
Transcriptomics Reveals an Energy-Saving Metabolic Switch in an Extremophilic Red Microalga Cyanidioschyzon merolae Under Nickel Stress

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
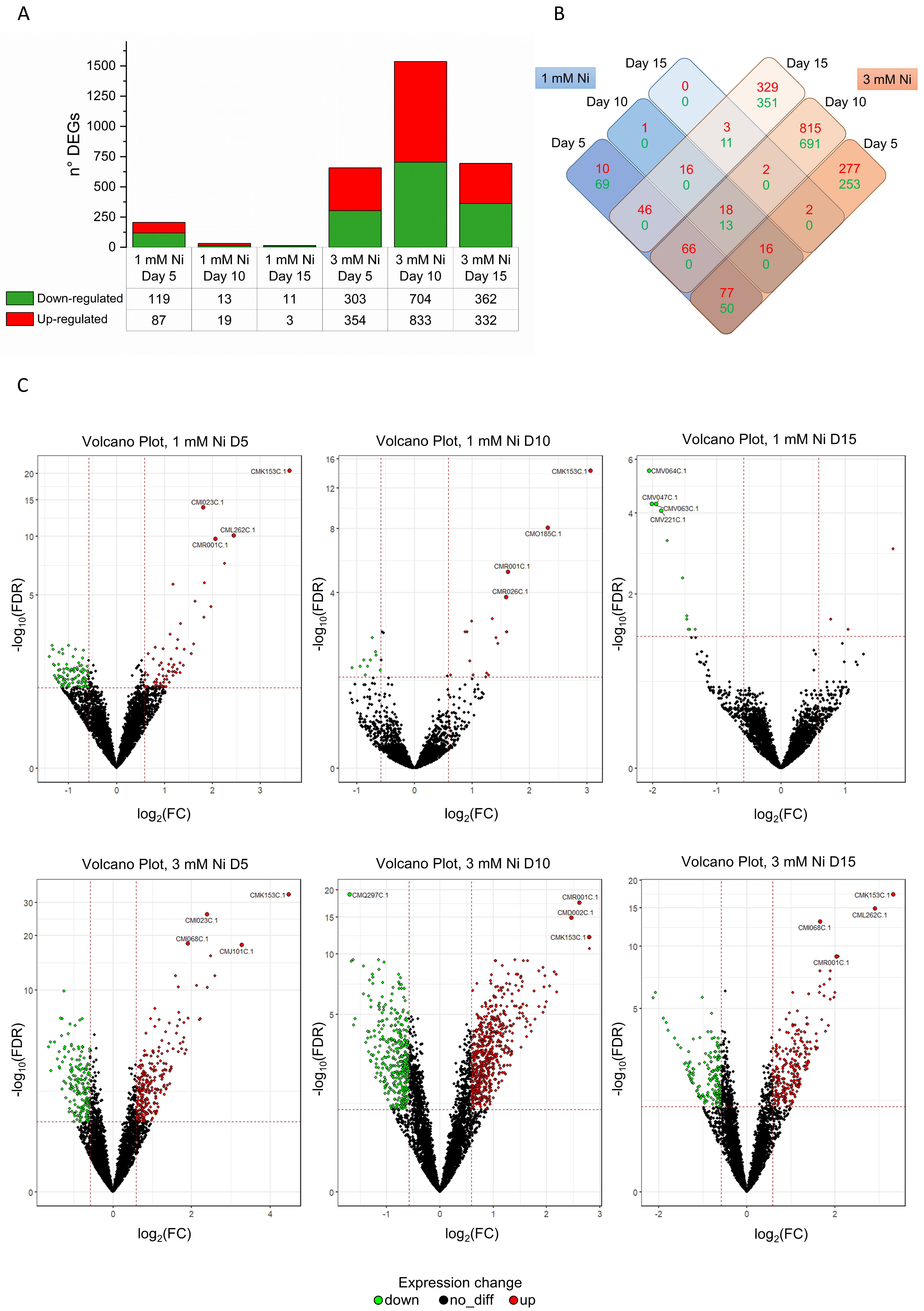
2.1. Dynamic Remodeling of C. merolae Transcriptome During Long-Term Ni Adaptation
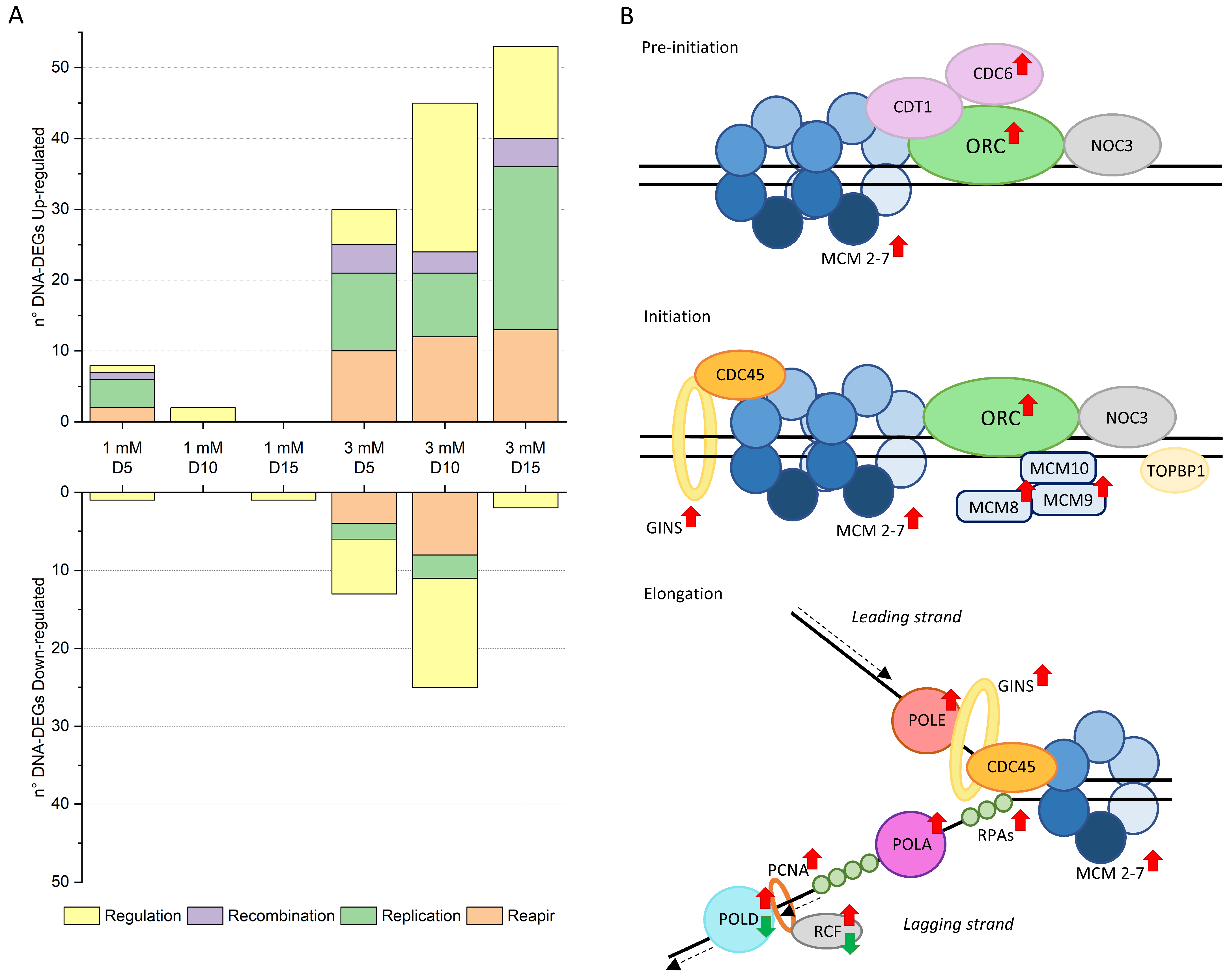
2.2. DNA Replication and Structure Remodeling During Ni Adaptation
2.3. Regulation of Oxidative Stress Responses
2.4. Protein Synthesis, Folding, and Degradation
2.5. Lipid Metabolism
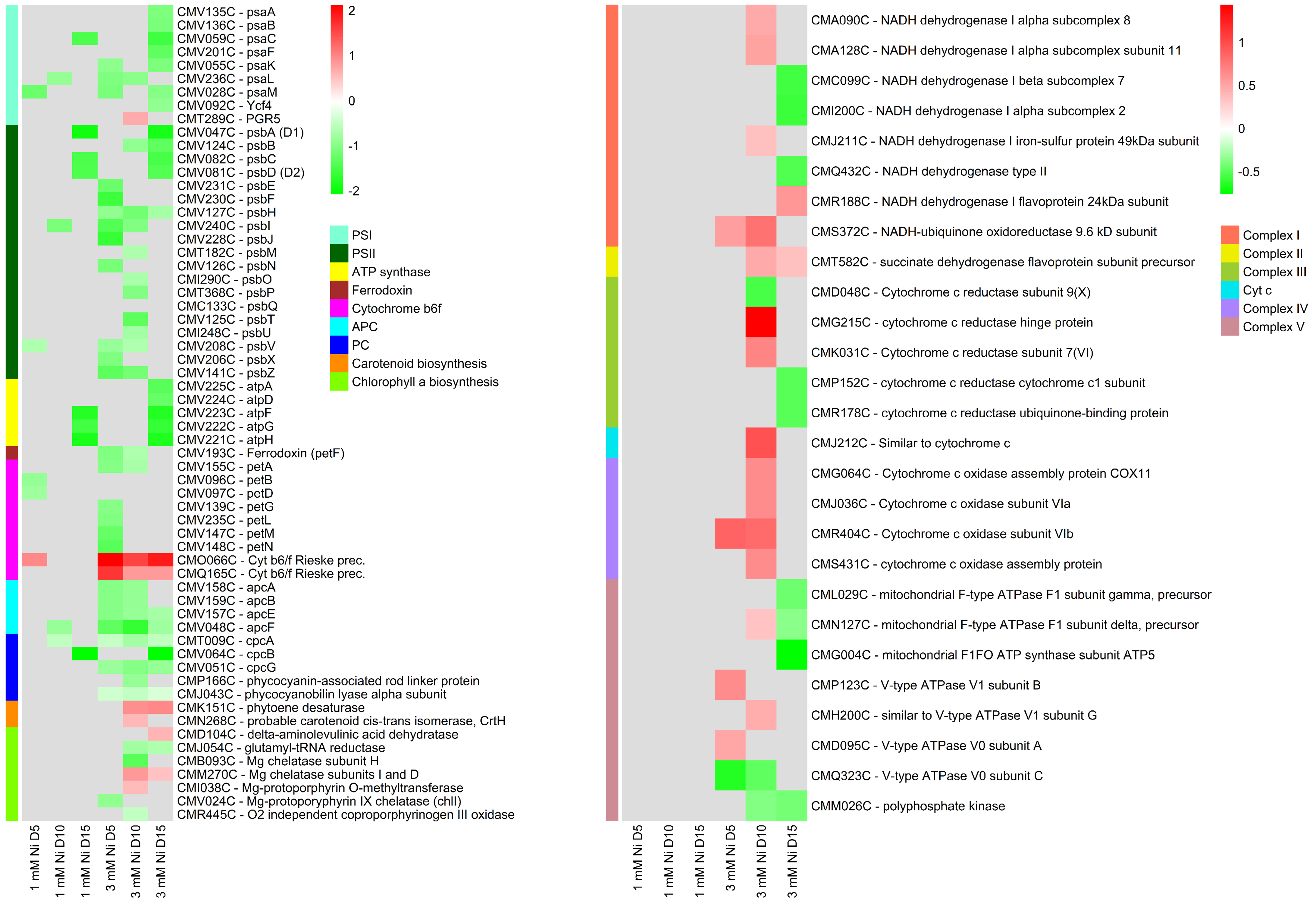
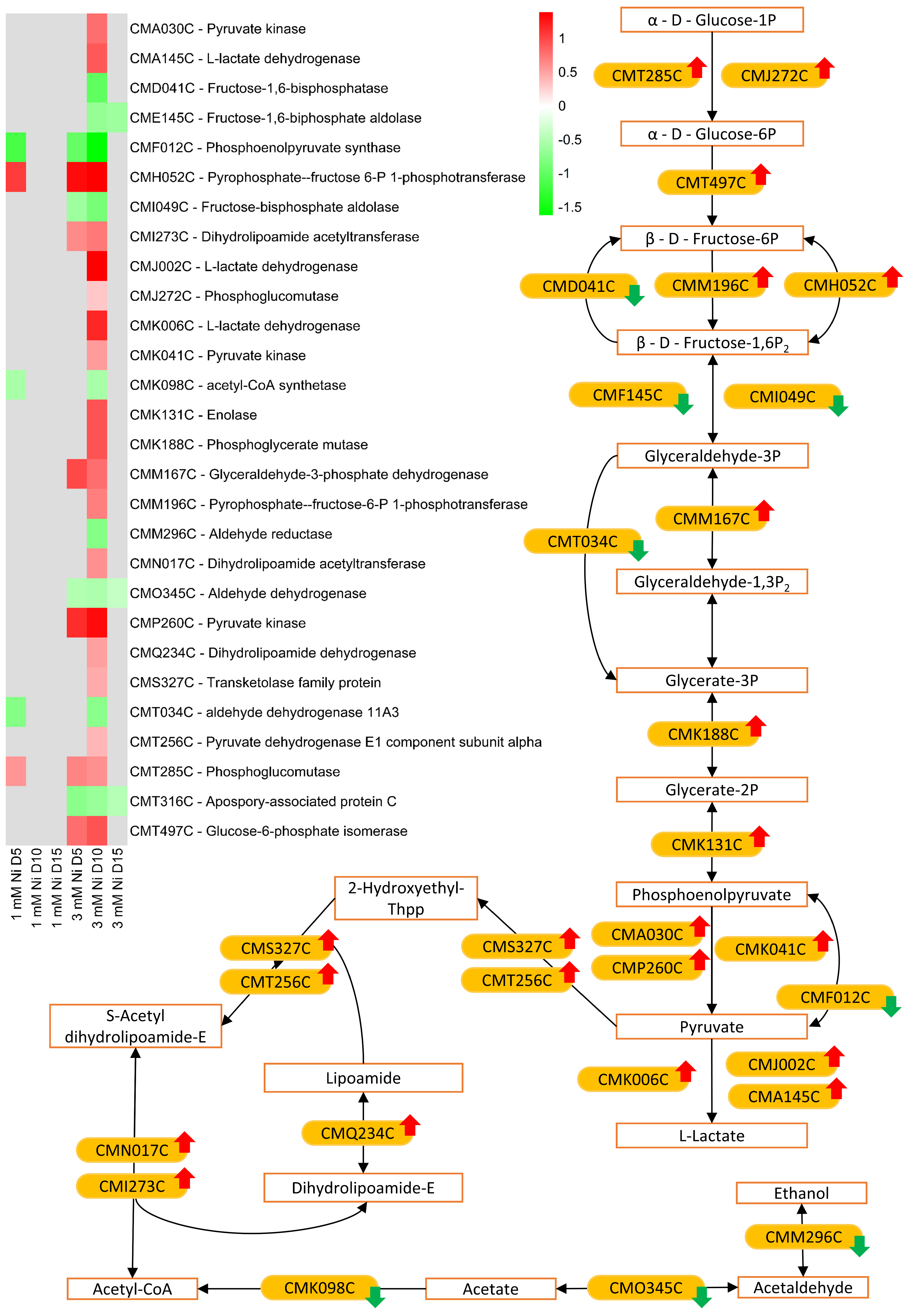
2.6. Energy Metabolism
2.7. Metal Transport
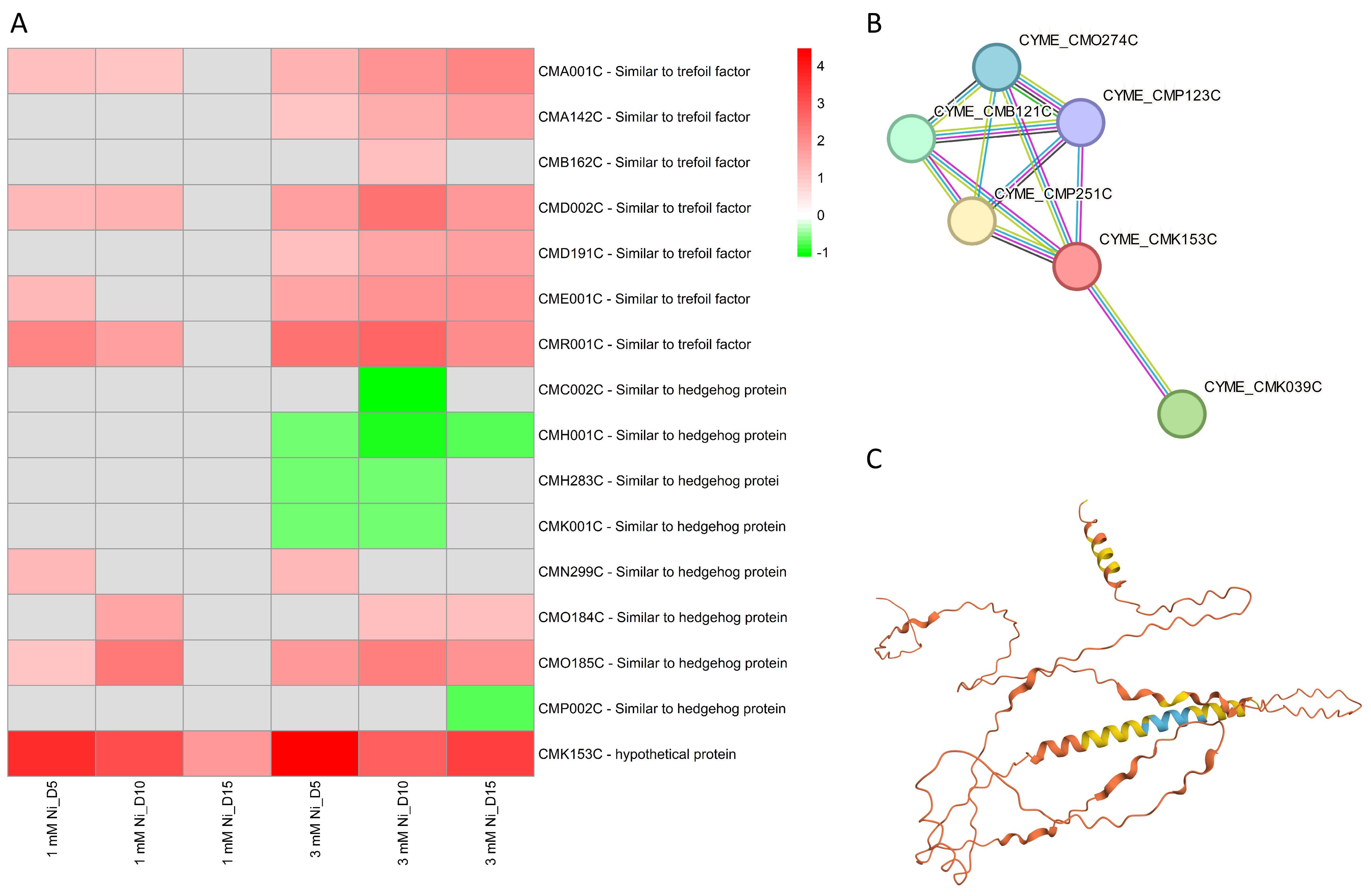
2.8. Other Molecular Components
3. Materials and Methods
3.1. Culture Conditions and Experimental Design
3.2. Cell Growth Measurements
3.3. Total RNA Extraction
3.4. Construction of RNA-Seq Libraries and Sequencing
3.5. Differentially Expressed Genes Analysis
3.6. Gene Ontology and KEGG Pathway Enrichment Analysis
3.7. Statistical and Bioinformatic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jamers, A.; Blust, R.; De Coen, W.; Griffin, J.L.; Jones, O.A.H. An Omics Based Assessment of Cadmium Toxicity in the Green Alga Chlamydomonas reinhardtii. Aquat. Toxicol. Amst. Neth. 2013, 126, 355–364. [Google Scholar] [CrossRef]
- Pillai, S.; Behra, R.; Nestler, H.; Suter, M.J.-F.; Sigg, L.; Schirmer, K. Linking Toxicity and Adaptive Responses across the Transcriptome, Proteome, and Phenotype of Chlamydomonas reinhardtii Exposed to Silver. Proc. Natl. Acad. Sci. USA 2014, 111, 3490–3495. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Lu, D.; Liu, C.; Hu, J.; Wang, P.; Dai, X. Toxic Effect of Nickel on Microalgae Phaeodactylum tricornutum (Bacillariophyceae). Ecotoxicol. Lond. Engl. 2022, 31, 746–760. [Google Scholar] [CrossRef] [PubMed]
- Nowicka, B. Heavy Metal-Induced Stress in Eukaryotic Algae-Mechanisms of Heavy Metal Toxicity and Tolerance with Particular Emphasis on Oxidative Stress in Exposed Cells and the Role of Antioxidant Response. Environ. Sci. Pollut. Res. Int. 2022, 29, 16860–16911. [Google Scholar] [CrossRef] [PubMed]
- De Luca, P.; Taddei, R.; Varano, L. Cyanidioschyzon merolae: A New Alga of Thermal Acidic Environments. Webbia 1978, 33, 37–44. [Google Scholar] [CrossRef]
- Seckbach, J. Overview on Cyanidian Biology. In Red Algae in the Genomic Age; Seckbach, J., Chapman, D.J., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 345–356. ISBN 978-90-481-3795-4. [Google Scholar]
- Miyagishima, S.; Wei, J.L.; Nozaki, H.; Hirooka, S. Cyanidiales: Evolution and Habitats. In Cyanidioschyzon merolae: A New Model Eukaryote for Cell and Organelle Biology; Kuroiwa, T., Miyagishima, S., Matsunaga, S., Sato, N., Nozaki, H., Tanaka, K., Misumi, O., Eds.; Springer: Singapore, 2017; pp. 3–15. ISBN 978-981-10-6101-1. [Google Scholar]
- Haniewicz, P.; Abram, M.; Nosek, L.; Kirkpatrick, J.; El-Mohsnawy, E.; Olmos, J.D.J.; Kouřil, R.; Kargul, J.M. Molecular Mechanisms of Photoadaptation of Photosystem I Supercomplex from an Evolutionary Cyanobacterial/Algal Intermediate. Plant Physiol. 2018, 176, 1433–1451. [Google Scholar] [CrossRef]
- Matsuzaki, M.; Misumi, O.; Shin-I, T.; Maruyama, S.; Takahara, M.; Miyagishima, S.-Y.; Mori, T.; Nishida, K.; Yagisawa, F.; Nishida, K.; et al. Genome Sequence of the Ultrasmall Unicellular Red Alga Cyanidioschyzon merolae 10D. Nature 2004, 428, 653–657. [Google Scholar] [CrossRef]
- Kuroiwa, T.; Kuroiwa, H.; Sakai, A.; Takahashi, H.; Toda, K.; Itoh, R. The Division Apparatus of Plastids and Mitochondria. Int. Rev. Cytol. 1998, 181, 1–41. [Google Scholar] [CrossRef]
- Kuroiwa, T. The Primitive Red Algae Cyanidium caldarium and Cyanidioschyzon merolae as Model System for Investigating the Dividing Apparatus of Mitochondria and Plastids. BioEssays 1998, 20, 344–354. [Google Scholar] [CrossRef]
- Imamura, S.; Kawase, Y.; Kobayashi, I.; Sone, T.; Era, A.; Miyagishima, S.-Y.; Shimojima, M.; Ohta, H.; Tanaka, K. Target of Rapamycin (TOR) Plays a Critical Role in Triacylglycerol Accumulation in Microalgae. Plant Mol. Biol. 2015, 89, 309–318. [Google Scholar] [CrossRef]
- Miyagishima, S.-Y.; Tanaka, K. The Unicellular Red Alga Cyanidioschyzon merolae—The Simplest Model of a Photosynthetic Eukaryote. Plant Cell Physiol. 2021, 62, 926–941. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.; Nield, J.; Hippler, M. The Composition and Structure of Photosystem I-Associated Antenna from Cyanidioschyzon merolae. Plant J. Cell Mol. Biol. 2010, 62, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Krupnik, T.; Kotabová, E.; van Bezouwen, L.S.; Mazur, R.; Garstka, M.; Nixon, P.J.; Barber, J.; Kaňa, R.; Boekema, E.J.; Kargul, J. A Reaction Center-Dependent Photoprotection Mechanism in a Highly Robust Photosystem II from an Extremophilic Red Alga, Cyanidioschyzon merolae. J. Biol. Chem. 2013, 288, 23529–23542. [Google Scholar] [CrossRef]
- Nikolova, D.; Weber, D.; Scholz, M.; Bald, T.; Scharsack, J.P.; Hippler, M. Temperature-Induced Remodeling of the Photosynthetic Machinery Tunes Photosynthesis in the Thermophilic Alga Cyanidioschyzon merolae. Plant Physiol. 2017, 174, 35–46. [Google Scholar] [CrossRef]
- Tian, L.; Liu, Z.; Wang, F.; Shen, L.; Chen, J.; Chang, L.; Zhao, S.; Han, G.; Wang, W.; Kuang, T.; et al. Isolation and Characterization of PSI–LHCI Super-Complex and Their Sub-Complexes from a Red Alga Cyanidioschyzon merolae. Photosynth. Res. 2017, 133, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Antoshvili, M.; Caspy, I.; Hippler, M.; Nelson, N. Structure and Function of Photosystem I in Cyanidioschyzon merolae. Photosynth. Res. 2019, 139, 499–508. [Google Scholar] [CrossRef]
- Abram, M.; Białek, R.; Szewczyk, S.; Karolczak, J.; Gibasiewicz, K.; Kargul, J. Remodeling of Excitation Energy Transfer in Extremophilic Red Algal PSI-LHCI Complex during Light Adaptation. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148093. [Google Scholar] [CrossRef]
- Chang, L.; Tian, L.; Ma, F.; Mao, Z.; Liu, X.; Han, G.; Wang, W.; Yang, Y.; Kuang, T.; Pan, J.; et al. Regulation of Photosystem I-Light-Harvesting Complex I from a Red Alga Cyanidioschyzon merolae in Response to Light Intensities. Photosynth. Res. 2020, 146, 287–297. [Google Scholar] [CrossRef]
- Piochi, M.; Mormone, A.; Strauss, H.; Balassone, G. The Acid Sulfate Zone and the Mineral Alteration Styles of the Roman Puteoli (Neapolitan Area, Italy): Clues on Fluid Fracturing Progression at the Campi Flegrei Volcano. Solid Earth 2019, 10, 1809–1831. [Google Scholar] [CrossRef]
- Marchetto, F.; Santaeufemia, S.; Lebiedzińska-Arciszewska, M.; Śliwińska, M.A.; Pich, M.; Kurek, E.; Naziębło, A.; Strawski, M.; Solymosi, D.; Szklarczyk, M.; et al. Dynamic Adaptation of the Extremophilic Red Microalga Cyanidioschyzon merolae to High Nickel Stress. Plant Physiol. Biochem. PPB 2024, 207, 108365. [Google Scholar] [CrossRef]
- Zheng, Q.; Cheng, Z.Z.; Yang, Z.M. HISN3 Mediates Adaptive Response of Chlamydomonas reinhardtii to Excess Nickel. Plant Cell Physiol. 2013, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Shultz, R.W.; Tatineni, V.M.; Hanley-Bowdoin, L.; Thompson, W.F. Genome-Wide Analysis of the Core DNA Replication Machinery in the Higher Plants Arabidopsis and Rice. Plant Physiol. 2007, 144, 1697–1714. [Google Scholar] [CrossRef]
- Seregin, I.V.; Kozhevnikova, A.D. Physiological Role of Nickel and Its Toxic Effects on Higher Plants. Russ. J. Plant Physiol. 2006, 53, 257–277. [Google Scholar] [CrossRef]
- Tye, B.K. MCM Proteins in DNA Replication. Annu. Rev. Biochem. 1999, 68, 649–686. [Google Scholar] [CrossRef] [PubMed]
- Drapkin, R.; Reardon, J.T.; Ansari, A.; Huang, J.C.; Zawel, L.; Ahn, K.; Sancar, A.; Reinberg, D. Dual Role of TFIIH in DNA Excision Repair and in Transcription by RNA Polymerase II. Nature 1994, 368, 769–772. [Google Scholar] [CrossRef]
- Pećina-Šlaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Harvey, S.H.; Krien, M.J.E.; O’Connell, M.J. Structural Maintenance of Chromosomes (SMC) Proteins, a Family of Conserved ATPases. Genome Biol. 2002, 3, REVIEWS3003. [Google Scholar] [CrossRef]
- Strunnikov, A.V.; Jessberger, R. Structural Maintenance of Chromosomes (SMC) Proteins. Eur. J. Biochem. 1999, 263, 6–13. [Google Scholar] [CrossRef]
- Potts, P.R.; Porteus, M.H.; Yu, H. Human SMC5/6 Complex Promotes Sister Chromatid Homologous Recombination by Recruiting the SMC1/3 Cohesin Complex to Double-Strand Breaks. EMBO J. 2006, 25, 3377–3388. [Google Scholar] [CrossRef]
- He, X.; Rines, D.R.; Espelin, C.W.; Sorger, P.K. Molecular Analysis of Kinetochore-Microtubule Attachment in Budding Yeast. Cell 2001, 106, 195–206. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Alhaithloul, H.A.S.; Parvin, K.; Bhuyan, M.H.M.B.; Tanveer, M.; Mohsin, S.M.; Nahar, K.; Soliman, M.H.; Mahmud, J.A.; Fujita, M. Polyamine Action under Metal/Metalloid Stress: Regulation of Biosynthesis, Metabolism, and Molecular Interactions. Int. J. Mol. Sci. 2019, 20, 3215. [Google Scholar] [CrossRef] [PubMed]
- Beauvais-Flück, R.; Slaveykova, V.I.; Cosio, C. Cellular Toxicity Pathways of Inorganic and Methyl Mercury in the Green Microalga Chlamydomonas reinhardtii. Sci. Rep. 2017, 7, 8034. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.J.A.; Budd, K.; Lefebvre, D.D. Biotransformation of Mercury in pH-Stat Cultures of Eukaryotic Freshwater Algae. Arch. Microbiol. 2007, 187, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.-P.; Ban, Y.; Inoue, H.; Matsuda, N.; Moriguchi, T. Spermidine Levels Are Implicated in Heavy Metal Tolerance in a Spermidine Synthase Overexpressing Transgenic European Pear by Exerting Antioxidant Activities. Transgenic Res. 2010, 19, 91–103. [Google Scholar] [CrossRef]
- Nahar, K.; Hasanuzzaman, M.; Alam, M.M.; Rahman, A.; Suzuki, T.; Fujita, M. Polyamine and Nitric Oxide Crosstalk: Antagonistic Effects on Cadmium Toxicity in Mung Bean Plants through Upregulating the Metal Detoxification, Antioxidant Defense and Methylglyoxal Detoxification Systems. Ecotoxicol. Environ. Saf. 2016, 126, 245–255. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Lin, H.-J. Polyamines in Microalgae: Something Borrowed, Something New. Mar. Drugs 2018, 17, 1. [Google Scholar] [CrossRef]
- Tajti, J.; Janda, T.; Majláth, I.; Szalai, G.; Pál, M. Comparative Study on the Effects of Putrescine and Spermidine Pre-Treatment on Cadmium Stress in Wheat. Ecotoxicol. Environ. Saf. 2018, 148, 546–554. [Google Scholar] [CrossRef]
- Krüger, A.; Vowinckel, J.; Mülleder, M.; Grote, P.; Capuano, F.; Bluemlein, K.; Ralser, M. Tpo1-Mediated Spermine and Spermidine Export Controls Cell Cycle Delay and Times Antioxidant Protein Expression during the Oxidative Stress Response. EMBO Rep. 2013, 14, 1113–1119. [Google Scholar] [CrossRef]
- Piotrowska-Niczyporuk, A.; Bajguz, A.; Zambrzycka, E.; Godlewska-Żyłkiewicz, B. Phytohormones as Regulators of Heavy Metal Biosorption and Toxicity in Green Alga Chlorella vulgaris (Chlorophyceae). Plant Physiol. Biochem. PPB 2012, 52, 52–65. [Google Scholar] [CrossRef]
- Jiang, D.; Hou, J.; Gao, W.; Tong, X.; Li, M.; Chu, X.; Chen, G. Exogenous Spermidine Alleviates the Adverse Effects of Aluminum Toxicity on Photosystem II through Improved Antioxidant System and Endogenous Polyamine Contents. Ecotoxicol. Environ. Saf. 2021, 207, 111265. [Google Scholar] [CrossRef]
- Park, C.-J.; Seo, Y.-S. Heat Shock Proteins: A Review of the Molecular Chaperones for Plant Immunity. Plant Pathol. J. 2015, 31, 323–333. [Google Scholar] [CrossRef]
- Hasan, M.K.; Cheng, Y.; Kanwar, M.K.; Chu, X.-Y.; Ahammed, G.J.; Qi, Z.-Y. Responses of Plant Proteins to Heavy Metal Stress-A Review. Front. Plant Sci. 2017, 8, 1492. [Google Scholar] [CrossRef]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of Plant Heat-Shock Proteins and Molecular Chaperones in the Abiotic Stress Response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Brackley, K.I.; Grantham, J. Activities of the Chaperonin Containing TCP-1 (CCT): Implications for Cell Cycle Progression and Cytoskeletal Organisation. Cell Stress Chaperones 2009, 14, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-M.; Rhee, J.-S.; Jeong, C.-B.; Seo, J.S.; Park, G.S.; Lee, Y.-M.; Lee, J.-S. Heavy Metals Induce Oxidative Stress and Trigger Oxidative Stress-Mediated Heat Shock Protein (Hsp) Modulation in the Intertidal Copepod Tigriopus japonicus. Comp. Biochem. Physiol. Toxicol. Pharmacol. CBP 2014, 166, 65–74. [Google Scholar] [CrossRef]
- Mori, N.; Moriyama, T.; Toyoshima, M.; Sato, N. Construction of Global Acyl Lipid Metabolic Map by Comparative Genomics and Subcellular Localization Analysis in the Red Alga Cyanidioschyzon merolae. Front. Plant Sci. 2016, 7, 958. [Google Scholar] [CrossRef]
- Sharma, P.; Lakra, N.; Goyal, A.; Ahlawat, Y.K.; Zaid, A.; Siddique, K.H.M. Drought and Heat Stress Mediated Activation of Lipid Signaling in Plants: A Critical Review. Front. Plant Sci. 2023, 14, 1216835. [Google Scholar] [CrossRef] [PubMed]
- Bilgin, D.D.; Zavala, J.A.; Zhu, J.; Clough, S.J.; Ort, D.R.; DeLucia, E.H. Biotic Stress Globally Downregulates Photosynthesis Genes. Plant Cell Environ. 2010, 33, 1597–1613. [Google Scholar] [CrossRef]
- Abdellaoui, N.; Kim, M.J.; Choi, T.J. Transcriptome Analysis of Gene Expression in Chlorella vulgaris under Salt Stress. World J. Microbiol. Biotechnol. 2019, 35, 141. [Google Scholar] [CrossRef]
- Grennan, A.K.; Ort, D.R. Cool Temperatures Interfere with D1 Synthesis in Tomato by Causing Ribosomal Pausing. Photosynth. Res. 2007, 94, 375–385. [Google Scholar] [CrossRef]
- Ingrisano, R.; Tosato, E.; Trost, P.; Gurrieri, L.; Sparla, F. Proline, Cysteine and Branched-Chain Amino Acids in Abiotic Stress Response of Land Plants and Microalgae. Plants Basel Switz. 2023, 12, 3410. [Google Scholar] [CrossRef]
- Hanikenne, M.; Krämer, U.; Demoulin, V.; Baurain, D. A Comparative Inventory of Metal Transporters in the Green Alga Chlamydomonas reinhardtii and the Red Alga Cyanidioschizon merolae. Plant Physiol. 2005, 137, 428–446. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Tampé, R. Structural and Mechanistic Principles of ABC Transporters. Annu. Rev. Biochem. 2020, 89, 605–636. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-U.; Song, W.-Y.; Hong, D.; Ko, D.; Yamaoka, Y.; Jang, S.; Yim, S.; Lee, E.; Khare, D.; Kim, K.; et al. Plant ABC Transporters Enable Many Unique Aspects of a Terrestrial Plant’s Lifestyle. Mol. Plant 2016, 9, 338–355. [Google Scholar] [CrossRef]
- Fu, H.-L.; Meng, Y.; Ordóñez, E.; Villadangos, A.F.; Bhattacharjee, H.; Gil, J.A.; Mateos, L.M.; Rosen, B.P. Properties of Arsenite Efflux Permeases (Acr3) from Alkaliphilus metalliredigens and Corynebacterium glutamicum. J. Biol. Chem. 2009, 284, 19887–19895. [Google Scholar] [CrossRef] [PubMed]
- Brautigam, C.A.; Deka, R.K.; Ouyang, Z.; Machius, M.; Knutsen, G.; Tomchick, D.R.; Norgard, M.V. Biophysical and Bioinformatic Analyses Implicate the Treponema pallidum Tp34 Lipoprotein (Tp0971) in Transition Metal Homeostasis. J. Bacteriol. 2012, 194, 6771–6781. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, S.J.; Min, W.K.; Cha, S.; Song, J.T.; Seo, H.S. COP1 Controls Salt Stress Tolerance by Modulating Sucrose Content. Plant Signal. Behav. 2022, 17, 2096784. [Google Scholar] [CrossRef]
- García de la Torre, V.S.; Majorel-Loulergue, C.; Rigaill, G.J.; Alfonso-González, D.; Soubigou-Taconnat, L.; Pillon, Y.; Barreau, L.; Thomine, S.; Fogliani, B.; Burtet-Sarramegna, V.; et al. Wide Cross-Species RNA-Seq Comparison Reveals Convergent Molecular Mechanisms Involved in Nickel Hyperaccumulation across Dicotyledons. New Phytol. 2021, 229, 994–1006. [Google Scholar] [CrossRef]
- Meier, S.K.; Adams, N.; Wolf, M.; Balkwill, K.; Muasya, A.M.; Gehring, C.A.; Bishop, J.M.; Ingle, R.A. Comparative RNA-Seq Analysis of Nickel Hyperaccumulating and Non-Accumulating Populations of Senecio coronatus (Asteraceae). Plant J. 2018, 95, 1023–1038. [Google Scholar] [CrossRef]
- Volpicella, M.; Leoni, C.; Manzari, C.; Chiara, M.; Picardi, E.; Piancone, E.; Italiano, F.; D’Erchia, A.; Trotta, M.; Horner, D.S.; et al. Transcriptomic Analysis of Nickel Exposure in Sphingobium Sp. Ba1 Cells Using RNA-Seq. Sci. Rep. 2017, 7, 8262. [Google Scholar] [CrossRef] [PubMed]
- Czajka, K.M.; Nkongolo, K.K. Whole Genome Expression Analysis of Trembling Aspen (Populus tremuloides) Genotypes Exposed to Increasing Concentrations of Nickel. Plant Gene 2023, 34, 100416. [Google Scholar] [CrossRef]
- Allen, M.B. Studies with Cyanidium caldarium, an Anomalously Pigmented Chlorophyte. Arch. Für Mikrobiol. 1959, 32, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinforma. Oxf. Engl. 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinforma. Oxf. Engl. 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and Accurate Filtering of Ribosomal RNAs in Metatranscriptomic Data. Bioinform. Oxf. Engl. 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python Framework to Work with High-Throughput Sequencing Data. Bioinform. Oxf. Engl. 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An Integrated Web Application for Differential Expression and Pathway Analysis of RNA-Seq Data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Andreeva, A.; Florentino, L.C.; Chuguransky, S.R.; Grego, T.; Hobbs, E.; Pinto, B.L.; Orr, A.; Paysan-Lafosse, T.; Ponamareva, I.; et al. InterPro: The Protein Sequence Classification Resource in 2025. Nucleic Acids Res. 2024, 53, D444–D456. [Google Scholar] [CrossRef]
- McGinnis, S.; Madden, T.L. BLAST: At the Core of a Powerful and Diverse Set of Sequence Analysis Tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING Database in 2023: Protein-Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]

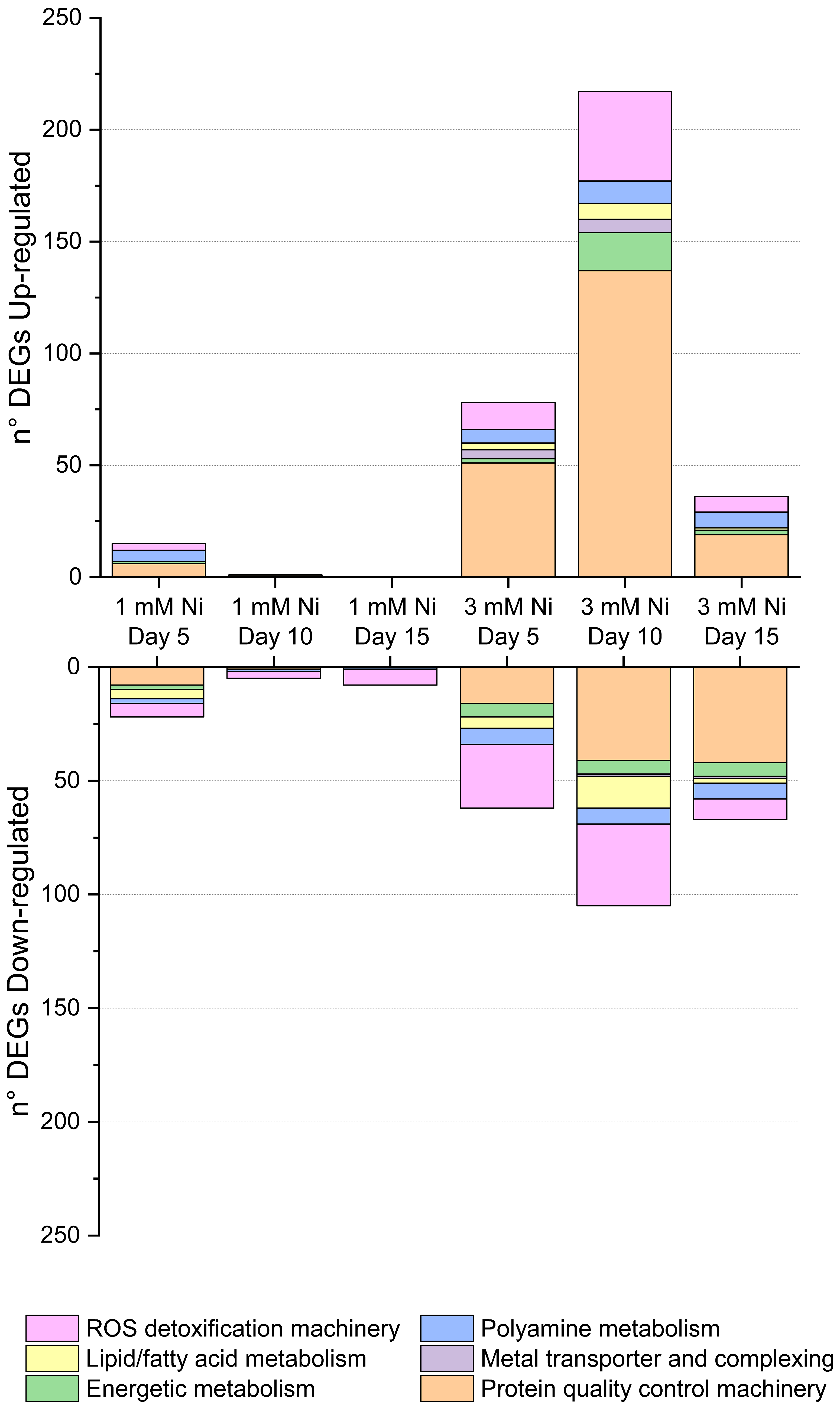

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santaeufemia, S.; Marchetto, F.; Romano, P.; Adamska, D.; Goryca, K.; Palatini, J.; Kargul, J. Transcriptomics Reveals an Energy-Saving Metabolic Switch in an Extremophilic Red Microalga Cyanidioschyzon merolae Under Nickel Stress. Int. J. Mol. Sci. 2025, 26, 4813. https://doi.org/10.3390/ijms26104813
Santaeufemia S, Marchetto F, Romano P, Adamska D, Goryca K, Palatini J, Kargul J. Transcriptomics Reveals an Energy-Saving Metabolic Switch in an Extremophilic Red Microalga Cyanidioschyzon merolae Under Nickel Stress. International Journal of Molecular Sciences. 2025; 26(10):4813. https://doi.org/10.3390/ijms26104813
Chicago/Turabian StyleSantaeufemia, Sergio, Francesca Marchetto, Patrizia Romano, Dorota Adamska, Krzysztof Goryca, Jeffrey Palatini, and Joanna Kargul. 2025. "Transcriptomics Reveals an Energy-Saving Metabolic Switch in an Extremophilic Red Microalga Cyanidioschyzon merolae Under Nickel Stress" International Journal of Molecular Sciences 26, no. 10: 4813. https://doi.org/10.3390/ijms26104813
APA StyleSantaeufemia, S., Marchetto, F., Romano, P., Adamska, D., Goryca, K., Palatini, J., & Kargul, J. (2025). Transcriptomics Reveals an Energy-Saving Metabolic Switch in an Extremophilic Red Microalga Cyanidioschyzon merolae Under Nickel Stress. International Journal of Molecular Sciences, 26(10), 4813. https://doi.org/10.3390/ijms26104813