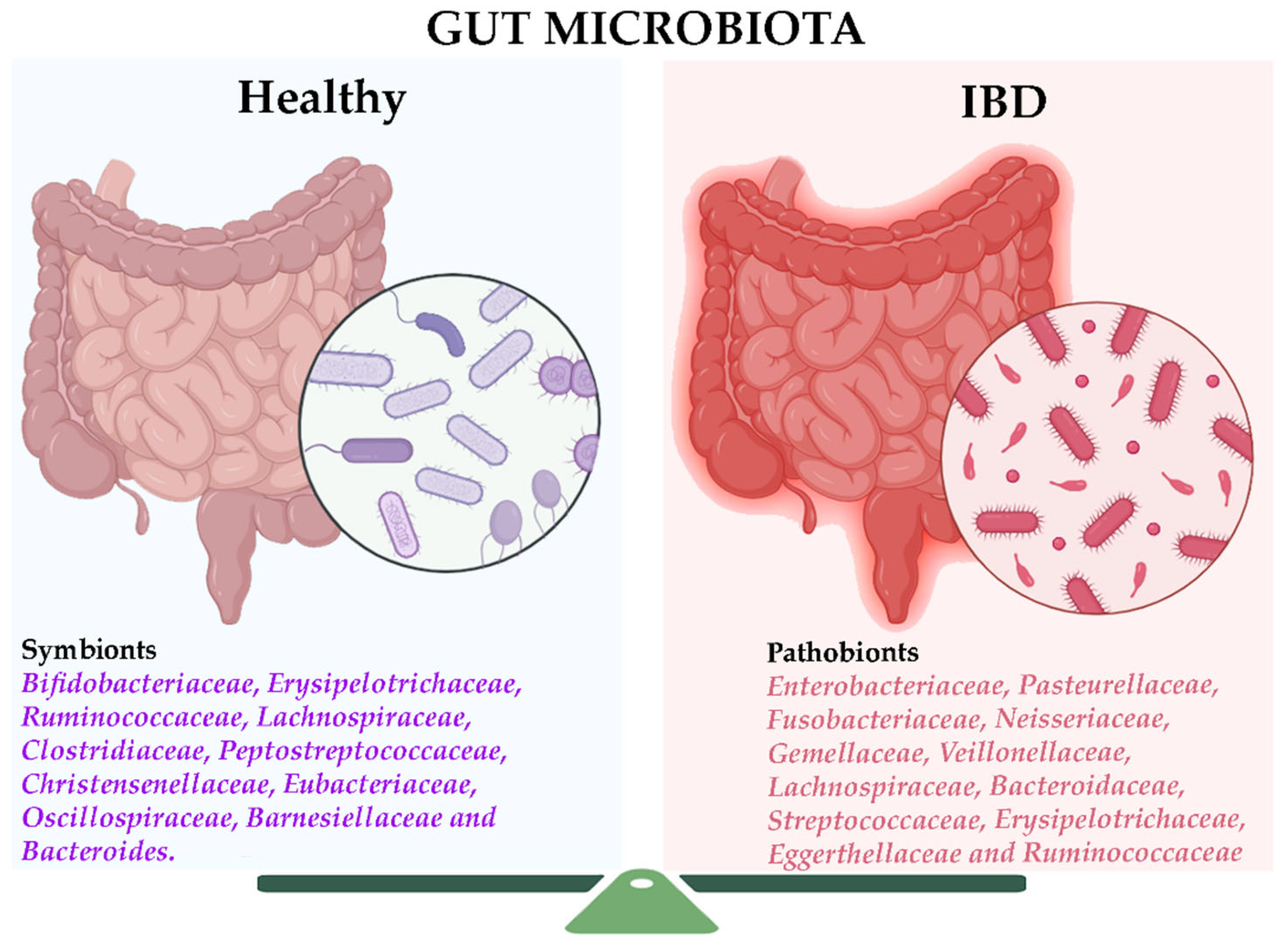
2. The Gut Microbiome in Inflammatory Bowel Disease
2.1. Bacterial Markers in IBD
- (i)
- (ii)
- (iii)
- (iv)
- (v)
- (vi)
- An increase in virulence factors and antibiotic resistance, possibly linked to the rise of Enterobacteriaceae and Bacteroides.
2.2. Non-Bacterial Markers in IBD
3. The Gut Microbiome in Colorectal Cancer
3.1. Bacterial Markers in CRC
3.2. Non-Bacterial Markers in CRC
4. The Gut Microbiome in Celiac Disease
4.1. Bacterial Markers in CeD

{kind=link}
{kind=link}
{kind=link}
| Study | Complexity of Study | Sequencing Method | Highlighted Bacterial Markers | |
|---|---|---|---|---|
| Costigan et al., 2024 [139] | One cohort with CeD before and after a GFD N = 36 fecal samples. One cohort HVs * N = 36 fecal samples. | Shotgun | CeD microbiota | |
| Before GFD | After GFD (12 months) | |||
| Increased vs. control Escherichia coli, Enterobacter, and Peptostreptococcus | Increased Blautia wexlerae Reduced Bifidobacteria | |||
| Kelley et al. (2025) [142] | One cohort of CeD progressors. Children and one cohort of age-matched healthy children (1–5 years old). N = 5–16 fecal samples. | 16S | Microbial markers in CeD progressor at 1 year old | |
| Increased: Firmicutes, Ruminococcus bromii, Dialister invisus, Bifidobacterium dentium, Clostridium, Lachnospiraceae, Alistipes, and Faecalibacterium prausnitzii. | Under-representation of Lactobacillus or Eubacterium. | |||
| Altered microbiota years before the onset of CeD. | ||||
| Rentala et al. (2017) [145] | One cohort of non-CeD children with a high genetic risk of CeD. N = 27 fecal samples. | 16S | Fecal microbiota composition differences between | |
| Children who later developed CD | Children without disease or associated autoantibodies | |||
| No statistically significant | ||||
| Bodkhe et al. (2019) [146] | One cohort of non-GFD CeD patients (Marsh > 2) (N = 23); one cohort of FDRs of CeD patients (N = 15), and one no-CeD control cohort (N = 24). Duodenal biopsy and fecal samples. | 16S | FDR patients showed | |
| Over-representation: Parvimonas, Granulicatella, Gemella, Bifidobacterium, Anaerostipes, and Actinomyces. | Under-representation: Akkermansia, Dorea, Lactobacillus, and Haemophilus. | |||
| El Mouzan et al. (2022) [147] | One cohort of children with CeD (N = 40), one cohort with healthy (N = 20), and non-CeD children (N = 19). Mucosal and fecal samples. | Shotgun | CeD children’s mucosa | CeD/non-CeD stools |
| Over-representation: Bifidobacterium angulatum and Roseburia intestinalis. | 169 bacterial species with significantly different abundances between individual types. | |||
| Olivares et al. (2018) [148] | One cohort of full-term newborns with at least one first-degree relative with CeD (N = 127). Fecal samples. | 16S | Pathogenic bacteria in newborns with a high risk of CeD. | |
| Increased: Clostridium perfringens, Clostridium difficile, and Escherichia coli. | ||||
| Sanchez et al. (2013) [152] | One cohort of active CeD patients (N = 32), non-active CeD patients (N = 17), and controls (N = 8). Duodenal biopsy. | 16S | Active CeD children | |
| Increased: Proteobacteria Enterobacteriaceae, Staphylococcaceae (Klebsiella oxytoca, Staphylococcus epidermidis, and Staphylococcus pasteuri) | Decreased: Firmicutes and Streptococcaceae. | |||
| D’Argenio et al. (2016) [153] | Cohorts of active CeD adults (N = 20), CeD adults in GFD (N = 6), and controls (N = 16). Duodenal biopsy. | 16S | Active CeD patients | |
| Increased: Proteobacteria, Neisseria genus (Neisseria flavescens was the most abundant Neisseria species in the duodenum). | Decreased: Firmicutes and Actinobacteria. | |||
| Wacklin et al., 2014 [156] | CeD patients in GFD with (N = 18) or without persistent symptoms (N = 18). | 16S | GFD-treated patients with persistent symptoms | |
| Increased: Proteobacteria (p = 0.04) | Decreased: Bacteroidetes and Firmicutes (p = 0.05). Microbial richness. | |||
4.2. Non-Bacterial Markers in CeD
5. Advantages and Limitations of Sequencing Methodologies in Microbiome Studies
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hou, K.; Wu, Z.-X.; Chen, X.-Y.; Wang, J.-Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in Health and Diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Garavaglia, B.; Vallino, L.; Amoruso, A.; Pane, M.; Ferraresi, A.; Isidoro, C. The Role of Gut Microbiota, Immune System, and Autophagy in the Pathogenesis of Inflammatory Bowel Disease: Molecular Mechanisms and Therapeutic Approaches. Asp. Mol. Med. 2024, 4, 100056. [Google Scholar] [CrossRef]
- Pedroza Matute, S.; Iyavoo, S. Exploring the Gut Microbiota: Lifestyle Choices, Disease Associations, and Personal Genomics. Front. Nutr. 2023, 10, 1225120. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Huang, C.; Xu, J.; Xu, H.; Liu, L.; Zhao, H.; Wang, J.; Huang, W.; Peng, W.; Chen, Y.; et al. Gut Microbiota Is a Potential Biomarker in Inflammatory Bowel Disease. Front. Nutr. 2022, 8, 818902. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Peng, J.; Cai, P.; Xia, Y.; Yi, C.; Shang, A.; Akanyibah, F.A.; Mao, F. The Emerging Role of the Gut Microbiota and Its Application in Inflammatory Bowel Disease. Biomed. Pharmacother. 2024, 179, 117302. [Google Scholar] [CrossRef] [PubMed]
- Noecker, C.; McNally, C.P.; Eng, A.; Borenstein, E. High-Resolution Characterization of the Human Microbiome. Transl. Res. 2017, 179, 7–23. [Google Scholar] [CrossRef]
- Schirmer, M.; Garner, A.; Vlamakis, H.; Xavier, R.J. Microbial Genes and Pathways in Inflammatory Bowel Disease. Nat. Rev. Microbiol. 2019, 17, 497–511. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-Omics of the Gut Microbial Ecosystem in Inflammatory Bowel Diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Liang, J.Q.; Li, T.; Nakatsu, G.; Chen, Y.-X.; Yau, T.O.; Chu, E.; Wong, S.; Szeto, C.H.; Ng, S.C.; Chan, F.K.L.; et al. A Novel Faecal Lachnoclostridium Marker for the Non-Invasive Diagnosis of Colorectal Adenoma and Cancer. Gut 2020, 69, 1248–1257. [Google Scholar] [CrossRef]
- Chassaing, B.; Koren, O.; Carvalho, F.A.; Ley, R.E.; Gewirtz, A.T. AIEC Pathobiont Instigates Chronic Colitis in Susceptible Hosts by Altering Microbiota Composition. Gut 2014, 63, 1069–1080. [Google Scholar] [CrossRef]
- Palmela, C.; Chevarin, C.; Xu, Z.; Torres, J.; Sevrin, G.; Hirten, R.; Barnich, N.; Ng, S.C.; Colombel, J.-F. Adherent-Invasive Escherichia Coli in Inflammatory Bowel Disease. Gut 2018, 67, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Suda, W.; Luo, C.; Kawaguchi, T.; Motoo, I.; Narushima, S.; Kiguchi, Y.; Yasuma, K.; Watanabe, E.; Tanoue, T.; et al. Ectopic Colonization of Oral Bacteria in the Intestine Drives T H 1 Cell Induction and Inflammation. Science 2017, 358, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Su, X.; Zhang, C.; Chen, W.; Wang, Y.; Yang, X.; Liu, D.; Zhang, Y.; Yang, R. Klebsiella pneumoniae Induces Inflammatory Bowel Disease Through Caspase-11–Mediated IL18 in the Gut Epithelial Cells. Cell Mol. Gastroenterol. Hepatol. 2023, 15, 613–632. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Ishimoto, T.; Fu, L.; Zhang, J.; Zhang, Z.; Liu, Y. The Gut Microbiota in Inflammatory Bowel Disease. Front. Cell Infect. Microbiol. 2022, 12, 733992. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal Microbe-Derived Butyrate Induces the Differentiation of Colonic Regulatory T Cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Tufail, M.A.; Schmitz, R.A. Exploring the Probiotic Potential of Bacteroides spp. Within One Health Paradigm. Probiotics Antimicrob. Proteins 2025, 17, 681–704. [Google Scholar] [CrossRef]
- Chu, H.; Khosravi, A.; Kusumawardhani, I.P.; Kwon, A.H.K.; Vasconcelos, A.C.; Cunha, L.D.; Mayer, A.E.; Shen, Y.; Wu, W.-L.; Kambal, A.; et al. Gene-Microbiota Interactions Contribute to the Pathogenesis of Inflammatory Bowel Disease. Science 2016, 352, 1116–1120. [Google Scholar] [CrossRef]
- Yang, J.-Y.; Kim, M.-S.; Kim, E.; Cheon, J.H.; Lee, Y.-S.; Kim, Y.; Lee, S.-H.; Seo, S.-U.; Shin, S.-H.; Choi, S.S.; et al. Enteric Viruses Ameliorate Gut Inflammation via Toll-like Receptor 3 and Toll-like Receptor 7-Mediated Interferon-β Production. Immunity 2016, 44, 889–900. [Google Scholar] [CrossRef]
- Adiliaghdam, F.; Amatullah, H.; Digumarthi, S.; Saunders, T.L.; Rahman, R.-U.; Wong, L.P.; Sadreyev, R.; Droit, L.; Paquette, J.; Goyette, P.; et al. Human Enteric Viruses Autonomously Shape Inflammatory Bowel Disease Phenotype through Divergent Innate Immunomodulation. Sci. Immunol. 2022, 7, eabn6660. [Google Scholar] [CrossRef]
- Cadwell, K.; Patel, K.K.; Maloney, N.S.; Liu, T.-C.; Ng, A.C.Y.; Storer, C.E.; Head, R.D.; Xavier, R.; Stappenbeck, T.S.; Virgin, H.W. Virus-Plus-Susceptibility Gene Interaction Determines Crohn’s Disease Gene Atg16L1 Phenotypes in Intestine. Cell 2010, 141, 1135–1145. [Google Scholar] [CrossRef]
- Tarris, G.; de Rougemont, A.; Charkaoui, M.; Michiels, C.; Martin, L.; Belliot, G. Enteric Viruses and Inflammatory Bowel Disease. Viruses 2021, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Limon, J.J.; Tang, J.; Li, D.; Wolf, A.J.; Michelsen, K.S.; Funari, V.; Gargus, M.; Nguyen, C.; Sharma, P.; Maymi, V.I.; et al. Malassezia Is Associated with Crohn’s Disease and Exacerbates Colitis in Mouse Models. Cell Host Microbe 2019, 25, 377–388.e6. [Google Scholar] [CrossRef]
- Sivignon, A.; de Vallée, A.; Barnich, N.; Denizot, J.; Darcha, C.; Pignède, G.; Vandekerckove, P.; Darfeuille-Michaud, A. Saccharomyces Cerevisiae CNCM I-3856 Prevents Colitis Induced by AIEC Bacteria in the Transgenic Mouse Model Mimicking Crohn’s Disease. Inflamm. Bowel Dis. 2015, 21, 276–286. [Google Scholar] [CrossRef]
- Hu, Q.; Yu, L.; Zhai, Q.; Zhao, J.; Tian, F. Anti-Inflammatory, Barrier Maintenance, and Gut Microbiome Modulation Effects of Saccharomyces Cerevisiae QHNLD8L1 on DSS-Induced Ulcerative Colitis in Mice. Int. J. Mol. Sci. 2023, 24, 6721. [Google Scholar] [CrossRef]
- Duncan, S.H.; Conti, E.; Ricci, L.; Walker, A.W. Links between Diet, Intestinal Anaerobes, Microbial Metabolites and Health. Biomedicines 2023, 11, 1338. [Google Scholar] [CrossRef] [PubMed]
- Jansen, D.; Matthijnssens, J. The Emerging Role of the Gut Virome in Health and Inflammatory Bowel Disease: Challenges, Covariates and a Viral Imbalance. Viruses 2023, 15, 173. [Google Scholar] [CrossRef]
- Dubik, M.; Pilecki, B.; Moeller, J.B. Commensal Intestinal Protozoa—Underestimated Members of the Gut Microbial Community. Biology 2022, 11, 1742. [Google Scholar] [CrossRef] [PubMed]
- Cabezas-Cruz, A.; Bermúdez-Humarán, L.G. Exploring the Relationship between Faecalibacterium duncaniae and Escherichia coli in Inflammatory Bowel Disease (IBD): Insights and Implications. Comput. Struct. Biotechnol. J. 2024, 23, 1–9. [Google Scholar] [CrossRef]
- Sezgin, E.; Terlemez, G.; Bozkurt, B.; Bengi, G.; Akpinar, H.; Büyüktorun, İ. Quantitative Real-Time PCR Analysis of Bacterial Biomarkers Enable Fast and Accurate Monitoring in Inflammatory Bowel Disease. PeerJ 2022, 10, e14217. [Google Scholar] [CrossRef]
- Aldars-García, L.; Chaparro, M.; Gisbert, J.P. Systematic Review: The Gut Microbiome and Its Potential Clinical Application in Inflammatory Bowel Disease. Microorganisms 2021, 9, 977. [Google Scholar] [CrossRef]
- Hu, J.; Cheng, S.; Yao, J.; Lin, X.; Li, Y.; Wang, W.; Weng, J.; Zou, Y.; Zhu, L.; Zhi, M. Correlation between Altered Gut Microbiota and Elevated Inflammation Markers in Patients with Crohn’s Disease. Front. Immunol. 2022, 13, 947313. [Google Scholar] [CrossRef] [PubMed]
- Jauregui-Amezaga, A.; Smet, A. The Microbiome in Inflammatory Bowel Disease. J. Clin. Med. 2024, 13, 4622. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Denson, L.; Vlamakis, H.; Franzosa, E.A.; Thomas, S.; Gotman, N.M.; Rufo, P.; Baker, S.S.; Sauer, C.; Markowitz, J.; et al. Compositional and Temporal Changes in the Gut Microbiome of Pediatric Ulcerative Colitis Patients Are Linked to Disease Course. Cell Host Microbe 2018, 24, 600–610.e4. [Google Scholar] [CrossRef]
- Ulas, M.; Hussey, S.; Broderick, A.; Fitzpatrick, E.; Dunne, C.; Cooper, S.; Dominik, A.; Bourke, B. FISH–Flow Cytometry Reveals Microbiome-Wide Changes in Post-Translational Modification and Altered Microbial Abundance Among Children with Inflammatory Bowel Disease. Pathogens 2024, 13, 1102. [Google Scholar] [CrossRef]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A Microbial Signature for Crohn’s Disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut Microbiome Structure and Metabolic Activity in Inflammatory Bowel Disease. Nat. Microbiol. 2018, 4, 293–305. [Google Scholar] [CrossRef]
- Vich Vila, A.; Imhann, F.; Collij, V.; Jankipersadsing, S.A.; Gurry, T.; Mujagic, Z.; Kurilshikov, A.; Bonder, M.J.; Jiang, X.; Tigchelaar, E.F.; et al. Gut Microbiota Composition and Functional Changes in Inflammatory Bowel Disease and Irritable Bowel Syndrome. Sci. Transl. Med. 2018, 10, eaap8914. [Google Scholar] [CrossRef]
- Nishino, K.; Nishida, A.; Inoue, R.; Kawada, Y.; Ohno, M.; Sakai, S.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Kawahara, M.; et al. Analysis of Endoscopic Brush Samples Identified Mucosa-Associated Dysbiosis in Inflammatory Bowel Disease. J. Gastroenterol. 2018, 53, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, B.; Juillerat, P.; Øyås, O.; Ramon, C.; Bravo, F.D.; Franc, Y.; Fournier, N.; Michetti, P.; Mueller, C.; Geuking, M.; et al. Microbial Network Disturbances in Relapsing Refractory Crohn’s Disease. Nat. Med. 2019, 25, 323–336. [Google Scholar] [CrossRef]
- Zhang, Y.; Thomas, J.P.; Korcsmaros, T.; Gul, L. Integrating Multi-Omics to Unravel Host-Microbiome Interactions in Inflammatory Bowel Disease. Cell Rep. Med. 2024, 5, 101738. [Google Scholar] [CrossRef]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The Treatment-Naive Microbiome in New-Onset Crohn’s Disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Chen, E.Z.; Baldassano, R.N.; Otley, A.R.; Griffiths, A.M.; Lee, D.; Bittinger, K.; Bailey, A.; Friedman, E.S.; Hoffmann, C.; et al. Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host Microbe 2015, 18, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Gouba, N.; Hien, Y.E.; Guissou, M.L.; Fonkou, M.D.M.; Traoré, Y.; Tarnagda, Z. Digestive Tract Mycobiota and Microbiota and the Effects on the Immune System. Hum. Microb. J. 2019, 12, 100056. [Google Scholar] [CrossRef]
- Anjana; Tiwari, S.K. Bacteriocin-Producing Probiotic Lactic Acid Bacteria in Controlling Dysbiosis of the Gut Microbiota. Front. Cell Infect. Microbiol. 2022, 12, 851140. [Google Scholar] [CrossRef]
- Yoo, J.; Groer, M.; Dutra, S.; Sarkar, A.; McSkimming, D. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef]
- Mentella, M.C.; Scaldaferri, F.; Pizzoferrato, M.; Gasbarrini, A.; Miggiano, G.A.D. Nutrition, IBD and Gut Microbiota: A Review. Nutrients 2020, 12, 944. [Google Scholar] [CrossRef]
- Dahal, R.H.; Kim, S.; Kim, Y.K.; Kim, E.S.; Kim, J. Insight into Gut Dysbiosis of Patients with Inflammatory Bowel Disease and Ischemic Colitis. Front. Microbiol. 2023, 14, 1174832. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.-Y.; Park, J.-L.; Yeo, M.-K.; Kang, S.-B.; Kim, J.-M.; Kim, J.S.; Kim, S.-Y. Diagnosis of Crohn’s Disease and Ulcerative Colitis Using the Microbiome. BMC Microbiol. 2023, 23, 336. [Google Scholar] [CrossRef]
- Park, Y.; Ahn, J.B.; Kim, D.H.; Park, I.S.; Son, M.; Kim, J.H.; Ma, H.W.; Kim, S.W.; Cheon, J.H. Integrated Analysis of Microbiome and Metabolome Reveals Disease-Specific Profiles in Inflammatory Bowel Diseases and Intestinal Behçet’s Disease. Int. J. Mol. Sci. 2024, 25, 6697. [Google Scholar] [CrossRef]
- Quaglio, A.E.; Magro, D.O.; Imbrizi, M.; De Oliveira, E.C.; Di Stasi, L.C.; Sassaki, L.Y. Creeping Fat and Gut Microbiota in Crohn’s Disease. World J. Gastroenterol. 2025, 31, 102042. [Google Scholar] [CrossRef]
- Lin, S.; Zhang, X.; Zhu, X.; Jiao, J.; Wu, Y.; Li, Y.; Zhao, L. Fusobacterium nucleatum Aggravates Ulcerative Colitis through Promoting Gut Microbiota Dysbiosis and Dysmetabolism. J. Periodontol. 2023, 94, 405–418. [Google Scholar] [CrossRef]
- Kandasamy, S.; Letchumanan, V.; Hong, K.W.; Chua, K.-O.; Ab Mutalib, N.S.; Ng, A.L.O.; Ming, L.C.; Lim, H.X.; Thurairajasingam, S.; Law, J.W.-F.; et al. The Role of Human Gut Microbe Ruminococcus gnavus in Inflammatory Diseases. Prog. Microbes Mol. Biol. 2023, 6, 12672–12677. [Google Scholar] [CrossRef]
- Yu, S.; Ge, X.; Xu, H.; Tan, B.; Tian, B.; Shi, Y.; Dai, Y.; Li, Y.; Hu, S.; Qian, J. Gut Microbiome and Mycobiome in Inflammatory Bowel Disease Patients with Clostridioides difficile Infection. Front. Cell Infect. Microbiol. 2023, 13, 1129043. [Google Scholar] [CrossRef]
- Raygoza Garay, J.A.; Turpin, W.; Lee, S.-H.; Smith, M.I.; Goethel, A.; Griffiths, A.M.; Moayyedi, P.; Espin-Garcia, O.; Abreu, M.; Aumais, G.L.; et al. Gut Microbiome Composition Is Associated with Future Onset of Crohn’s Disease in Healthy First-Degree Relatives. Gastroenterology 2023, 165, 670–681. [Google Scholar] [CrossRef]
- Zakerska-Banaszak, O.; Tomczak, H.; Gabryel, M.; Baturo, A.; Wolko, L.; Michalak, M.; Malinska, N.; Mankowska-Wierzbicka, D.; Eder, P.; Dobrowolska, A.; et al. Dysbiosis of Gut Microbiota in Polish Patients with Ulcerative Colitis: A Pilot Study. Sci. Rep. 2021, 11, 2166. [Google Scholar] [CrossRef]
- Krela-Kaźmierczak, I.; Zakerska-Banaszak, O.; Skrzypczak-Zielińska, M.; Łykowska-Szuber, L.; Szymczak-Tomczak, A.; Zawada, A.; Rychter, A.M.; Ratajczak, A.E.; Skoracka, K.; Skrzypczak, D.; et al. Where Do We Stand in the Behavioral Pathogenesis of Inflammatory Bowel Disease? The Western Dietary Pattern and Microbiota—A Narrative Review. Nutrients 2022, 14, 2520. [Google Scholar] [CrossRef]
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 2019, 7247238. [Google Scholar] [CrossRef]
- Olbjørn, C.; Småstuen, M.C.; Moen, A.E.F. Targeted Analysis of the Gut Microbiome for Diagnosis, Prognosis and Treatment Individualization in Pediatric Inflammatory Bowel Disease. Microorganisms 2022, 10, 1273. [Google Scholar] [CrossRef]
- El Mouzan, M.; Al Sarkhy, A.; Assiri, A. Gut Microbiota Predicts the Diagnosis of Ulcerative Colitis in Saudi Children. World J. Clin. Pediatr. 2024, 13, 90755. [Google Scholar] [CrossRef]
- Dong, L.-N.; Wang, M.; Guo, J.; Wang, J.-P. Role of Intestinal Microbiota and Metabolites in Inflammatory Bowel Disease. Chin. Med. J. 2019, 132, 1610–1614. [Google Scholar] [CrossRef]
- Doherty, M.K.; Ding, T.; Koumpouras, C.; Telesco, S.E.; Monast, C.; Das, A.; Brodmerkel, C.; Schloss, P.D. Fecal Microbiota Signatures Are Associated with Response to Ustekinumab Therapy among Crohn’s Disease Patients. mBio 2018, 9, 10–1128. [Google Scholar] [CrossRef]
- Wiredu Ocansey, D.K.; Hang, S.; Yuan, X.; Qian, H.; Zhou, M.; Valerie Olovo, C.; Zhang, X.; Mao, F. The Diagnostic and Prognostic Potential of Gut Bacteria in Inflammatory Bowel Disease. Gut Microbes 2023, 15, 2176118. [Google Scholar] [CrossRef]
- Xia, Y.; Wang, J.; Fang, X.; Dou, T.; Han, L.; Yang, C. Combined Analysis of Metagenomic Data Revealed Consistent Changes of Gut Microbiome Structure and Function in Inflammatory Bowel Disease. J. Appl. Microbiol. 2021, 131, 3018–3031. [Google Scholar] [CrossRef]
- Konjar, Š.; Pavšič, M.; Veldhoen, M. Regulation of Oxygen Homeostasis at the Intestinal Epithelial Barrier Site. Int. J. Mol. Sci. 2021, 22, 9170. [Google Scholar] [CrossRef]
- Roy, S.; Dhaneshwar, S. Role of Prebiotics, Probiotics, and Synbiotics in Management of Inflammatory Bowel Disease: Current Perspectives. World J. Gastroenterol. 2023, 29, 2078–2100. [Google Scholar] [CrossRef]
- Nogal, A.; Valdes, A.M.; Menni, C. The Role of Short-Chain Fatty Acids in the Interplay between Gut Microbiota and Diet in Cardio-Metabolic Health. Gut Microbes 2021, 13, 2325–2340. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhi, F. Lower Level of Bacteroides in the Gut Microbiota Is Associated with Inflammatory Bowel Disease: A Meta-Analysis. Biomed. Res. Int. 2016, 2016, 5828959. [Google Scholar] [CrossRef]
- Ruuskanen, M.O.; Åberg, F.; Männistö, V.; Havulinna, A.S.; Méric, G.; Liu, Y.; Loomba, R.; Vázquez-Baeza, Y.; Tripathi, A.; Valsta, L.M.; et al. Links between Gut Microbiome Composition and Fatty Liver Disease in a Large Population Sample. Gut Microbes 2021, 13, 1–22. [Google Scholar] [CrossRef]
- Corrêa-Oliveira, R.; Fachi, J.L.; Vieira, A.; Sato, F.T.; Vinolo, M.A.R. Regulation of Immune Cell Function by Short-chain Fatty Acids. Clin Transl Immunol. 2016, 5, e73. [Google Scholar] [CrossRef]
- Topalova-Dimitrova, A.; Dimitrov, I.V.; Nikolov, R. Lower Vitamin D Levels Are Associated with the Pathogenesis of Inflammatory Bowel Diseases. Medicine 2023, 102, e35505. [Google Scholar] [CrossRef]
- Zheng, X.; Zhu, Y.; Zhao, Z.; Chu, Y.; Yang, W. The Role of Amino Acid Metabolism in Inflammatory Bowel Disease and Other Inflammatory Diseases. Front. Immunol. 2023, 14, 1284133. [Google Scholar] [CrossRef]
- Clinton, N.A.; Hameed, S.A.; Agyei, E.K.; Jacob, J.C.; Oyebanji, V.O.; Jabea, C.E. Crosstalk between the Intestinal Virome and Other Components of the Microbiota, and Its Effect on Intestinal Mucosal Response and Diseases. J. Immunol. Res. 2022, 2022, 7883945. [Google Scholar] [CrossRef]
- Neurath, M.F.; Artis, D.; Becker, C. The Intestinal Barrier: A Pivotal Role in Health, Inflammation, and Cancer. Lancet Gastroenterol. Hepatol. 2025, 10, 573–592. [Google Scholar] [CrossRef]
- Salvador-Erro, J.; Pastor, Y.; Gamazo, C. Targeting Enterotoxins: Advancing Vaccine Development for Enterotoxigenic Escherichia coli ETEC. Toxins 2025, 17, 71. [Google Scholar] [CrossRef]
- Rogers, A.P.; Mileto, S.J.; Lyras, D. Impact of Enteric Bacterial Infections at and beyond the Epithelial Barrier. Nat. Rev. Microbiol. 2023, 21, 260–274. [Google Scholar] [CrossRef]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 508738. [Google Scholar] [CrossRef]
- Shin, Y.; Han, S.; Kwon, J.; Ju, S.; Choi, T.; Kang, I.; Kim, S. Roles of Short-Chain Fatty Acids in Inflammatory Bowel Disease. Nutrients 2023, 15, 4466. [Google Scholar] [CrossRef]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Wu, Z.; Nybom, P.; Magnusson, K.-E. Distinct Effects of Vibrio Cholerae Haemagglutinin/Protease on the Structure and Localization of the Tight Junction-Associated Proteins Occludin and ZO-1. Cell Microbiol. 2000, 2, 11–17. [Google Scholar] [CrossRef]
- Chen, W.; Wang, D.; Deng, X.; Zhang, H.; Dong, D.; Su, T.; Lu, Q.; Jiang, C.; Ni, Q.; Cui, Y.; et al. Bile Acid Profiling as an Effective Biomarker for Staging in Pediatric Inflammatory Bowel Disease. Gut Microbes 2024, 16, 2323231. [Google Scholar] [CrossRef]
- Hou, Y.; Li, J.; Ying, S. Tryptophan Metabolism and Gut Microbiota: A Novel Regulatory Axis Integrating the Microbiome, Immunity, and Cancer. Metabolites 2023, 13, 1166. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Z.; Zabed, H.M.; Yun, J.; Zhang, G.; Qi, X. An Insight into the Roles of Dietary Tryptophan and Its Metabolites in Intestinal Inflammation and Inflammatory Bowel Disease. Mol. Nutr. Food Res. 2021, 65, 2000461. [Google Scholar] [CrossRef]
- Dehghani, T.; Gholizadeh, O.; Daneshvar, M.; Nemati, M.M.; Akbarzadeh, S.; Amini, P.; Afkhami, H.; Kohansal, M.; Javanmard, Z.; Poortahmasebi, V. Association Between Inflammatory Bowel Disease and Viral Infections. Curr. Microbiol. 2023, 80, 195. [Google Scholar] [CrossRef]
- Neil, J.A.; Cadwell, K. The Intestinal Virome and Immunity. J. Immunol. 2018, 201, 1615–1624. [Google Scholar] [CrossRef]
- Cao, Z.; Sugimura, N.; Burgermeister, E.; Ebert, M.P.; Zuo, T.; Lan, P. The Gut Virome: A New Microbiome Component in Health and Disease. EBioMedicine 2022, 81, 104113. [Google Scholar] [CrossRef]
- Tiamani, K.; Luo, S.; Schulz, S.; Xue, J.; Costa, R.; Khan Mirzaei, M.; Deng, L. The Role of Virome in the Gastrointestinal Tract and Beyond. FEMS Microbiol. Rev. 2022, 46, fuac027. [Google Scholar] [CrossRef]
- Shuwen, H.; Kefeng, D. Intestinal Phages Interact with Bacteria and Are Involved in Human Diseases. Gut Microbes 2022, 14, 2113717. [Google Scholar] [CrossRef]
- Tian, X.; Li, S.; Wang, C.; Zhang, Y.; Feng, X.; Yan, Q.; Guo, R.; Wu, F.; Wu, C.; Wang, Y.; et al. Gut Virome-Wide Association Analysis Identifies Cross-Population Viral Signatures for Inflammatory Bowel Disease. Microbiome 2024, 12, 130. [Google Scholar] [CrossRef]
- Zuo, T.; Lu, X.-J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; et al. Gut Mucosal Virome Alterations in Ulcerative Colitis. Gut 2019, 68, 1169–1179. [Google Scholar] [CrossRef]
- Cao, Z.; Fan, D.; Sun, Y.; Huang, Z.; Li, Y.; Su, R.; Zhang, F.; Li, Q.; Yang, H.; Zhang, F.; et al. The Gut Ileal Mucosal Virome Is Disturbed in Patients with Crohn’s Disease and Exacerbates Intestinal Inflammation in Mice. Nat. Commun. 2024, 15, 1638. [Google Scholar] [CrossRef]
- Ding, Y.; Wan, M.; Li, Z.; Ma, X.; Zhang, W.; Xu, M. Comparison of the Gut Virus Communities between Patients with Crohn’s Disease and Healthy Individuals. Front. Microbiol. 2023, 14, 1190172. [Google Scholar] [CrossRef]
- Chehoud, C.; Albenberg, L.G.; Judge, C.; Hoffmann, C.; Grunberg, S.; Bittinger, K.; Baldassano, R.N.; Lewis, J.D.; Bushman, F.D.; Wu, G.D. Fungal Signature in the Gut Microbiota of Pediatric Patients With Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1948–1956. [Google Scholar] [CrossRef] [PubMed]
- Hoarau, G.; Mukherjee, P.K.; Gower-Rousseau, C.; Hager, C.; Chandra, J.; Retuerto, M.A.; Neut, C.; Vermeire, S.; Clemente, J.; Colombel, J.F.; et al. Bacteriome and Mycobiome Interactions Underscore Microbial Dysbiosis in Familial Crohn’s Disease. mBio 2016, 7, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Jangi, S.; Hsia, K.; Zhao, N.; Kumamoto, C.A.; Friedman, S.; Singh, S.; Michaud, D.S. Dynamics of the Gut Mycobiome in Patients With Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2024, 22, 821–830.e7. [Google Scholar] [CrossRef] [PubMed]
- Liguori, G.; Lamas, B.; Richard, M.L.; Brandi, G.; da Costa, G.; Hoffmann, T.W.; Di Simone, M.P.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Fungal Dysbiosis in Mucosa-Associated Microbiota of Crohn’s Disease Patients. J. Crohns Colitis 2016, 10, 296–305. [Google Scholar] [CrossRef]
- Cimická, J.; Riegert, J.; Kavková, M.; Černá, K. Intestinal Mycobiome Associated with Diagnosis of Inflammatory Bowel Disease Based on Tissue Biopsies. Med. Mycol. 2022, 60, myab076. [Google Scholar] [CrossRef]
- Richard, M.L.; Sokol, H. The Gut Mycobiota: Insights into Analysis, Environmental Interactions and Role in Gastrointestinal Diseases. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 331–345. [Google Scholar] [CrossRef]
- Catalán-Serra, I.; Thorsvik, S.; Beisvag, V.; Bruland, T.; Underhill, D.; Sandvik, A.K.; Granlund, A. van B. Fungal Microbiota Composition in Inflammatory Bowel Disease Patients: Characterization in Different Phenotypes and Correlation with Clinical Activity and Disease Course. Inflamm. Bowel Dis. 2024, 30, 1164–1177. [Google Scholar] [CrossRef]
- Zhang, L.; Zhan, H.; Xu, W.; Yan, S.; Ng, S.C. The Role of Gut Mycobiome in Health and Diseases. Ther. Adv. Gastroenterol. 2021, 14, 17562848211047130. [Google Scholar] [CrossRef]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.-P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal Microbiota Dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef]
- Beheshti-Maal, A.; Shahrokh, S.; Ansari, S.; Mirsamadi, E.S.; Yadegar, A.; Mirjalali, H.; Zali, M.R. Gut Mycobiome: The Probable Determinative Role of Fungi in IBD Patients. Mycoses 2021, 64, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.C.; Itzkowitz, S.H. Colorectal Cancer in Inflammatory Bowel Disease: Mechanisms and Management. Gastroenterology 2022, 162, 715–730.e3. [Google Scholar] [CrossRef]
- Wang, N.; Fang, J.-Y. Fusobacterium nucleatum, a Key Pathogenic Factor and Microbial Biomarker for Colorectal Cancer. Trends Microbiol. 2023, 31, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Thiele Orberg, E.; Fan, H.; Tam, A.J.; Dejea, C.M.; Destefano Shields, C.E.; Wu, S.; Chung, L.; Finard, B.B.; Wu, X.; Fathi, P.; et al. The Myeloid Immune Signature of Enterotoxigenic Bacteroides fragilis-Induced Murine Colon Tumorigenesis. Mucosal Immunol. 2017, 10, 421–433. [Google Scholar] [CrossRef]
- Long, X.; Wong, C.C.; Tong, L.; Chu, E.S.H.; Ho Szeto, C.; Go, M.Y.Y.; Coker, O.O.; Chan, A.W.H.; Chan, F.K.L.; Sung, J.J.Y.; et al. Peptostreptococcus anaerobius Promotes Colorectal Carcinogenesis and Modulates Tumour Immunity. Nat. Microbiol. 2019, 4, 2319–2330. [Google Scholar] [CrossRef]
- Liu, N.-N.; Jiao, N.; Tan, J.-C.; Wang, Z.; Wu, D.; Wang, A.-J.; Chen, J.; Tao, L.; Zhou, C.; Fang, W.; et al. Multi-Kingdom Microbiota Analyses Identify Bacterial–Fungal Interactions and Biomarkers of Colorectal Cancer across Cohorts. Nat. Microbiol. 2022, 7, 238–250. [Google Scholar] [CrossRef]
- Janney, A.; Powrie, F.; Mann, E.H. Host–Microbiota Maladaptation in Colorectal Cancer. Nature 2020, 585, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, I.; Tap, J.; Roudot-Thoraval, F.; Roperch, J.P.; Letulle, S.; Langella, P.; Corthier, G.; Tran Van Nhieu, J.; Furet, J.P. Microbial Dysbiosis in Colorectal Cancer (CRC) Patients. PLoS ONE 2011, 6, e16393. [Google Scholar] [CrossRef]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Böhm, J.; Brunetti, F.; Habermann, N.; et al. Potential of Fecal Microbiota for Early-Stage Detection of Colorectal Cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Sanapareddy, N.; Legge, R.M.; Jovov, B.; McCoy, A.; Burcal, L.; Araujo-Perez, F.; Randall, T.A.; Galanko, J.; Benson, A.; Sandler, R.S.; et al. Increased Rectal Microbial Richness Is Associated with the Presence of Colorectal Adenomas in Humans. ISME J. 2012, 6, 1858–1868. [Google Scholar] [CrossRef]
- Yu, J.; Feng, Q.; Wong, S.H.; Zhang, D.; Liang, Q.Y.; Qin, Y.; Tang, L.; Zhao, H.; Stenvang, J.; Li, Y.; et al. Metagenomic Analysis of Faecal Microbiome as a Tool towards Targeted Non-Invasive Biomarkers for Colorectal Cancer. Gut 2017, 66, 70–78. [Google Scholar] [CrossRef]
- Katsuura, G.; Gottschall, P.E.; Dahl, R.R.; Arimura, A. Adrenocorticotropin Release Induced by Intracerebroventricular Injection of Recombinant Human Interleukin-1 in Rats: Possible Involvement of Prostaglandin. Endocrinology 1988, 122, 1773–1779. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Coker, O.O.; Nakatsu, G.; Wu, W.K.K.; Zhao, L.; Chen, Z.; Chan, F.K.L.; Kristiansen, K.; Sung, J.J.Y.; Wong, S.H.; et al. Multi-Cohort Analysis of Colorectal Cancer Metagenome Identified Altered Bacteria across Populations and Universal Bacterial Markers. Microbiome 2018, 6, 70. [Google Scholar] [CrossRef] [PubMed]
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Meta-Analysis of Fecal Metagenomes Reveals Global Microbial Signatures That Are Specific for Colorectal Cancer. Nat. Med. 2019, 25, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.M.; Manghi, P.; Asnicar, F.; Pasolli, E.; Armanini, F.; Zolfo, M.; Beghini, F.; Manara, S.; Karcher, N.; Pozzi, C.; et al. Metagenomic Analysis of Colorectal Cancer Datasets Identifies Cross-Cohort Microbial Diagnostic Signatures and a Link with Choline Degradation. Nat. Med. 2019, 25, 667–678. [Google Scholar] [CrossRef]
- Nakatsu, G.; Li, X.; Zhou, H.; Sheng, J.; Wong, S.H.; Wu, W.K.K.; Ng, S.C.; Tsoi, H.; Dong, Y.; Zhang, N.; et al. Gut Mucosal Microbiome across Stages of Colorectal Carcinogenesis. Nat. Commun. 2015, 6, 8727. [Google Scholar] [CrossRef]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut Microbiome Development along the Colorectal Adenoma–Carcinoma Sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef]
- Flemer, B.; Lynch, D.B.; Brown, J.M.R.; Jeffery, I.B.; Ryan, F.J.; Claesson, M.J.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. Tumour-Associated and Non-Tumour-Associated Microbiota in Colorectal Cancer. Gut 2017, 66, 633–643. [Google Scholar] [CrossRef]
- Shah, M.S.; DeSantis, T.Z.; Weinmaier, T.; McMurdie, P.J.; Cope, J.L.; Altrichter, A.; Yamal, J.-M.; Hollister, E.B. Leveraging Sequence-Based Faecal Microbial Community Survey Data to Identify a Composite Biomarker for Colorectal Cancer. Gut 2018, 67, 882–891. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic Analysis Identifies Association of Fusobacterium with Colorectal Carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef]
- Negrut, R.L.; Cote, A.; Maghiar, A.M. Exploring the Potential of Oral Microbiome Biomarkers for Colorectal Cancer Diagnosis and Prognosis: A Systematic Review. Microorganisms 2023, 11, 1586. [Google Scholar] [CrossRef]
- Kudra, A.; Muszyński, D.; Sobocki, B.K.; Atzeni, A.; Carbone, L.; Kaźmierczak-Siedlecka, K.; Połom, K.; Kalinowski, L. Insights into Oral Microbiome and Colorectal Cancer—On the Way of Searching New Perspectives. Front. Cell Infect. Microbiol. 2023, 13, 1159822. [Google Scholar] [CrossRef] [PubMed]
- Flemer, B.; Warren, R.D.; Barrett, M.P.; Cisek, K.; Das, A.; Jeffery, I.B.; Hurley, E.; O‘Riordain, M.; Shanahan, F.; O‘Toole, P.W. The Oral Microbiota in Colorectal Cancer Is Distinctive and Predictive. Gut 2018, 67, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Kong, C.; Yang, Y.; Cai, S.; Li, X.; Cai, G.; Ma, Y. Human Oral Microbiome Dysbiosis as a Novel Non-Invasive Biomarker in Detection of Colorectal Cancer. Theranostics 2020, 10, 11595–11606. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.-H.; Gao, Q.-Y.; Cai, G.-X.; Sun, X.-M.; Sun, X.-M.; Zou, T.-H.; Chen, H.-M.; Yu, S.-Y.; Qiu, Y.-W.; Gu, W.-Q.; et al. Fecal Clostridium symbiosum for Noninvasive Detection of Early and Advanced Colorectal Cancer: Test and Validation Studies. EBioMedicine 2017, 25, 32–40. [Google Scholar] [CrossRef]
- Yang, T.-W.; Lee, W.-H.; Tu, S.-J.; Huang, W.-C.; Chen, H.-M.; Sun, T.-H.; Tsai, M.-C.; Wang, C.-C.; Chen, H.-Y.; Huang, C.-C.; et al. Enterotype-Based Analysis of Gut Microbiota along the Conventional Adenoma-Carcinoma Colorectal Cancer Pathway. Sci. Rep. 2019, 9, 10923. [Google Scholar] [CrossRef]
- Chen, F.; Dai, X.; Zhou, C.-C.; Li, K.; Zhang, Y.; Lou, X.-Y.; Zhu, Y.-M.; Sun, Y.-L.; Peng, B.-X.; Cui, W. Integrated Analysis of the Faecal Metagenome and Serum Metabolome Reveals the Role of Gut Microbiome-Associated Metabolites in the Detection of Colorectal Cancer and Adenoma. Gut 2022, 71, 1315–1325. [Google Scholar] [CrossRef]
- Li, G.; Jin, Y.; Chen, B.; Lin, A.; Wang, E.; Xu, F.; Hu, G.; Xiao, C.; Liu, H.; Hou, X.; et al. Exploring the Relationship between the Gut Mucosal Virome and Colorectal Cancer: Characteristics and Correlations. Cancers 2023, 15, 3555. [Google Scholar] [CrossRef]
- Hannigan, G.D.; Duhaime, M.B.; Ruffin, M.T.; Koumpouras, C.C.; Schloss, P.D. Diagnostic Potential and Interactive Dynamics of the Colorectal Cancer Virome. mBio 2018, 9, 10–1128. [Google Scholar] [CrossRef]
- Nakatsu, G.; Zhou, H.; Wu, W.K.K.; Wong, S.H.; Coker, O.O.; Dai, Z.; Li, X.; Szeto, C.-H.; Sugimura, N.; Lam, T.Y.-T.; et al. Alterations in Enteric Virome Are Associated with Colorectal Cancer and Survival Outcomes. Gastroenterology 2018, 155, 529–541.e5. [Google Scholar] [CrossRef]
- Luan, C.; Xie, L.; Yang, X.; Miao, H.; Lv, N.; Zhang, R.; Xiao, X.; Hu, Y.; Liu, Y.; Wu, N.; et al. Dysbiosis of Fungal Microbiota in the Intestinal Mucosa of Patients with Colorectal Adenomas. Sci. Rep. 2015, 5, 7980. [Google Scholar] [CrossRef]
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Enteric Fungal Microbiota Dysbiosis and Ecological Alterations in Colorectal Cancer. Gut 2019, 68, 654–662. [Google Scholar] [CrossRef]
- Caminero, A.; Galipeau, H.J.; McCarville, J.L.; Johnston, C.W.; Bernier, S.P.; Russell, A.K.; Jury, J.; Herran, A.R.; Casqueiro, J.; Tye-Din, J.A.; et al. Duodenal Bacteria from Patients with Celiac Disease and Healthy Subjects Distinctly Affect Gluten Breakdown and Immunogenicity. Gastroenterology 2016, 151, 670–683. [Google Scholar] [CrossRef] [PubMed]
- Sciurti, M.; Fornaroli, F.; Gaiani, F.; Bonaguri, C.; Leandro, G.; Di Mario, F.; De’ Angelis, G.L. Genetic Susceptibilty and Celiac Disease: What Role Do HLA Haplotypes Play? Acta Biomed. 2018, 89, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A. A Potential Link between Environmental Triggers and Autoimmunity. Autoimmune Dis. 2014, 2014, 437231. [Google Scholar] [CrossRef] [PubMed]
- Mowat, A.M.; Agace, W.W. Regional Specialization within the Intestinal Immune System. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef]
- Schiepatti, A.; Bacchi, S.; Biagi, F.; Panelli, S.; Betti, E.; Corazza, G.R.; Capelli, E.; Ciccocioppo, R. Relationship between Duodenal Microbiota Composition, Clinical Features at Diagnosis, and Persistent Symptoms in Adult Coeliac Disease. Dig. Liver Dis. 2021, 53, 972–979. [Google Scholar] [CrossRef]
- Catassi, G.; Lener, E.; Grattagliano, M.M.; Motuz, S.; Zavarella, M.A.; Bibbò, S.; Cammarota, G.; Gasbarrini, A.; Ianiro, G.; Catassi, C. The Role of Microbiome in the Development of Gluten-Related Disorders. Best. Pr. Res. Clin. Gastroenterol. 2024, 72, 101951. [Google Scholar] [CrossRef]
- Costigan, C.M.; Warren, F.J.; Duncan, A.P.; Hoad, C.L.; Lewis, N.; Hill, T.; Crooks, C.J.; Morgan, P.S.; Ciacci, C.; Iovino, P.; et al. One Year of Gluten-Free Diet Impacts Gut Function and Microbiome in Celiac Disease. Clin. Gastroenterol. Hepatol. 2024. [Google Scholar] [CrossRef]
- Caminero, A.; McCarville, J.L.; Zevallos, V.F.; Pigrau, M.; Yu, X.B.; Jury, J.; Galipeau, H.J.; Clarizio, A.V.; Casqueiro, J.; Murray, J.A.; et al. Lactobacilli Degrade Wheat Amylase Trypsin Inhibitors to Reduce Intestinal Dysfunction Induced by Immunogenic Wheat Proteins. Gastroenterology 2019, 156, 2266–2280. [Google Scholar] [CrossRef]
- Zevallos, V.F.; Raker, V.; Tenzer, S.; Jimenez-Calvente, C.; Ashfaq-Khan, M.; Rüssel, N.; Pickert, G.; Schild, H.; Steinbrink, K.; Schuppan, D. Nutritional Wheat Amylase-Trypsin Inhibitors Promote Intestinal Inflammation via Activation of Myeloid Cells. Gastroenterology 2017, 152, 1100–1113.e12. [Google Scholar] [CrossRef] [PubMed]
- Kelley, K.; Dogru, D.; Huang, Q.; Yang, Y.; Palm, N.W.; Altindis, E.; Ludvigsson, J. Children Who Develop Celiac Disease Are Predicted to Exhibit Distinct Metabolic Pathways among Their Gut Microbiota Years before Diagnosis. Microbiol. Spectr. 2025, 13, e01468-24. [Google Scholar] [CrossRef]
- Di Vincenzo, F.; Del Gaudio, A.; Petito, V.; Lopetuso, L.R.; Scaldaferri, F. Gut Microbiota, Intestinal Permeability, and Systemic Inflammation: A Narrative Review. Intern. Emerg. Med. 2024, 19, 275–293. [Google Scholar] [CrossRef]
- Salvi, P.S.; Cowles, R.A. Butyrate and the Intestinal Epithelium: Modulation of Proliferation and Inflammation in Homeostasis and Disease. Cells 2021, 10, 1775. [Google Scholar] [CrossRef]
- Rintala, A.; Riikonen, I.; Toivonen, A.; Pietilä, S.; Munukka, E.; Pursiheimo, J.-P.; Elo, L.L.; Arikoski, P.; Luopajärvi, K.; Schwab, U.; et al. Early Fecal Microbiota Composition in Children Who Later Develop Celiac Disease and Associated Autoimmunity. Scand. J. Gastroenterol. 2018, 53, 403–409. [Google Scholar] [CrossRef]
- Bodkhe, R.; Shetty, S.A.; Dhotre, D.P.; Verma, A.K.; Bhatia, K.; Mishra, A.; Kaur, G.; Pande, P.; Bangarusamy, D.K.; Santosh, B.P.; et al. Comparison of Small Gut and Whole Gut Microbiota of First-Degree Relatives with Adult Celiac Disease Patients and Controls. Front. Microbiol. 2019, 10, 137–140. [Google Scholar] [CrossRef] [PubMed]
- El Mouzan, M.; Al-Hussaini, A.; Serena, G.; Assiri, A.; Al Sarkhy, A.; Al Mofarreh, M.; Alasmi, M.; Fasano, A. Microbiota Profile of New-Onset Celiac Disease in Children in Saudi Arabia. Gut Pathog. 2022, 14, 37. [Google Scholar] [CrossRef] [PubMed]
- Olivares, M.; Benítez-Páez, A.; de Palma, G.; Capilla, A.; Nova, E.; Castillejo, G.; Varea, V.; Marcos, A.; Garrote, J.A.; Polanco, I.; et al. Increased Prevalence of Pathogenic Bacteria in the Gut Microbiota of Infants at Risk of Developing Celiac Disease: The PROFICEL Study. Gut Microbes 2018, 9, 551–558. [Google Scholar] [CrossRef]
- Nadal, I.; Donant, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Imbalance in the Composition of the Duodenal Microbiota of Children with Coeliac Disease. J. Med. Microbiol. 2007, 56, 1669–1674. [Google Scholar] [CrossRef]
- Di Cagno, R.; De Angelis, M.; De Pasquale, I.; Ndagijimana, M.; Vernocchi, P.; Ricciuti, P.; Gagliardi, F.; Laghi, L.; Crecchio, C.; Guerzoni, M.E.; et al. Duodenal and Faecal Microbiota of Celiac Children: Molecular, Phenotype and Metabolome Characterization. BMC Microbiol. 2011, 11, 219. [Google Scholar] [CrossRef]
- Collado, M.C.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Imbalances in Faecal and Duodenal Bifidobacterium Species Composition in Active and Non-Active Coeliac Disease. BMC Microbiol. 2008, 8, 232. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, E.; Donat, E.; Ribes-Koninckx, C.; Fernández-Murga, M.L.; Sanz, Y. Duodenal-Mucosal Bacteria Associated with Celiac Disease in Children. Appl. Environ. Microbiol. 2013, 79, 5472–5479. [Google Scholar] [CrossRef]
- D’Argenio, V.; Casaburi, G.; Precone, V.; Pagliuca, C.; Colicchio, R.; Sarnataro, D.; Discepolo, V.; Kim, S.M.; Russo, I.; Del Vecchio Blanco, G.; et al. Metagenomics Reveals Dysbiosis and a Potentially Pathogenic N. flavescens Strain in Duodenum of Adult Celiac Patients. Am. J. Gastroenterol. 2016, 111, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Caminero, A.; McCarville, J.L.; Galipeau, H.J.; Deraison, C.; Bernier, S.P.; Constante, M.; Rolland, C.; Meisel, M.; Murray, J.A.; Yu, X.B.; et al. Duodenal Bacterial Proteolytic Activity Determines Sensitivity to Dietary Antigen through Protease-Activated Receptor-2. Nat. Commun. 2019, 10, 1198. [Google Scholar] [CrossRef] [PubMed]
- Wacklin, P.; Kaukinen, K.; Tuovinen, E.; Collin, P.; Lindfors, K.; Partanen, J.; Mäki, M.; Mättö, J. The Duodenal Microbiota Composition of Adult Celiac Disease Patients Is Associated with the Clinical Manifestation of the Disease. Inflamm. Bowel Dis. 2013, 19, 934–941. [Google Scholar] [CrossRef]
- Wacklin, P.; Laurikka, P.; Lindfors, K.; Collin, P.; Salmi, T.; Lähdeaho, M.-L.; Saavalainen, P.; Mäki, M.; Mättö, J.; Kurppa, K.; et al. Altered Duodenal Microbiota Composition in Celiac Disease Patients Suffering From Persistent Symptoms on a Long-Term Gluten-Free Diet. Am. J. Gastroenterol. 2014, 109, 1933–1941. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut Biogeography of the Bacterial Microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef]
- Sánchez, E.; De Palma, G.; Capilla, A.; Nova, E.; Pozo, T.; Castillejo, G.; Varea, V.; Marcos, A.; Garrote, J.A.; Polanco, I.; et al. Influence of Environmental and Genetic Factors Linked to Celiac Disease Risk on Infant Gut Colonization by Bacteroides Species. Appl. Environ. Microbiol. 2011, 77, 5316–5323. [Google Scholar] [CrossRef]
- De Palma, G.; Capilla, A.; Nova, E.; Castillejo, G.; Varea, V.; Pozo, T.; Garrote, J.A.; Polanco, I.; López, A.; Ribes-Koninckx, C.; et al. Influence of Milk-Feeding Type and Genetic Risk of Developing Coeliac Disease on Intestinal Microbiota of Infants: The PROFICEL Study. PLoS ONE 2012, 7, e30791. [Google Scholar] [CrossRef]
- Lionetti, E.; Catassi, C. The Role of Environmental Factors in the Development of Celiac Disease: What Is New? Diseases 2015, 3, 282–293. [Google Scholar] [CrossRef]
- Szajewska, H.; Shamir, R.; Chmielewska, A.; Pieścik-Lech, M.; Auricchio, R.; Ivarsson, A.; Kolacek, S.; Koletzko, S.; Korponay-Szabo, I.; Mearin, M.L.; et al. Systematic Review with Meta-Analysis: Early Infant Feeding and Coeliac Disease--Update 2015. Aliment. Pharmacol. Ther. 2015, 41, 1038–1054. [Google Scholar] [CrossRef]
- Benítez-Páez, A.; Olivares, M.; Szajewska, H.; Pieścik-Lech, M.; Polanco, I.; Castillejo, G.; Nuñez, M.; Ribes-Koninckx, C.; Korponay-Szabó, I.R.; Koletzko, S.; et al. Breast-Milk Microbiota Linked to Celiac Disease Development in Children: A Pilot Study From the PreventCD Cohort. Front. Microbiol. 2020, 11, 1335. [Google Scholar] [CrossRef] [PubMed]
- Leonard, M.M.; Karathia, H.; Pujolassos, M.; Troisi, J.; Valitutti, F.; Subramanian, P.; Camhi, S.; Kenyon, V.; Colucci, A.; Serena, G.; et al. Multi-Omics Analysis Reveals the Influence of Genetic and Environmental Risk Factors on Developing Gut Microbiota in Infants at Risk of Celiac Disease. Microbiome 2020, 8, 130. [Google Scholar] [CrossRef] [PubMed]
- Hov, J.R.; Zhong, H.; Qin, B.; Anmarkrud, J.A.; Holm, K.; Franke, A.; Lie, B.A.; Karlsen, T.H. The Influence of the Autoimmunity-Associated Ancestral HLA Haplotype AH8.1 on the Human Gut Microbiota: A Cross-Sectional Study. PLoS ONE 2015, 10, e0133804. [Google Scholar] [CrossRef]
- Lindfors, K.; Lin, J.; Lee, H.-S.; Hyöty, H.; Nykter, M.; Kurppa, K.; Liu, E.; Koletzko, S.; Rewers, M.; Hagopian, W.; et al. Metagenomics of the Faecal Virome Indicate a Cumulative Effect of Enterovirus and Gluten Amount on the Risk of Coeliac Disease Autoimmunity in Genetically at Risk Children: The TEDDY Study. Gut 2020, 69, 1416–1422. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento, L.; Galvan, J.A.; Cabrera-Rode, E.; Aira, L.; Correa, C.; Sariego, S.; Fonseca, M.; Cubas-Dueñas, I.; Hung, L.H.; Resik, S.; et al. Type 1 Diabetes Associated and Tissue Transglutaminase Autoantibodies in Patients without Type 1 Diabetes and Coeliac Disease with Confirmed Viral Infections. J. Med. Virol. 2012, 84, 1049–1053. [Google Scholar] [CrossRef]
- Kahrs, C.R.; Chuda, K.; Tapia, G.; Stene, L.C.; Mårild, K.; Rasmussen, T.; Rønningen, K.S.; Lundin, K.E.A.; Kramna, L.; Cinek, O.; et al. Enterovirus as Trigger of Coeliac Disease: Nested Case-Control Study within Prospective Birth Cohort. BMJ 2019, 364, l231. [Google Scholar] [CrossRef]
- Bouziat, R.; Hinterleitner, R.; Brown, J.J.; Stencel-Baerenwald, J.E.; Ikizler, M.; Mayassi, T.; Meisel, M.; Kim, S.M.; Discepolo, V.; Pruijssers, A.J.; et al. Reovirus Infection Triggers Inflammatory Responses to Dietary Antigens and Development of Celiac Disease. Science 2017, 356, 44–50. [Google Scholar] [CrossRef]
- Brown, J.J.; Jabri, B.; Dermody, T.S. A Viral Trigger for Celiac Disease. PLoS Pathog. 2018, 14, e1007181. [Google Scholar] [CrossRef]
- El Mouzan, M.; Al-Hussaini, A.; Fanelli, B.; Assiri, A.; AlSaleem, B.; Al Mofarreh, M.; Al Sarkhy, A.; Alasmi, M. Fungal Dysbiosis in Children with Celiac Disease. Dig. Dis. Sci. 2022, 67, 216–223. [Google Scholar] [CrossRef]
- D’Argenio, V.; Casaburi, G.; Precone, V.; Pagliuca, C.; Colicchio, R.; Sarnataro, D.; Discepolo, V.; Kim, S.M.; Russo, I.; Del Vecchio Blanco, G.; et al. No Change in the Mucosal Gut Microbiome Is Associated with Celiac Disease-Specific Microbiome Alteration in Adult Patients. Am. J. Gastroenterol. 2016, 111, 1659–1661. [Google Scholar] [CrossRef]
- Harnett, J.; Myers, S.P.; Rolfe, M. Significantly Higher Faecal Counts of the Yeasts Candida and Saccharomyces Identified in People with Coeliac Disease. Gut Pathog. 2017, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Pelizzaro, F.; Cardin, R.; Sarasini, G.; Minotto, M.; Carlotto, C.; Fassan, M.; Palo, M.; Farinati, F.; Zingone, F. Crosstalk between MicroRNAs and Oxidative Stress in Coeliac Disease. Inflamm. Intest. Dis. 2024, 9, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Kunst, C.; Schmid, S.; Michalski, M.; Tümen, D.; Buttenschön, J.; Müller, M.; Gülow, K. The Influence of Gut Microbiota on Oxidative Stress and the Immune System. Biomedicines 2023, 11, 1388. [Google Scholar] [CrossRef]
- Laudadio, I.; Fulci, V.; Palone, F.; Stronati, L.; Cucchiara, S.; Carissimi, C. Quantitative Assessment of Shotgun Metagenomics and 16S RDNA Amplicon Sequencing in the Study of Human Gut Microbiome. OMICS 2018, 22, 248–254. [Google Scholar] [CrossRef]
- Gao, B.; Chi, L.; Zhu, Y.; Shi, X.; Tu, P.; Li, B.; Yin, J.; Gao, N.; Shen, W.; Schnabl, B. An Introduction to next Generation Sequencing Bioinformatic Analysis in Gut Microbiome Studies. Biomolecules 2021, 11, 530. [Google Scholar] [CrossRef] [PubMed]
- Wensel, C.R.; Pluznick, J.L.; Salzberg, S.L.; Sears, C.L. Next-Generation Sequencing: Insights to Advance Clinical Investigations of the Microbiome. J. Clin. Investig. 2022, 132, e154944. [Google Scholar] [CrossRef]
- Schirmer, M.; Franzosa, E.A.; Lloyd-Price, J.; McIver, L.J.; Schwager, R.; Poon, T.W.; Ananthakrishnan, A.N.; Andrews, E.; Barron, G.; Lake, K.; et al. Dynamics of Metatranscription in the Inflammatory Bowel Disease Gut Microbiome. Nat. Microbiol. 2018, 3, 337–346. [Google Scholar] [CrossRef]


| Study | Complexity of Study | Sequencing Method | Highlighted Bacterial Markers | |
|---|---|---|---|---|
| Gevers D et al., 2014 [41] | One cohort N = 668 biopsies and stools from pediatric CD (447) and controls (221). | Shotgun | Increased in CD vs. control | Decreased in CD vs. control |
| Enterobacteriaceae, Pasteurellaceae, Fusobacteriaceae, Neisseriaceae, Gemellaceae, and Veillonellaceae | Bifidobacteriaceae, Erysipelotrichaceae, Bacteroidales (Bacteroides), Clostridiales (Coprococcus, Ruminococcus, Roseburia, Blautia, Faecalibacterium), including members from Ruminococcaceae and Lachnospiraceae. | |||
| Lewis JD et al., 2015 [42] | One cohort N = 112 stools from pediatrics CD (86) and controls (26). | Shotgun | Increased in CD vs. control | Decreased in CD vs. control |
| Escherichia, Klebsiella, Enterococcus, and Veillonella | Prevotella, Eubacterium, Odoribacter, Akkermansia, Roseburia, Parabacteroides, Alistipes, Coprococcus, Dorea, and Ruminococcus. | |||
| Schirmer M et al., 2018 [33] | One cohort N = 405 biopsies and stools from new-onset pediatric UC patients. | 16S | Increased in severe or moderate vs. mild CD | Decreased in severe or moderate vs. mild CD |
| Veillonela dispar, Aggregatibacter segnis, Campylobacter, Lachnospiraceae, Veillonela parvula, Haemophilus parainfluenzae, and Megasphaera (all representing bacteria typical of the oral cavity). | Ruminococcaceae and Lachnospiraceae families. | |||
| Pascal V et al., 2017 [35] | Four discovery cohorts N = 178 stools from CD (34), UC (33), and controls (111). | 16S | Increased in CD vs. control | Decreased in CD vs. control |
| Blautia, Escherichia, Fusobacterium, Dialister, Sutterella, and Collinsella. | Ruminococcus, Coprococcus, Roseburia, Ocillospira, Faecalibacterium, Lachnospira, Turicibacter, Clostridium, Parabacteroides, Anaerostipes, and Methanobrevibacter | |||
| Increased in CD vs. non-CD | Decreased in CD vs. non-CD | |||
| Fusobacterium and Escherichia. | Christensenellaceae, Peptostreptococcaceae. Faecalibacterium, Anaerostipes, Methanobrevibacter, and Collinsella. | |||
| Vich Vila A, et al., 2018 [37] | Three discovery cohorts N = 1380 stools from CD (208), UC (126), IBD-unclassified (21), and controls (1025). | Shotgun | Increased in CD vs. control | Decreased in CD vs. control |
| Enterobacteriaceae, Lachnospiraceae, Bacteroidaceae, Streptococcaceae, Erysipelotrichaceae, and Eggerthellaceae. | Lachnospiraceae, Ruminococcaceae, Clostridiaceae, Eubacteriaceae, Oscillospiraceae, Erysipelotrichaceae, and Peptostreptococcaceae. | |||
| Increased in UC vs. controls | Decreased in UC vs. controls | |||
| Bacteroidaceae. | Lachnospiraceae, Ruminococcaceae, Clostridiaceae, and Peptostreptococcaceae. | |||
| Franzosa EA et al., 2018 [36] | One discovery cohort N = 155 stools from CD (68), UC (53), and non-IBD control (34) patients. Validation with one additional cohort N = 65 stools from CD (20), UC (23), and non-IBD control (22) patients. | Shotgun | Increased in IBD vs. non-IBD | Decreased in IBD vs. non-IBD |
| Roseburia | Ruminococcus, Dorea, Eubacterium, Roseburia, Coprococcus, Faecalibacterium, Lachnospiraceae bacterium, Anaerostipes, Alistipes, Bacteroidales, Barnesiella, Oscillibacter, Holdemania, Subdoligranulum, Adlercreutzia, Gordonibacter, and Anaerotruncus. The species Roseburia hominis, Dorea formicigenerans, and Ruminococcus obeum exhibited the strongest effects. | |||
| Increased in UC vs. non-IBD: | Increased in CD vs. non-IBD: | |||
| Bifidobacterium breve and Clostridium symbiosum | Dorea, Lactobacillus, Pediococcus (2), Blautia, Clostridium, Ruminoccocus, Lachnospiraceae bacterium (2), and Escherichia (2). The species Ruminococcus gnavus, Escherichia coli, and Clostridium clostridioforme exhibited the strongest effects. | |||
| Nishino K et al., 2018 [38] | One cohort N = 174 mucus samples from CD (26), UC (43), and non-IBD control (14) patients. | 16S | Increased in CD vs. non-IBD | Decreased in CD vs. non-IBD |
| Proteobacteria such as Escherichia, Ruminococcus gnavus, Cetobacterium, Actinobacillus, and Enterococcus. | Faecalibacterium, Coprococcus, Prevotella, and Roseburia | |||
| Increased in CD vs. UC | Increased in UC vs. CD | |||
| Escherichia, Ruminococcus gnavus, Clostridium, Cetobacterium, and Peptostreptococcus. | Faecalibacterium, Blautia, Bifidobacterium, Roseburia, and Citrobacter. | |||
| Yilmaz B et al., 2019 [39] | One discovery cohort N = 1255 biopsies from IBD and non-IBD control patients. Validation with one additional cohort N = 1846 biopsies from IBD and non-IBD control patients | 16S | Increased in CD vs. non-IBD | Decreased in CD vs. non-IBD |
| Blautia, Ruminococcaceae, and Enterobacteriaceae. | Bifidobacterium, Collinsella, Barnesiellaceae, Butyricimonas, Rikenellaceae, Clostridiales, Lachnospiraceae, Coprococcus, Lachnospira, Roseburia, Ruminococcaceae, Faecalibacterium, and Erysipelotrichaceae. | |||
| Increased in UC vs. non-IBD | Decreased in UC vs. non-IBD | |||
| Bifidobacterium, Collinsela, Odoribacter, Blautia, Lachnospiraceae, and Ruminococcaceae. | Bacteroidetes, Parabacteroides, Clostridiaceae, and Tenericutes phylum. | |||
| Lloyd-Price J et al., 2019 [8] | One cohort N = 1595 fecal samples from 130 CD, UC, and control patients. | Shotgun | Increased in CD vs. non-CD | Decreased in CD vs. non-CD |
| Escherichia coli, Ruminococcus torques, and Bacteroides fragilis. | Faecalibacterium prausnitzii, Roseburia hominis, Subdoligranulum, Bacteroides thetaiotaomicron, and Coprococcus comes. | |||
| Increased in UC vs. non-UC | ||||
| Ruminococcus gnavus and Veillonela. | ||||
| Mevlut Ulas et al., 2024 [34] | One cohort N = 49 mucus samples from CD (13), UC (8), and non-IBD control (28) patients. | 16S | Increased in CD and UC vs. non-IBD | |
| Faecalibacterium prausnitzii, Bacteroidales/Bacteroidota, and Gammaproteobacteria | ||||
| Study | Complexity of Study | Sequencing Method | Highlighted Bacterial Markers | |
|---|---|---|---|---|
| Nakatsu G et al., 2015 [116] | One cohort N = 160 mucosal biopsies of adenoma and adenoma-adjacent mucosae (47), carcinoma and carcinoma-adjacent mucosae (52), and controls (61). Validation with two additional previously published cohorts. | 16S | Increased in CRC vs. control | Decreased in CRC vs. control |
| Fusobacterium, Bacteroides fragilis, Parvimonas, Peptostreptococcus, Gemella, and Leptotrichia | Bacteroides, Blautia, Sutterella, Faecalibacterium prausnitzii, Collinsella aerofaciens, and Alistipes putredinis | |||
| Feng Q et al., 2015 [117] | One cohort N = 156 fecal metagenomes from colorectal adenoma, carcinoma, and healthy controls. | Shotgun | Increased in CRC vs. advanced adenoma or control | Decreased in carcinoma or adenoma vs. control: |
| Bacteroides and Parabacteroides spp., Alistipes putredinis, Bilophila wadsworthia, Lachnospiraceae sp., Escherichia coli, and oral anaerobes Fusobacterium sp., Pavimonas micra, Gemella morbillorum, and Peptostreptococcus stomatis. | Bifidobactium animalis and Streptococcus thermophilus. | |||
| Yu J et al., 2017 [111] | Two discovery cohorts N = 168 fecal metagenomes from CRC (90) and controls (78). Validation with two additional previously published cohorts. | Shotgun | Increased in CRC vs. control | Decreased in CRC vs. control |
| Parvimonas micra, Solobacterium moorei, Fusobacterium nucleatum, and Peptostreptococcus stomatis. | Eubacterium ventriosum. | |||
| Flemer B et al., 2017 [118] | One cohort N = 179 fecal and/or mucosal metagenomes from patients with CRC (102), polyps (21), and healthy controls (56). Validation with two additional previously published cohorts | 16S | Increased in CRC vs. control (mucosal) | |
| Bacteroides, Roseburia, Ruminococcus, Oscillibacter, and the oral pathogens Porphyromonas, Peptostreptococcus, Parvimonas, and Fusobacterium | ||||
| Dai Z et al., 2018 [113] | Four cohorts N = 526 fecal metagenomes from CRC (255) and controls (271). | Shotgun | Seven CRC-enriched bacteria vs. control | Twenty CRC-depleted species with the largest fold change from a total of 69 CRC-depleted bacteria |
| Porphyromonas asaccharolytica, Fusobacterium nucleatum, Prevotella intermedia, Parvimonas micra, Bacteroides fragilis, Alistipes finegoldii, and Thermanaerovibrio acidaminovorans. | Ehrlichia ruminantium, Bartonella bacilliformis, Eubacterium eligens, Acinetobacter sp. ADP1, Mycoplasma canadense, Weissella cibaria, Dictyoglomus thermophilum, Thermosipho africanus, Thermodesulfobacterium geofontis, Campylobacter iguaniorum, Spiroplasma diminutum, Candidatus Phytoplasma australiense, Mycoplasma capricolum, Arcobacter sp., L Streptococcus sp. I−G2, Staphylococcus argenteus, Streptococcus thermophilus, SR1 bacterium RAAC1, Streptococcus salivarius, and Bifidobacterium catenulatum. | |||
| Shah MS et al., 2018 [114,119] | Nine previously published cohorts N = 509 total fecal samples from CRC (195), colorectal adenoma (79), and controls (235) | 16S | Increased in adenoma vs. control | Decreased in adenoma vs. control |
| Prevotella, Methanosphaera, Succinovibrio species, Haemophilus parainfluenzae, and the strains of the Synergistes family DSM 25858 and Methanosphaera stadtmanae DSM 3091. | Akkermansia muciniphila. | |||
| CRC increased bacteria vs. control | Increased in CRC and adenoma vs. control | |||
| Peptostreptococcus anerobius, Parvimonas, Porphyromonas, Akkermansia muciniphila, Fusobacterium sp., Parabacteroides distasonis, Streptococcus anginosus, Porphyromonas asaccharolytica ATCC 25260, Parvimonas micra ATCC 33270, Pantoea agglomerans, and Proteobacteria. | Ruminococcus, Lactobacillus, and Enterobacteriaceae. | |||
| Wirbel J et al., 2019 [114] | Eight discovery cohorts N = 768 total fecal metagenomes from CRC (285) and control (290) from five cohorts. Validation with three additional independent previously published cohorts comprising metagenomes from CRC (101) and controls (102). | Shotgun | A core set of 29 species increased in CRC vs. control | |
| Parvimonas micra, Gemella morbillorum, Peptostreptococcus stomatis, F. nucleatum subspecies animalis, Dialister, Unknown Porphyromonas, Solobacterium moorei, Porphyromonas uenonis, Clostridium symbiosum, Clostridiales, Hungatella hathewayi, Prevotella intermedia, Porphyromonas somerae, Porphyromonas asaccharolytica, F. nucleatum subspecies nucleatum, Parvimonas species, Prevotella nigrescens, Porphyromonas, Ruminococcus torques, F. nucleatum subspecies vincentii, Fusobacterium species oral taxon 370, Peptostreptococcaceae, Anaerococcus obesiensis/vaginalis, Anaerotruncus, P. uenonis, Clostridiales, Porphyromonas, Clostridium bolteae/clostridioforme, and Subdoligranulum species. | ||||
| Thomas AM et al., 2019 [115] | Nine discovery cohorts N = 969 total fecal metagenomes from CRC (313), adenoma (143), and control (308) from 7 cohorts. Validation with two additional independent previously published cohorts comprising metagenomes from CRC (100) and controls (105). | Shotgun | A core set of 26 species increased in CRC vs. control metagenomes | |
| Fusobacterium nucleatum, Parvimonas micra, Parvimonas spp., Gemella morbillorum, Peptostreptococcus stomatis, Solobacterium moorei, Porphyromonas asaccharolytica, Clostridium symbiosum, Anaerococcus vaginalis, Prevotella intermedia, Bacteroides fragilis, Porphyromonas somerae, Anaerococcus obesiensis, Porphyromonas uenonis, Peptostreptococcus anaerobius, Streptococcus constellatus, Granulicatella adiacens, Methanobrevibacter smithii, Eikenella corrodens, Ruminococcus torques, Peptostreptococcus spp., Streptococcus gallolyticus, Methanobrevibacter spp., Actinomyces cardiffensis, Campylobacter ureolyticus, and Anaerotruncus spp. Biomarkers enriched in the majority of the datasets are underlined. | ||||
| Liang JQ et al., 2019 [9] | Two cohorts N = 1012 fecal metagenomes from CRC (274), adenoma (353), and controls (385). | Shotgun | Increased in CRC or adenoma vs. control | |
| Fusobacterium nucleatum, Clostridium hathewayi, and Lachnoclostridium. | ||||
| Sequencing Methodology | Advantages | Disadvantages |
|---|---|---|
| 16S rRNA |
|
|
| Shotgun |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
San-Martin, M.I.; Chamizo-Ampudia, A.; Sanchiz, Á.; Ferrero, M.Á.; Martínez-Blanco, H.; Rodríguez-Aparicio, L.B.; Navasa, N. Microbiome Markers in Gastrointestinal Disorders: Inflammatory Bowel Disease, Colorectal Cancer, and Celiac Disease. Int. J. Mol. Sci. 2025, 26, 4818. https://doi.org/10.3390/ijms26104818
San-Martin MI, Chamizo-Ampudia A, Sanchiz Á, Ferrero MÁ, Martínez-Blanco H, Rodríguez-Aparicio LB, Navasa N. Microbiome Markers in Gastrointestinal Disorders: Inflammatory Bowel Disease, Colorectal Cancer, and Celiac Disease. International Journal of Molecular Sciences. 2025; 26(10):4818. https://doi.org/10.3390/ijms26104818
Chicago/Turabian StyleSan-Martin, M. Isabel, Alejandro Chamizo-Ampudia, África Sanchiz, Miguel Ángel Ferrero, Honorina Martínez-Blanco, Leandro Benito Rodríguez-Aparicio, and Nicolás Navasa. 2025. "Microbiome Markers in Gastrointestinal Disorders: Inflammatory Bowel Disease, Colorectal Cancer, and Celiac Disease" International Journal of Molecular Sciences 26, no. 10: 4818. https://doi.org/10.3390/ijms26104818
APA StyleSan-Martin, M. I., Chamizo-Ampudia, A., Sanchiz, Á., Ferrero, M. Á., Martínez-Blanco, H., Rodríguez-Aparicio, L. B., & Navasa, N. (2025). Microbiome Markers in Gastrointestinal Disorders: Inflammatory Bowel Disease, Colorectal Cancer, and Celiac Disease. International Journal of Molecular Sciences, 26(10), 4818. https://doi.org/10.3390/ijms26104818