
High-Fat Diet and Metabolic Diseases: A Comparative Analysis of Sex-Dependent Responses and Mechanisms

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Sex Differences in Metabolic Responses Associated with HFD and the Development of Metabolic Diseases
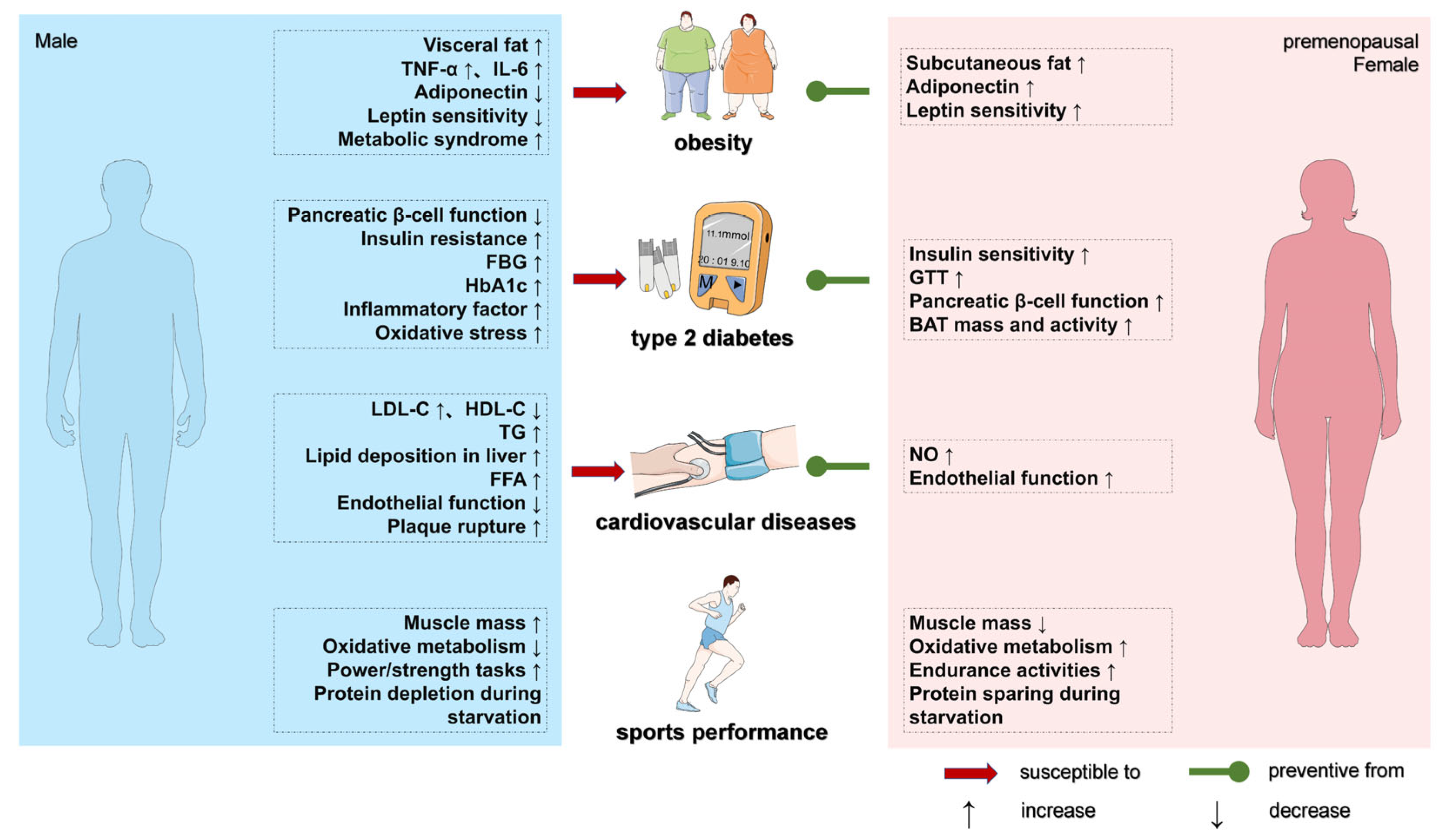
2.1. Sex Differences in Body Composition Abnormalities and Obesity Development
2.2. Sex Differences in Disorders of Glucose Metabolism and Development of T2DM
2.3. Sex Differences in HFD-Induced Lipid Metabolism Disorders and Cardiovascular Disease Development
2.4. Sex Differences in HFD-Induced Inflammation, Oxidative Stress, and Associated Disease Development
2.4.1. Inflammation
2.4.2. Oxidative Stress
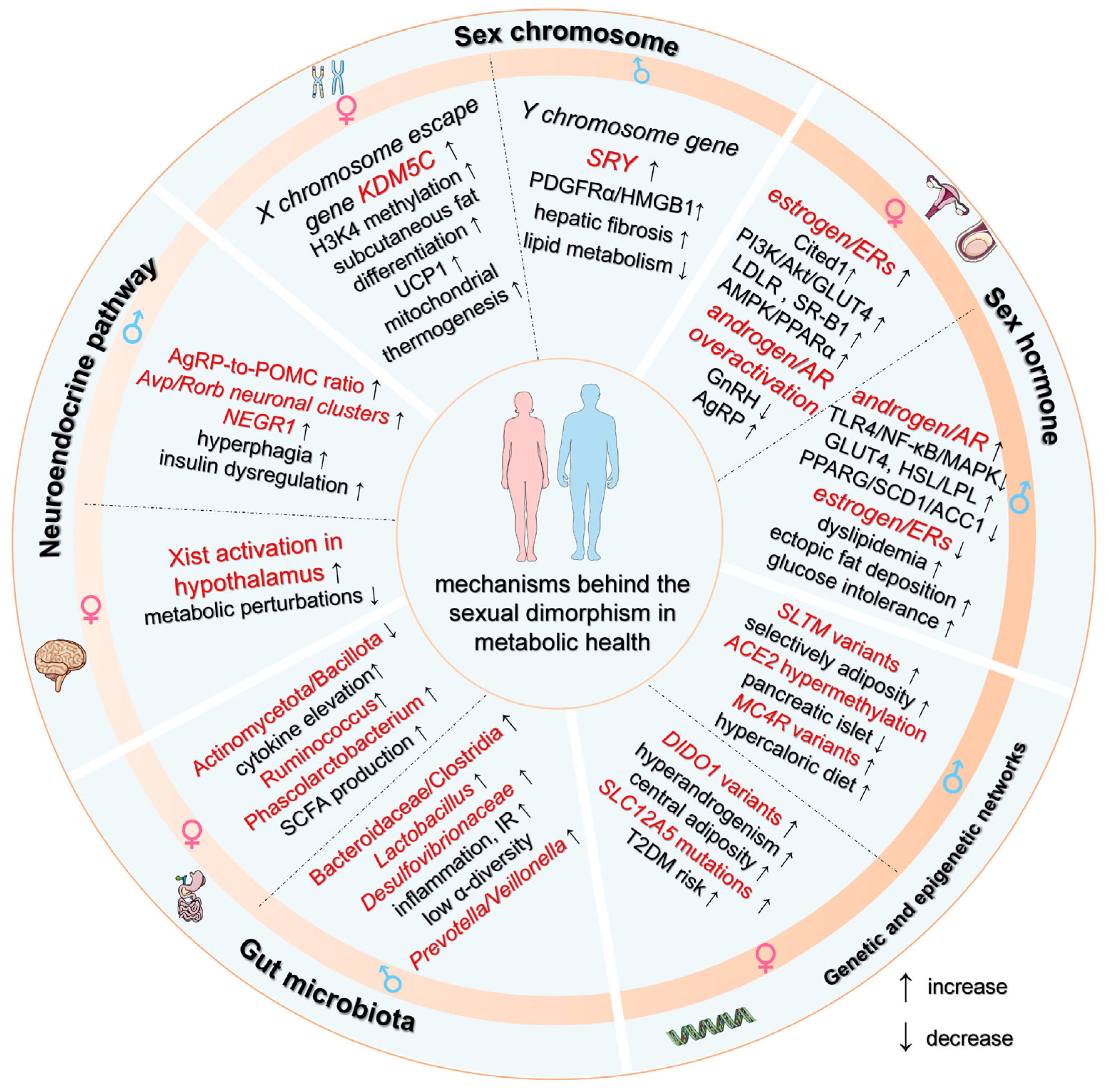
3. Mechanisms Behind the Sex Differences in the Development of Metabolic Diseases
3.1. Dosage Effects and Compensatory Mechanisms of Sex Chromosome Genes
3.2. Sex Hormone Receptor Signaling Dynamics
3.2.1. Androgens/Androgen Receptor
3.2.2. Estrogen and Estrogen Receptors
3.3. Genetic and Epigenetic Regulatory Networks
3.4. Gut Microbiota–Host Metabolic Crosstalk
3.5. Neuroendocrine Pathway Dimorphisms
4. Dietary and Pharmacotherapeutic Interventions Targeting Sex-Specific HFD-Induced Metabolic Diseases
5. Sex Differences in Exercise-Related Exercise Capacity and Metabolic Health
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Guidelines Approved by the Guidelines Review Committee. In Total Fat Intake for the Prevention of Unhealthy Weight Gain in Adults and Children: WHO Guideline; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- Cyr-Scully, A.; Howard, A.G.; Sanzone, E.; Meyer, K.A.; Du, S.; Zhang, B.; Wang, H.; Gordon-Larsen, P. Characterizing the urban diet: Development of an urbanized diet index. Nutr. J. 2022, 21, 55. [Google Scholar] [CrossRef]
- Shan, Z.; Rehm, C.D.; Rogers, G.; Ruan, M.; Wang, D.D.; Hu, F.B.; Mozaffarian, D.; Zhang, F.F.; Bhupathiraju, S.N. Trends in Dietary Carbohydrate, Protein, and Fat Intake and Diet Quality Among US Adults, 1999–2016. JAMA 2019, 322, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Retterstøl, K.; Rosqvist, F. Fat and fatty acids—A scoping review for Nordic Nutrition Recommendations 2023. Food Nutr. Res. 2024, 68, 9980. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Dalamaga, M.; Liatis, S. Update on the Obesity Epidemic: After the Sudden Rise, Is the Upward Trajectory Beginning to Flatten? Curr. Obes. Rep. 2023, 12, 514–527. [Google Scholar] [CrossRef]
- Talukdar, J.R.; Steen, J.P.; Goldenberg, J.Z.; Zhang, Q.; Vernooij, R.W.M.; Ge, L.; Zeraatkar, D.; Bała, M.M.; Ball, G.D.C.; Thabane, L.; et al. Saturated fat, the estimated absolute risk and certainty of risk for mortality and major cancer and cardiometabolic outcomes: An overview of systematic reviews. Syst. Rev. 2023, 12, 179. [Google Scholar] [CrossRef] [PubMed]
- Maki, K.C.; Dicklin, M.R.; Kirkpatrick, C.F. Saturated fats and cardiovascular health: Current evidence and controversies. J. Clin. Lipidol. 2021, 15, 765–772. [Google Scholar] [CrossRef]
- Mei, J.; Qian, M.; Hou, Y.; Liang, M.; Chen, Y.; Wang, C.; Zhang, J. Association of saturated fatty acids with cancer risk: A systematic review and meta-analysis. Lipids Health Dis. 2024, 23, 32. [Google Scholar] [CrossRef]
- Tan, B.L.; Norhaizan, M.E. Effect of High-Fat Diets on Oxidative Stress, Cellular Inflammatory Response and Cognitive Function. Nutrients 2019, 11, 2579. [Google Scholar] [CrossRef]
- Hases, L.; Stepanauskaite, L.; Birgersson, M.; Brusselaers, N.; Schuppe-Koistinen, I.; Archer, A.; Engstrand, L.; Williams, C. High-fat diet and estrogen modulate the gut microbiota in a sex-dependent manner in mice. Commun. Biol. 2023, 6, 20. [Google Scholar] [CrossRef]
- Lefebvre, C.; Tiffay, A.; Breemeersch, C.E.; Dreux, V.; Bôle-Feysot, C.; Guérin, C.; Breton, J.; Maximin, E.; Monnoye, M.; Déchelotte, P.; et al. Sex-dependent effects of a high fat diet on metabolic disorders, intestinal barrier function and gut microbiota in mouse. Sci. Rep. 2024, 14, 19835. [Google Scholar] [CrossRef]
- Tran, V.; Brettle, H.; Diep, H.; Dinh, Q.N.; O’Keeffe, M.; Fanson, K.V.; Sobey, C.G.; Lim, K.; Drummond, G.R.; Vinh, A.; et al. Sex-specific effects of a high fat diet on aortic inflammation and dysfunction. Sci. Rep. 2023, 13, 21644. [Google Scholar] [CrossRef] [PubMed]
- Medrikova, D.; Jilkova, Z.M.; Bardova, K.; Janovska, P.; Rossmeisl, M.; Kopecky, J. Sex differences during the course of diet-induced obesity in mice: Adipose tissue expandability and glycemic control. Int. J. Obes. 2012, 36, 262–272. [Google Scholar] [CrossRef] [PubMed]
- El Khoudary, S.R.; Aggarwal, B.; Beckie, T.M.; Hodis, H.N.; Johnson, A.E.; Langer, R.D.; Limacher, M.C.; Manson, J.E.; Stefanick, M.L.; Allison, M.A. Menopause Transition and Cardiovascular Disease Risk: Implications for Timing of Early Prevention: A Scientific Statement From the American Heart Association. Circulation 2020, 142, e506–e532. [Google Scholar] [CrossRef]
- Oya, J.; Nakagami, T.; Yamamoto, Y.; Fukushima, S.; Takeda, M.; Endo, Y.; Uchigata, Y. Effects of age on insulin resistance and secretion in subjects without diabetes. Intern. Med. 2014, 53, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Mauvais-Jarvis, F. Gender differences in glucose homeostasis and diabetes. Physiol. Behav. 2018, 187, 20–23. [Google Scholar] [CrossRef]
- Kautzky-Willer, A.; Harreiter, J.; Pacini, G. Sex and Gender Differences in Risk, Pathophysiology and Complications of Type 2 Diabetes Mellitus. Endocr. Rev. 2016, 37, 278–316. [Google Scholar] [CrossRef]
- Clayton, J.A. Studying both sexes: A guiding principle for biomedicine. FASEB J. 2016, 30, 519–524. [Google Scholar] [CrossRef]
- U.S. Government Accountability Office. Drug Safety: Most Drugs Withdrawn in Recent Years Had Greater Health R isks for Women. 19 January 2001. Available online: https://www.gao.gov/products/gao-01-286r (accessed on 9 February 2001).
- Tramunt, B.; Smati, S.; Grandgeorge, N.; Lenfant, F.; Arnal, J.F.; Montagner, A.; Gourdy, P. Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia 2020, 63, 453–461. [Google Scholar] [CrossRef]
- Goossens, G.H.; Jocken, J.W.E.; Blaak, E.E. Sexual dimorphism in cardiometabolic health: The role of adipose tissue, muscle and liver. Nat. Rev. Endocrinol. 2021, 17, 47–66. [Google Scholar] [CrossRef]
- Jiang, X.; Zhao, Y.; Yang, Q.; Wang, W.; Lin, T.; Qiu, Y. Gender differences in associations between obesity and hypertension, diabetes, dyslipidemia: Evidence from electronic health records of 3.5 million Chinese senior population. BMC Public Health 2025, 25, 405. [Google Scholar] [CrossRef]
- Nauli, A.M.; Matin, S. Why Do Men Accumulate Abdominal Visceral Fat? Front. Physiol. 2019, 10, 1486. [Google Scholar] [CrossRef]
- Kolb, H. Obese visceral fat tissue inflammation: From protective to detrimental? BMC Med. 2022, 20, 494. [Google Scholar] [CrossRef] [PubMed]
- McArdle, M.A.; Finucane, O.M.; Connaughton, R.M.; McMorrow, A.M.; Roche, H.M. Mechanisms of obesity-induced inflammation and insulin resistance: Insights into the emerging role of nutritional strategies. Front. Endocrinol. 2013, 4, 52. [Google Scholar] [CrossRef] [PubMed]
- Bovolini, A.; Garcia, J.; Andrade, M.A.; Duarte, J.A. Metabolic Syndrome Pathophysiology and Predisposing Factors. Int. J. Sports Med. 2021, 42, 199–214. [Google Scholar] [CrossRef]
- Karastergiou, K.; Smith, S.R.; Greenberg, A.S.; Fried, S.K. Sex differences in human adipose tissues—The biology of pear shape. Biol. Sex. Differ. 2012, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.; Duesman, S.J.; Patel, S.; Huynh, P.; Toh, P.; Shroff, S.; Das, A.; Chowhan, D.; Keller, B.; Alvarez, J.; et al. Sex-specific role of high-fat diet and stress on behavior, energy metabolism, and the ventromedial hypothalamus. Biol. Sex. Differ. 2024, 15, 55. [Google Scholar] [CrossRef]
- Bracht, J.R.; Vieira-Potter, V.J.; De Souza Santos, R.; Öz, O.K.; Palmer, B.F.; Clegg, D.J. The role of estrogens in the adipose tissue milieu. Ann. N. Y. Acad. Sci. 2020, 1461, 127–143. [Google Scholar] [CrossRef]
- Adam, S.; Maas, S.L.; Huchzermeier, R.; Rakateli, L.; Abschlag, K.; Hohl, M.; Liao, L.; Bartneck, M.; Teunissen, M.; Wouters, K.; et al. The calcium-sensing-receptor (CaSR) in adipocytes contributes to sex-differences in the susceptibility to high fat diet induced obesity and atherosclerosis. EBioMedicine 2024, 107, 105293. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef]
- James, D.E.; Stöckli, J.; Birnbaum, M.J. The aetiology and molecular landscape of insulin resistance. Nat. Rev. Mol. Cell Biol. 2021, 22, 751–771. [Google Scholar] [CrossRef]
- Du, W.; Jiang, S.; Yin, S.; Wang, R.; Zhang, C.; Yin, B.C.; Li, J.; Li, L.; Qi, N.; Zhou, Y.; et al. The microbiota-dependent tryptophan metabolite alleviates high-fat diet-induced insulin resistance through the hepatic AhR/TSC2/mTORC1 axis. Proc. Natl. Acad. Sci. USA 2024, 121, e2400385121. [Google Scholar] [CrossRef] [PubMed]
- Elkanawati, R.Y.; Sumiwi, S.A.; Levita, J. Impact of Lipids on Insulin Resistance: Insights from Human and Animal Studies. Drug Des. Dev. Ther. 2024, 18, 3337–3360. [Google Scholar] [CrossRef] [PubMed]
- Zeng, N.; Wu, F.; Lu, J.; Li, X.; Lin, S.; Zhou, L.; Wang, Z.; Wu, G.; Huang, Q.; Zheng, D.; et al. High-fat diet impairs gut barrier through intestinal microbiota-derived reactive oxygen species. Sci. China Life Sci. 2024, 67, 879–891. [Google Scholar] [CrossRef]
- Li, M.; Chi, X.; Wang, Y.; Setrerrahmane, S.; Xie, W.; Xu, H. Trends in insulin resistance: Insights into mechanisms and therapeutic strategy. Signal Transduct. Target. Ther. 2022, 7, 216. [Google Scholar] [CrossRef]
- Sakamoto, K.; Butera, M.A.; Zhou, C.; Maurizi, G.; Chen, B.; Ling, L.; Shawkat, A.; Patlolla, L.; Thakker, K.; Calle, V.; et al. Overnutrition causes insulin resistance and metabolic disorder through increased sympathetic nervous system activity. Cell Metab. 2025, 37, 121–137.E6. [Google Scholar] [CrossRef] [PubMed]
- Abdou, M.; Hafez, M.H.; Anwar, G.M.; Fahmy, W.A.; Abd Alfattah, N.M.; Salem, R.I.; Arafa, N. Effect of high protein and fat diet on postprandial blood glucose levels in children and adolescents with type 1 diabetes in Cairo, Egypt. Diabetes Metab. Syndr. 2021, 15, 7–12. [Google Scholar] [CrossRef]
- Smith, J.A.; Soares, R.N.; McMillan, N.J.; Jurrissen, T.J.; Martinez-Lemus, L.A.; Padilla, J.; Manrique-Acevedo, C. Young Women Are Protected Against Vascular Insulin Resistance Induced by Adoption of an Obesogenic Lifestyle. Endocrinology 2022, 163, bqac137. [Google Scholar] [CrossRef]
- Varlamov, O.; Bethea, C.L.; Roberts, C.T., Jr. Sex-specific differences in lipid and glucose metabolism. Front. Endocrinol. 2014, 5, 241. [Google Scholar] [CrossRef]
- Churuangsuk, C.; Lean, M.E.J.; Combet, E. Lower carbohydrate and higher fat intakes are associated with higher hemoglobin A1c: Findings from the UK National Diet and Nutrition Survey 2008–2016. Eur. J. Nutr. 2020, 59, 2771–2782. [Google Scholar] [CrossRef]
- Palmer, B.F.; Clegg, D.J. The sexual dimorphism of obesity. Mol. Cell Endocrinol. 2015, 402, 113–119. [Google Scholar] [CrossRef]
- Speksnijder, E.M.; Ten Noever de Brauw, G.V.; Malekzadeh, A.; Bisschop, P.H.; Stenvers, D.J.; Siegelaar, S.E. Effect of Postmenopausal Hormone Therapy on Glucose Regulation in Women With Type 1 or Type 2 Diabetes: A Systematic Review and Meta-analysis. Diabetes Care 2023, 46, 1866–1875. [Google Scholar] [CrossRef] [PubMed]
- Yazdkhasti, M.; Jafarabady, K.; Shafiee, A.; Omran, S.P.; Mahmoodi, Z.; Esmaeilzadeh, S.; Babaheidari, T.B.; Kabir, K.; Peisepar, M.; Bakhtiyari, M. The association between age of menopause and type 2 diabetes: A systematic review and meta-analysis. Nutr. Metab. 2024, 21, 87. [Google Scholar] [CrossRef] [PubMed]
- Lizcano, F.; Guzmán, G. Estrogen Deficiency and the Origin of Obesity during Menopause. Biomed. Res. Int. 2014, 2014, 757461. [Google Scholar] [CrossRef] [PubMed]
- Szukiewicz, D. Molecular Mechanisms for the Vicious Cycle between Insulin Resistance and the Inflammatory Response in Obesity. Int. J. Mol. Sci. 2023, 24, 9818. [Google Scholar] [CrossRef]
- Sjøberg, K.A.; Frøsig, C.; Kjøbsted, R.; Sylow, L.; Kleinert, M.; Betik, A.C.; Shaw, C.S.; Kiens, B.; Wojtaszewski, J.F.P.; Rattigan, S.; et al. Exercise Increases Human Skeletal Muscle Insulin Sensitivity via Coordinated Increases in Microvascular Perfusion and Molecular Signaling. Diabetes 2017, 66, 1501–1510. [Google Scholar] [CrossRef]
- Gupta, D.; Jetton, T.L.; LaRock, K.; Monga, N.; Satish, B.; Lausier, J.; Peshavaria, M.; Leahy, J.L. Temporal characterization of β cell-adaptive and -maladaptive mechanisms during chronic high-fat feeding in C57BL/6NTac mice. J. Biol. Chem. 2017, 292, 12449–12459. [Google Scholar] [CrossRef]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef]
- Eguchi, N.; Vaziri, N.D.; Dafoe, D.C.; Ichii, H. The Role of Oxidative Stress in Pancreatic β Cell Dysfunction in Diabetes. Int. J. Mol. Sci. 2021, 22, 1509. [Google Scholar] [CrossRef]
- Zhou, Z.; Ribas, V.; Rajbhandari, P.; Drew, B.G.; Moore, T.M.; Fluitt, A.H.; Reddish, B.R.; Whitney, K.A.; Georgia, S.; Vergnes, L.; et al. Estrogen receptor α protects pancreatic β-cells from apoptosis by preserving mitochondrial function and suppressing endoplasmic reticulum stress. J. Biol. Chem. 2018, 293, 4735–4751. [Google Scholar] [CrossRef]
- De Paoli, M.; Zakharia, A.; Werstuck, G.H. The Role of Estrogen in Insulin Resistance: A Review of Clinical and Preclinical Data. Am. J. Pathol. 2021, 191, 1490–1498. [Google Scholar] [CrossRef]
- Ribas, V.; Drew, B.G.; Zhou, Z.; Phun, J.; Kalajian, N.Y.; Soleymani, T.; Daraei, P.; Widjaja, K.; Wanagat, J.; de Aguiar Vallim, T.Q.; et al. Skeletal muscle action of estrogen receptor α is critical for the maintenance of mitochondrial function and metabolic homeostasis in females. Sci. Transl. Med. 2016, 8, 334ra54. [Google Scholar] [CrossRef] [PubMed]
- Moholdt, T.; Parr, E.B.; Devlin, B.L.; Giskeødegård, G.F.; Hawley, J.A. Effect of high-fat diet and morning or evening exercise on lipoprotein subfraction profiles: Secondary analysis of a randomised trial. Sci. Rep. 2023, 13, 4008. [Google Scholar] [CrossRef] [PubMed]
- Shinohata, R.; Shibakura, M.; Arao, Y.; Watanabe, S.; Hirohata, S.; Usui, S. A high-fat/high-cholesterol diet, but not high-cholesterol alone, increases free cholesterol and apoE-rich HDL serum levels in rats and upregulates hepatic ABCA1 expression. Biochimie 2022, 197, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Zhou, Q.; Dai, W.; Peng, H.; Zhou, S.; Tian, H.; Shen, L.; Han, H. Gender differences in metabolic syndrome and its components in southern china using a healthy lifestyle index: A cross-sectional study. BMC Public Health 2023, 23, 686. [Google Scholar] [CrossRef]
- Ambikairajah, A.; Walsh, E.; Cherbuin, N. Lipid profile differences during menopause: A review with meta-analysis. Menopause 2019, 26, 1327–1333. [Google Scholar] [CrossRef]
- McCoin, C.S.; Von Schulze, A.; Allen, J.; Fuller, K.N.Z.; Xia, Q.; Koestler, D.C.; Houchen, C.J.; Maurer, A.; Dorn, G.W., 2nd; Shankar, K.; et al. Sex modulates hepatic mitochondrial adaptations to high-fat diet and physical activity. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E298–E311. [Google Scholar] [CrossRef]
- Arcones, A.C.; Cruces-Sande, M.; Ramos, P.; Mayor, F., Jr.; Murga, C. Sex Differences in High Fat Diet-Induced Metabolic Alterations Correlate with Changes in the Modulation of GRK2 Levels. Cells 2019, 8, 1464. [Google Scholar] [CrossRef]
- Ryczkowska, K.; Adach, W.; Janikowski, K.; Banach, M.; Bielecka-Dabrowa, A. Menopause and women’s cardiovascular health: Is it really an obvious relationship? Arch. Med. Sci. 2023, 19, 458–466. [Google Scholar] [CrossRef]
- Ko, S.H.; Jung, Y. Energy Metabolism Changes and Dysregulated Lipid Metabolism in Postmenopausal Women. Nutrients 2021, 13, 4556. [Google Scholar] [CrossRef]
- Spitler, K.M.; Davies, B.S.J. Aging and plasma triglyceride metabolism. J. Lipid Res. 2020, 61, 1161–1167. [Google Scholar] [CrossRef]
- Nie, G.; Yang, X.; Wang, Y.; Liang, W.; Li, X.; Luo, Q.; Yang, H.; Liu, J.; Wang, J.; Guo, Q.; et al. The Effects of Menopause Hormone Therapy on Lipid Profile in Postmenopausal Women: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2022, 13, 850815. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.H.; Kim, H.S. Menopause-Associated Lipid Metabolic Disorders and Foods Beneficial for Postmenopausal Women. Nutrients 2020, 12, 202. [Google Scholar] [CrossRef] [PubMed]
- Szudzik, M.; Hutsch, T.; Chabowski, D.; Zajdel, M.; Ufnal, M. Normal caloric intake with high-fat diet induces metabolic dysfunction-associated steatotic liver disease and dyslipidemia without obesity in rats. Sci. Rep. 2024, 14, 22796. [Google Scholar] [CrossRef]
- Gu, Y.; Han, F.; Xue, M.; Wang, M.; Huang, Y. The benefits and risks of menopause hormone therapy for the cardiovascular system in postmenopausal women: A systematic review and meta-analysis. BMC Womens Health 2024, 24, 60. [Google Scholar] [CrossRef] [PubMed]
- Hodis, H.N.; Mack, W.J. Menopausal Hormone Replacement Therapy and Reduction of All-Cause Mortality and Cardiovascular Disease: It Is About Time and Timing. Cancer J. 2022, 28, 208–223. [Google Scholar] [CrossRef]
- Jiang, H.; Zhou, Y.; Nabavi, S.M.; Sahebkar, A.; Little, P.J.; Xu, S.; Weng, J.; Ge, J. Mechanisms of Oxidized LDL-Mediated Endothelial Dysfunction and Its Consequences for the Development of Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 925923. [Google Scholar] [CrossRef]
- Gui, Y.; Zheng, H.; Cao, R.Y. Foam Cells in Atherosclerosis: Novel Insights Into Its Origins, Consequences, and Molecular Mechanisms. Front. Cardiovasc. Med. 2022, 9, 845942. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Yerly, A.; van der Vorst, E.P.C.; Baumgartner, I.; Bernhard, S.M.; Schindewolf, M.; Döring, Y. Sex-specific and hormone-related differences in vascular remodelling in atherosclerosis. Eur. J. Clin. Investig. 2023, 53, e13885. [Google Scholar] [CrossRef]
- Ren, Z.; Guo, J.; Xiao, X.; Huang, J.; Li, M.; Cai, R.; Zeng, H. The Effect of Sex Differences on Endothelial Function and Circulating Endothelial Progenitor Cells in Hypertriglyceridemia. Cardiol. Res. Pract. 2020, 2020, 2132918. [Google Scholar] [CrossRef]
- Fairweather, D. Sex differences in inflammation during atherosclerosis. Clin. Med. Insights Cardiol. 2014, 8 (Suppl. S3), 49–59. [Google Scholar] [CrossRef] [PubMed]
- Vakhtangadze, T.; Singh Tak, R.; Singh, U.; Baig, M.S.; Bezsonov, E. Gender Differences in Atherosclerotic Vascular Disease: From Lipids to Clinical Outcomes. Front. Cardiovasc. Med. 2021, 8, 707889. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, Y.; Yao, C.; Zhao, X.; Xiong, W.; Rang, W. Trend and Prediction of Changes in the Disease Burden of Diet-related Ischemic Heart Disease in China, 1990–2021. Chin. Gen. Pract. 2025, 28, 305–312. (In Chinese) [Google Scholar]
- Regitz-Zagrosek, V.; Gebhard, C. Gender medicine: Effects of sex and gender on cardiovascular disease manifestation and outcomes. Nat. Rev. Cardiol. 2023, 20, 236–247. [Google Scholar] [CrossRef]
- Rexrode, K.M.; Madsen, T.E.; Yu, A.Y.X.; Carcel, C.; Lichtman, J.H.; Miller, E.C. The Impact of Sex and Gender on Stroke. Circ. Res. 2022, 130, 512–528. [Google Scholar] [CrossRef]
- Jia, Z.; Wang, Z.; Pan, H.; Zhang, J.; Wang, Q.; Zhou, C.; Liu, J. Crosstalk between fat tissue and muscle, brain, liver, and heart in obesity: Cellular and molecular perspectives. Eur. J. Med. Res. 2024, 29, 637. [Google Scholar] [CrossRef]
- Funcke, J.B.; Scherer, P.E. Beyond adiponectin and leptin: Adipose tissue-derived mediators of inter-organ communication. J. Lipid Res. 2019, 60, 1648–1684. [Google Scholar]
- Moghbeli, M.; Khedmatgozar, H.; Yadegari, M.; Avan, A.; Ferns, G.A.; Ghayour Mobarhan, M. Cytokines and the immune response in obesity-related disorders. Adv. Clin. Chem. 2021, 101, 135–168. [Google Scholar] [PubMed]
- Strizova, Z.; Benesova, I.; Bartolini, R.; Novysedlak, R.; Cecrdlova, E.; Foley, L.K.; Striz, I. M1/M2 macrophages and their overlaps—Myth or reality? Clin. Sci. 2023, 137, 1067–1093. [Google Scholar] [CrossRef]
- Li, Y.; Dong, X.; He, W.; Quan, H.; Chen, K.; Cen, C.; Wei, W. Ube2L6 Promotes M1 Macrophage Polarization in High-Fat Diet-Fed Obese Mice via ISGylation of STAT1 to Trigger STAT1 Activation. Obes. Facts 2024, 17, 24–36. [Google Scholar] [CrossRef]
- Duan, Z.; Luo, Y. Targeting macrophages in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 127. [Google Scholar] [CrossRef] [PubMed]
- Alfaddagh, A.; Martin, S.S.; Leucker, T.M.; Michos, E.D.; Blaha, M.J.; Lowenstein, C.J.; Jones, S.R.; Toth, P.P. Inflammation and cardiovascular disease: From mechanisms to therapeutics. Am. J. Prev. Cardiol. 2020, 4, 100130. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, X.; Zhang, H.G. Editorial: Cardiac Hypertrophy: From Compensation to Decompensation and Pharmacological Interventions. Front. Pharmacol. 2021, 12, 665936. [Google Scholar] [CrossRef]
- Braga Tibaes, J.R.; Azarcoya-Barrera, J.; Wollin, B.; Veida-Silva, H.; Makarowski, A.; Vine, D.; Tsai, S.; Jacobs, R.; Richard, C. Sex Differences Distinctly Impact High-Fat Diet-Induced Immune Dysfunction in Wistar Rats. J. Nutr. 2022, 152, 1347–1357. [Google Scholar] [CrossRef]
- Peng, Y.; Zhou, M.; Yang, H.; Qu, R.; Qiu, Y.; Hao, J.; Bi, H.; Guo, D. Regulatory Mechanism of M1/M2 Macrophage Polarization in the Development of Autoimmune Diseases. Mediators Inflamm. 2023, 2023, 8821610. [Google Scholar] [CrossRef] [PubMed]
- Shu, Z.; Zhang, G.; Zhu, X.; Xiong, W. Estrogen receptor α mediated M1/M2 macrophages polarization plays a critical role in NASH of female mice. Biochem. Biophys. Res. Commun. 2022, 596, 63–70. [Google Scholar] [CrossRef]
- Liu, H.; Liu, K.; Bodenner, D.L. Estrogen receptor inhibits interleukin-6 gene expression by disruption of nuclear factor kappaB transactivation. Cytokine 2005, 31, 251–257. [Google Scholar] [CrossRef]
- Kurmann, L.; Azzarito, G.; Leeners, B.; Rosselli, M.; Dubey, R.K. 17β-Estradiol Abrogates TNF-α-Induced Human Brain Vascular Pericyte Migration by Downregulating miR-638 via ER-β. Int. J. Mol. Sci. 2024, 25, 11416. [Google Scholar] [CrossRef]
- Bandala, C.; Cárdenas-Rodríguez, N.; Reyes-Long, S.; Cortés-Algara, A.; Contreras-García, I.J.; Cruz-Hernández, T.R.; Alfaro-Rodriguez, A.; Cortes-Altamirano, J.L.; Perez-Santos, M.; Anaya-Ruiz, M.; et al. Estrogens as a Possible Therapeutic Strategy for the Management of Neuroinflammation and Neuroprotection in COVID-19. Curr. Neuropharmacol. 2023, 21, 2110–2125. [Google Scholar] [CrossRef]
- Ketchem, J.M.; Bowman, E.J.; Isales, C.M. Male sex hormones, aging, and inflammation. Biogerontology 2023, 24, 1–25. [Google Scholar] [CrossRef]
- Koning, T.; Calaf, G.M. Association of Inflammation and Immune Cell Infiltration with Estrogen Receptor Alpha in an Estrogen and Ionizing Radiation-Induced Breast Cancer Model. Int. J. Mol. Sci. 2024, 25, 8604. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.T.; Liao, Q.Q.; Tian, H.Y.; Yu, D.J.; Xie, T.; Sun, X.L.; Zhou, X.M.; Han, Y.X.; Zhao, Y.J.; El-Kassas, M.; et al. Estrogen: The forgotten player in metaflammation. Front. Pharmacol. 2024, 15, 1478819. [Google Scholar] [CrossRef] [PubMed]
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Steppan, J.; Jandu, S.; Wang, H.; Kang, S.; Savage, W.; Narayanan, R.; Nandakumar, K.; Santhanam, L. Commonly used mouse strains have distinct vascular properties. Hypertens. Res. 2020, 43, 1175–1181. [Google Scholar] [CrossRef]
- Iorga, A.; Cunningham, C.M.; Moazeni, S.; Ruffenach, G.; Umar, S.; Eghbali, M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol. Sex. Differ. 2017, 8, 33. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Marseglia, L.; Manti, S.; D’Angelo, G.; Nicotera, A.; Parisi, E.; Di Rosa, G.; Gitto, E.; Arrigo, T. Oxidative stress in obesity: A critical component in human diseases. Int. J. Mol. Sci. 2014, 16, 378–400. [Google Scholar] [CrossRef]
- Bej, E.; Cesare, P.; Volpe, A.R.; d’Angelo, M.; Castelli, V. Oxidative Stress and Neurodegeneration: Insights and Therapeutic Strategies for Parkinson’s Disease. Neurol. Int. 2024, 16, 502–517. [Google Scholar] [CrossRef]
- Jin, S.; Kang, P.M. A Systematic Review on Advances in Management of Oxidative Stress-Associated Cardiovascular Diseases. Antioxidants 2024, 13, 923. [Google Scholar] [CrossRef]
- Chaudhary, P.; Janmeda, P.; Docea, A.O.; Yeskaliyeva, B.; Abdull Razis, A.F.; Modu, B.; Calina, D.; Sharifi-Rad, J. Oxidative stress, free radicals and antioxidants: Potential crosstalk in the pathophysiology of human diseases. Front. Chem. 2023, 11, 1158198. [Google Scholar] [CrossRef]
- Tiberi, J.; Cesarini, V.; Stefanelli, R.; Canterini, S.; Fiorenza, M.T.; La Rosa, P. Sex differences in antioxidant defence and the regulation of redox homeostasis in physiology and pathology. Mech. Ageing Dev. 2023, 211, 111802. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Castro, J.; Pulido-Moran, M.; Moreno-Fernandez, J.; Kajarabille, N.; de Paco, C.; Garrido-Sanchez, M.; Prados, S.; Ochoa, J.J. Gender specific differences in oxidative stress and inflammatory signaling in healthy term neonates and their mothers. Pediatr. Res. 2016, 80, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.; Liu, Y.; Zhou, S.; Zhou, E.; Wang, Y. Protective Effects of Estrogen on Cardiovascular Disease Mediated by Oxidative Stress. Oxid. Med. Cell. Longev. 2021, 2021, 5523516. [Google Scholar] [CrossRef] [PubMed]
- Borrás, C.; Ferrando, M.; Inglés, M.; Gambini, J.; Lopez-Grueso, R.; Edo, R.; Mas-Bargues, C.; Pellicer, A.; Viña, J. Estrogen Replacement Therapy Induces Antioxidant and Longevity-Related Genes in Women after Medically Induced Menopause. Oxid. Med. Cell. Longev. 2021, 2021, 8101615. [Google Scholar] [CrossRef]
- Sahoo, D.K.; Samanta, L.; Kesari, K.K.; Mukherjee, S. Editorial: Hormonal imbalance-associated oxidative stress and protective benefits of nutritional antioxidants. Front. Endocrinol. 2024, 15, 1368580. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Maithili Karpaga Selvi, N.; Sridhar, M.G.; Swaminathan, R.P.; Sripradha, R. Curcumin Attenuates Oxidative Stress and Activation of Redox-Sensitive Kinases in High Fructose- and High-Fat-Fed Male Wistar Rats. Sci. Pharm. 2015, 83, 159–175. [Google Scholar] [CrossRef]
- Hong, Y.; Boiti, A.; Vallone, D.; Foulkes, N.S. Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants 2024, 13, 312. [Google Scholar] [CrossRef]
- Darbandi, M.; Darbandi, S.; Agarwal, A.; Sengupta, P.; Durairajanayagam, D.; Henkel, R.; Sadeghi, M.R. Reactive oxygen species and male reproductive hormones. Reprod. Biol. Endocrinol. 2018, 16, 87. [Google Scholar] [CrossRef]
- Tostes, R.C.; Carneiro, F.S.; Carvalho, M.H.; Reckelhoff, J.F. Reactive oxygen species: Players in the cardiovascular effects of testosterone. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R1–R14. [Google Scholar] [CrossRef]
- Lavorando, E.; Owens, M.C.; Liu, K.F. Comparing the roles of sex chromosome-encoded protein homologs in gene regulation. Genes. Dev. 2024, 38, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Tukiainen, T.; Villani, A.C.; Yen, A.; Rivas, M.A.; Marshall, J.L.; Satija, R.; Aguirre, M.; Gauthier, L.; Fleharty, M.; Kirby, A.; et al. Landscape of X chromosome inactivation across human tissues. Nature 2017, 550, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Reue, K. Sex differences in obesity: X chromosome dosage as a risk factor for increased food intake, adiposity and co-morbidities. Physiol. Behav. 2017, 176, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; McClusky, R.; Itoh, Y.; Reue, K.; Arnold, A.P. X and Y chromosome complement influence adiposity and metabolism in mice. Endocrinology 2013, 154, 1092–1104. [Google Scholar] [CrossRef]
- White, M.; Zacharin, M.R.; Fawcett, S.; McGillivray, G. Klinefelter Syndrome: What should we tell prospective parents? Prenat. Diagn. 2023, 43, 240–249. [Google Scholar] [CrossRef]
- Granato, S.; Barbaro, G.; Di Giorgio, M.R.; Rossi, F.M.; Marzano, C.; Impronta, F.; Spaziani, M.; Anzuini, A.; Lenzi, A.; Radicioni, A.F. Epicardial fat: The role of testosterone and lipid metabolism in a cohort of patients with Klinefelter syndrome. Metabolism 2019, 95, 21–26. [Google Scholar] [CrossRef]
- Demirci, I.; Haymana, C.; Candemir, B.; Yuksel, B.; Eser, M.; Meric, C.; Akin, S.; Gulcelik, N.E.; Sonmez, A. Triglyceride-glucose index levels in patients with Klinefelter syndrome and its relationship with endothelial dysfunction and insulin resistance: A cross-sectional observational study. Arch. Endocrinol. Metab. 2023, 67, 378–384. [Google Scholar] [CrossRef]
- Krzyścin, M.; Sowińska-Przepiera, E.; Gruca-Stryjak, K.; Soszka-Przepiera, E.; Syrenicz, I.; Przepiera, A.; Bumbulienė, Ž.; Syrenicz, A. Are Young People with Turner Syndrome Who Have Undergone Treatment with Growth and Sex Hormones at Higher Risk of Metabolic Syndrome and Its Complications? Biomedicines 2024, 12, 1034. [Google Scholar] [CrossRef]
- Link, J.C.; Wiese, C.B.; Chen, X.; Avetisyan, R.; Ronquillo, E.; Ma, F.; Guo, X.; Yao, J.; Allison, M.; Chen, Y.I.; et al. X chromosome dosage of histone demethylase KDM5C determines sex differences in adiposity. J. Clin. Investig. 2020, 130, 5688–5702. [Google Scholar] [CrossRef]
- Qian, S.; Wang, Y.; Ma, H.; Zhang, L. Expansion and Functional Divergence of Jumonji C-Containing Histone Demethylases: Significance of Duplications in Ancestral Angiosperms and Vertebrates. Plant Physiol. 2015, 168, 1321–1337. [Google Scholar] [CrossRef]
- Godfrey, A.K.; Naqvi, S.; Chmátal, L.; Chick, J.M.; Mitchell, R.N.; Gygi, S.P.; Skaletsky, H.; Page, D.C. Quantitative analysis of Y-Chromosome gene expression across 36 human tissues. Genome Res. 2020, 30, 860–873. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D.; Marin, R.; Toledo-Flores, D.; Froidevaux, L.; Liechti, A.; Waters, P.D.; Grützner, F.; Kaessmann, H. Origins and functional evolution of Y chromosomes across mammals. Nature 2014, 508, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, S.B.; Jensen, C.H.; Schneider, M.; Thomassen, M.; Kruse, T.A.; Laborda, J.; Sheikh, S.P.; Andersen, D.C. Membrane-tethered delta-like 1 homolog (DLK1) restricts adipose tissue size by inhibiting preadipocyte proliferation. Diabetes 2012, 61, 2814–2822. [Google Scholar] [CrossRef] [PubMed]
- Vergnes, L.; Wiese, C.B.; Zore, T.; Riestenberg, C.; Avetisyan, R.; Reue, K. Gene Regulation and Mitochondrial Activity During White and Brown Adipogenesis Are Modulated by KDM5 Histone Demethylase. J. Endocr. Soc. 2024, 8, bvae029. [Google Scholar] [CrossRef]
- Long, J.; Su, Y.X.; Deng, H.C. Lipoapoptosis pathways in pancreatic β-cells and the anti-apoptosis mechanisms of adiponectin. Horm. Metab. Res. 2014, 46, 722–727. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, F.; Luo, Q.; Wu, X.; Liu, Z.; Chen, H.; Huang, Y. Inhibition of XIAP increases carboplatin sensitivity in ovarian cancer. OncoTargets Ther. 2018, 11, 8751–8759. [Google Scholar] [CrossRef]
- Jiang, C.; Yi, X.P.; Shen, H.; Li, Y.X. Targeting X-linked inhibitor of apoptosis protein inhibits pancreatic cancer cell growth through p-Akt depletion. World J. Gastroenterol. 2012, 18, 2956–2965. [Google Scholar] [CrossRef]
- Zhao, Y.; Stankovic, S.; Koprulu, M.; Wheeler, E.; Day, F.R.; Lango Allen, H.; Kerrison, N.D.; Pietzner, M.; Loh, P.R.; Wareham, N.J.; et al. GIGYF1 loss of function is associated with clonal mosaicism and adverse metabolic health. Nat. Commun. 2021, 12, 4178. [Google Scholar] [CrossRef]
- Wu, X.N.; Wang, M.Z.; Zhang, N.; Zhang, W.; Dong, J.; Ke, M.Y.; Xiang, J.X.; Ma, F.; Xue, F.; Hou, J.J.; et al. Sex-determining region Y gene promotes liver fibrosis and accounts for sexual dimorphism in its pathophysiology. J. Hepatol. 2024, 80, 928–940. [Google Scholar] [CrossRef]
- Wang, P.; Li, Q.; Wu, L.; Yu, X.; Zheng, Y.; Liu, J.; Yao, J.; Liu, Z.; Fan, S.; Li, Y. Association between the weight-adjusted-waist index and testosterone deficiency in adult males: A cross-sectional study. Sci. Rep. 2024, 14, 25574. [Google Scholar] [CrossRef]
- Santosa, S.; Bush, N.C.; Jensen, M.D. Acute Testosterone Deficiency Alters Adipose Tissue Fatty Acid Storage. J. Clin. Endocrinol. Metab. 2017, 102, 3056–3064. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.; Madden-Doyle, L.; Jayasena, C.; McIlroy, M.; Sherlock, M.; O’Reilly, M.W. Mechanisms in endocrinology: Hypogonadism and metabolic health in men-novel insights into pathophysiology. Eur. J. Endocrinol. 2024, 191, R1–R17. [Google Scholar] [CrossRef] [PubMed]
- Mauvais-Jarvis, F.; Bairey Merz, N.; Barnes, P.J.; Brinton, R.D.; Carrero, J.J.; DeMeo, D.L.; De Vries, G.J.; Epperson, C.N.; Govindan, R.; Klein, S.L.; et al. Sex and gender: Modifiers of health, disease, and medicine. Lancet 2020, 396, 565–582. [Google Scholar] [CrossRef]
- Naamneh Elzenaty, R.; du Toit, T.; Flück, C.E. Basics of androgen synthesis and action. Best. Pract. Res. Clin. Endocrinol. Metab. 2022, 36, 101665. [Google Scholar] [CrossRef]
- Apaiajai, N.; Chunchai, T.; Jaiwongkam, T.; Kerdphoo, S.; Chattipakorn, S.C.; Chattipakorn, N. Testosterone Deprivation Aggravates Left-Ventricular Dysfunction in Male Obese Insulin-Resistant Rats via Impairing Cardiac Mitochondrial Function and Dynamics Proteins. Gerontology 2018, 64, 333–343. [Google Scholar] [CrossRef]
- Leciejewska, N.; Pruszynska-Oszmalek, E.; Bien, J.; Nogowski, L.; Kolodziejski, P.A. Effect of ostarine (enobosarm/GTX024), a selective androgen receptor modulator, on adipocyte metabolism in Wistar rats. J. Physiol. Pharmacol. 2019, 70, 525–533. [Google Scholar]
- Raza, M.T.; Sharif, S.; Khan, Z.A.; Naz, S.; Mushtaq, S.; Umer, A. Frequency of Hypogonadism in Type 2 Diabetes Mellitus Patients with and without Coronary Artery Disease. Cureus 2019, 11, e6500. [Google Scholar] [CrossRef]
- Ruth, K.S.; Day, F.R.; Tyrrell, J.; Thompson, D.J.; Wood, A.R.; Mahajan, A.; Beaumont, R.N.; Wittemans, L.; Martin, S.; Busch, A.S.; et al. Using human genetics to understand the disease impacts of testosterone in men and women. Nat. Med. 2020, 26, 252–258. [Google Scholar] [CrossRef]
- Dhindsa, S.; Ghanim, H.; Batra, M.; Kuhadiya, N.D.; Abuaysheh, S.; Sandhu, S.; Green, K.; Makdissi, A.; Hejna, J.; Chaudhuri, A.; et al. Insulin Resistance and Inflammation in Hypogonadotropic Hypogonadism and Their Reduction After Testosterone Replacement in Men with Type 2 Diabetes. Diabetes Care 2016, 39, 82–91. [Google Scholar] [CrossRef]
- Yassin, A.; Haider, A.; Haider, K.S.; Caliber, M.; Doros, G.; Saad, F.; Garvey, W.T. Testosterone Therapy in Men With Hypogonadism Prevents Progression from Prediabetes to Type 2 Diabetes: Eight-Year Data From a Registry Study. Diabetes Care 2019, 42, 1104–1111. [Google Scholar] [CrossRef]
- Saad, F.; Yassin, A.; Doros, G.; Haider, A. Effects of long-term treatment with testosterone on weight and waist size in 411 hypogonadal men with obesity classes I-III: Observational data from two registry studies. Int. J. Obes. 2016, 40, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.C.; Lin, H.Y.; Liu, N.C.; Sparks, J.D.; Yeh, S.; Fang, L.Y.; Chen, L.; Chang, C. Neuronal androgen receptor regulates insulin sensitivity via suppression of hypothalamic NF-κB-mediated PTP1B expression. Diabetes 2013, 62, 411–423. [Google Scholar] [CrossRef]
- Yang, L.; Zhou, R.; Tong, Y.; Chen, P.; Shen, Y.; Miao, S.; Liu, X. Neuroprotection by dihydrotestosterone in LPS-induced neuroinflammation. Neurobiol. Dis. 2020, 140, 104814. [Google Scholar] [CrossRef] [PubMed]
- Antinozzi, C.; Marampon, F.; Corinaldesi, C.; Vicini, E.; Sgrò, P.; Vannelli, G.B.; Lenzi, A.; Crescioli, C.; Di Luigi, L. Testosterone insulin-like effects: An in vitro study on the short-term metabolic effects of testosterone in human skeletal muscle cells. J. Endocrinol. Investig. 2017, 40, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Moreira-Pais, A.; Ferreira, R.; Neves, J.S.; Vitorino, R.; Moreira-Gonçalves, D.; Nogueira-Ferreira, R. Sex differences on adipose tissue remodeling: From molecular mechanisms to therapeutic interventions. J. Mol. Med. 2020, 98, 483–493. [Google Scholar] [CrossRef]
- Zhang, D.; Fan, J.; Liu, H.; Qiu, G.; Cui, S. Testosterone enhances taurine synthesis by upregulating androgen receptor and cysteine sulfinic acid decarboxylase expressions in male mouse liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2023, 324, G295–G304. [Google Scholar] [CrossRef]
- Wei, W.; Lyu, X.; Markhard, A.L.; Fu, S.; Mardjuki, R.E.; Cavanagh, P.E.; Zeng, X.; Rajniak, J.; Lu, N.; Xiao, S.; et al. PTER is a N-acetyltaurine hydrolase that regulates feeding and obesity. Nature 2024, 633, 182–188. [Google Scholar] [CrossRef]
- Zhu, L.; Zhou, J.; Pan, Y.; Lv, J.; Liu, Y.; Yu, S.; Zhang, Y. Glucagon-like peptide-1 receptor expression and its functions are regulated by androgen. Biomed. Pharmacother. 2019, 120, 109555. [Google Scholar] [CrossRef]
- Xu, W.; Qadir, M.M.F.; Nasteska, D.; Mota de Sa, P.; Gorvin, C.M.; Blandino-Rosano, M.; Evans, C.R.; Ho, T.; Potapenko, E.; Veluthakal, R.; et al. Architecture of androgen receptor pathways amplifying glucagon-like peptide-1 insulinotropic action in male pancreatic β cells. Cell Rep. 2023, 42, 112529. [Google Scholar] [CrossRef]
- Sanchez-Garrido, M.A.; Tena-Sempere, M. Metabolic dysfunction in polycystic ovary syndrome: Pathogenic role of androgen excess and potential therapeutic strategies. Mol. Metab. 2020, 35, 100937. [Google Scholar] [CrossRef]
- Liu, L.; Liu, S.; Song, Q.; Luo, D.; Su, Y.; Qi, X.; Wang, Q.; Ning, J.; Lv, Y.; Guan, Q. Association of Metabolic Obesity Phenotypes and Total Testosterone in Chinese Male Population. Diabetes Metab. Syndr. Obes. 2021, 14, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Allard, C.; Morford, J.J.; Xu, W.; Liu, S.; Molinas, A.J.; Butcher, S.M.; Fine, N.H.; Blandino-Rosano, M.; Sure, V.N.; et al. Androgen excess in pancreatic β cells and neurons predisposes female mice to type 2 diabetes. JCI Insight 2018, 3, e98607. [Google Scholar] [CrossRef]
- Brown, R.E.; Wilkinson, D.A.; Imran, S.A.; Caraty, A.; Wilkinson, M. Hypothalamic kiss1 mRNA and kisspeptin immunoreactivity are reduced in a rat model of polycystic ovary syndrome (PCOS). Brain Res. 2012, 1467, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nohara, K.; Laque, A.; Allard, C.; Münzberg, H.; Mauvais-Jarvis, F. Central mechanisms of adiposity in adult female mice with androgen excess. Obesity 2014, 22, 1477–1484. [Google Scholar] [CrossRef]
- Han, Q.; Wang, J.; Li, W.; Chen, Z.J.; Du, Y. Androgen-induced gut dysbiosis disrupts glucolipid metabolism and endocrinal functions in polycystic ovary syndrome. Microbiome 2021, 9, 101. [Google Scholar] [CrossRef]
- Osei-Ntansah, A.; Oliver, T.; Lofton, T.; Falzarano, C.; Carr, K.; Huang, R.; Wilson, A.; Damaser, E.; Harvey, G.; Rahman, M.A.; et al. Liver Androgen Receptor Knockout Improved High-fat Diet Induced Glucose Dysregulation in Female Mice but Not Male Mice. J. Endocr. Soc. 2024, 8, bvae021. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of Estrogens in the Regulation of Liver Lipid Metabolism. Adv. Exp. Med. Biol. 2017, 1043, 227–256. [Google Scholar]
- Alemany, M. Estrogens and the regulation of glucose metabolism. World J. Diabetes 2021, 12, 1622–1654. [Google Scholar] [CrossRef]
- Nappi, R.E.; Chedraui, P.; Lambrinoudaki, I.; Simoncini, T. Menopause: A cardiometabolic transition. Lancet Diabetes Endocrinol. 2022, 10, 442–456. [Google Scholar] [CrossRef]
- Reue, K.; Wiese, C.B. Illuminating the Mechanisms Underlying Sex Differences in Cardiovascular Disease. Circ. Res. 2022, 130, 1747–1762. [Google Scholar] [CrossRef]
- de Oliveira, M.C.; Gilglioni, E.H.; de Boer, B.A.; Runge, J.H.; de Waart, D.R.; Salgueiro, C.L.; Ishii-Iwamoto, E.L.; Oude Elferink, R.P.; Gaemers, I.C. Bile acid receptor agonists INT747 and INT777 decrease oestrogen deficiency-related postmenopausal obesity and hepatic steatosis in mice. Biochim. Biophys. Acta 2016, 1862, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, G.E.; Hans, D.; Gonzalez Rodriguez, E.; Vollenweider, P.; Waeber, G.; Marques-Vidal, P.; Lamy, O. Menopausal Hormone Therapy Is Associated with Reduced Total and Visceral Adiposity: The OsteoLaus Cohort. J. Clin. Endocrinol. Metab. 2018, 103, 1948–1957. [Google Scholar] [CrossRef] [PubMed]
- González-García, I.; García-Clavé, E.; Cebrian-Serrano, A.; Le Thuc, O.; Contreras, R.E.; Xu, Y.; Gruber, T.; Schriever, S.C.; Legutko, B.; Lintelmann, J.; et al. Estradiol regulates leptin sensitivity to control feeding via hypothalamic Cited1. Cell Metab. 2023, 35, 438–455.E7. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Della Torre, S.; Stell, A.; Cook, J.; Brown, M.; Maggi, A. Tetradian oscillation of estrogen receptor α is necessary to prevent liver lipid deposition. Proc. Natl. Acad. Sci. USA 2012, 109, 11806–11811. [Google Scholar] [CrossRef]
- Fukata, Y.; Yu, X.; Imachi, H.; Nishiuchi, T.; Lyu, J.; Seo, K.; Takeuchi, A.; Iwama, H.; Masugata, H.; Hoshikawa, H.; et al. 17β-Estradiol regulates scavenger receptor class BI gene expression via protein kinase C in vascular endothelial cells. Endocrine 2014, 46, 644–650. [Google Scholar] [CrossRef]
- Zhu, L.; Shi, J.; Luu, T.N.; Neuman, J.C.; Trefts, E.; Yu, S.; Palmisano, B.T.; Wasserman, D.H.; Linton, M.F.; Stafford, J.M. Hepatocyte estrogen receptor alpha mediates estrogen action to promote reverse cholesterol transport during Western-type diet feeding. Mol. Metab. 2018, 8, 106–116. [Google Scholar] [CrossRef]
- Sutjarit, N.; Sueajai, J.; Boonmuen, N.; Sornkaew, N.; Suksamrarn, A.; Tuchinda, P.; Zhu, W.; Weerachayaphorn, J.; Piyachaturawat, P. Curcuma comosa reduces visceral adipose tissue and improves dyslipidemia in ovariectomized rats. J. Ethnopharmacol. 2018, 215, 167–175. [Google Scholar] [CrossRef]
- Hammes, S.R.; Levin, E.R. Impact of estrogens in males and androgens in females. J. Clin. Investig. 2019, 129, 1818–1826. [Google Scholar] [CrossRef]
- Lee, C.; Kim, J.; Han, J.; Oh, D.; Kim, M.; Jeong, H.; Kim, T.J.; Kim, S.W.; Kim, J.N.; Seo, Y.S.; et al. Formyl peptide receptor 2 determines sex-specific differences in the progression of nonalcoholic fatty liver disease and steatohepatitis. Nat. Commun. 2022, 13, 578. [Google Scholar] [CrossRef]
- Axsom, J.E.; Schmidt, H.D.; Matura, L.A.; Libonati, J.R. The Influence of Epigenetic Modifications on Metabolic Changes in White Adipose Tissue and Liver and Their Potential Impact in Exercise. Front. Physiol. 2021, 12, 686270. [Google Scholar] [CrossRef]
- Mannar, V.; Boro, H.; Patel, D.; Agstam, S.; Dalvi, M.; Bundela, V. Epigenetics of the Pathogenesis and Complications of Type 2 Diabetes Mellitus. touchREVIEWS Endocrinol. 2023, 19, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Lin, Z.J.; Li, C.C.; Lin, X.; Shan, S.K.; Guo, B.; Zheng, M.H.; Li, F.; Yuan, L.Q.; Li, Z.H. Epigenetic regulation in metabolic diseases: Mechanisms and advances in clinical study. Signal Transduct. Target. Ther. 2023, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Xia, Y.; Mei, S.; Ye, Z.; Song, B.; Yan, X.; Lin, F.; Rao, T.; Yu, W.; Mei, C.; et al. Histone H3K27 methyltransferase EZH2 regulates apoptotic and inflammatory responses in sepsis-induced AKI. Theranostics 2023, 13, 1860–1875. [Google Scholar] [CrossRef] [PubMed]
- Kaisinger, L.R.; Kentistou, K.A.; Stankovic, S.; Gardner, E.J.; Day, F.R.; Zhao, Y.; Mörseburg, A.; Carnie, C.J.; Zagnoli-Vieira, G.; Puddu, F.; et al. Large-scale exome sequence analysis identifies sex- and age-specific determinants of obesity. Cell Genom. 2023, 3, 100362. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, A.; Zhou, W.; Li, B.; Zhang, L.; Rudolf, A.M.; Jin, Z.; Hambly, C.; Wang, G.; Speakman, J.R. Maternal dietary fat during lactation shapes single nucleus transcriptomic profile of postnatal offspring hypothalamus in a sexually dimorphic manner in mice. Nat. Commun. 2024, 15, 2382. [Google Scholar] [CrossRef]
- Shi, Y.; Ma, J.; Li, S.; Liu, C.; Liu, Y.; Chen, J.; Liu, N.; Liu, S.; Huang, H. Sex difference in human diseases: Mechanistic insights and clinical implications. Signal Transduct. Target. Ther. 2024, 9, 238. [Google Scholar] [CrossRef]
- Horstmann, A.; Kovacs, P.; Kabisch, S.; Boettcher, Y.; Schloegl, H.; Tönjes, A.; Stumvoll, M.; Pleger, B.; Villringer, A. Common genetic variation near MC4R has a sex-specific impact on human brain structure and eating behavior. PLoS ONE 2013, 8, e74362. [Google Scholar] [CrossRef]
- Claussnitzer, M.; Dankel, S.N.; Kim, K.H.; Quon, G.; Meuleman, W.; Haugen, C.; Glunk, V.; Sousa, I.S.; Beaudry, J.L.; Puviindran, V.; et al. FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N. Engl. J. Med. 2015, 373, 895–907. [Google Scholar] [CrossRef]
- Adak, A.; Khan, M.R. An insight into gut microbiota and its functionalities. Cell Mol. Life Sci. 2019, 76, 473–493. [Google Scholar] [CrossRef]
- Lemons, J.M.S.; Liu, L. Chewing the Fat with Microbes: Lipid Crosstalk in the Gut. Nutrients 2022, 14, 573. [Google Scholar] [CrossRef]
- Fan, S.; Chen, S.; Lin, L. Research progress of gut microbiota and obesity caused by high-fat diet. Front. Cell Infect. Microbiol. 2023, 13, 1139800. [Google Scholar] [CrossRef]
- Krautkramer, K.A.; Fan, J.; Bäckhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 2021, 19, 77–94. [Google Scholar] [CrossRef]
- Friedman, E.S.; Li, Y.; Shen, T.D.; Jiang, J.; Chau, L.; Adorini, L.; Babakhani, F.; Edwards, J.; Shapiro, D.; Zhao, C.; et al. FXR-Dependent Modulation of the Human Small Intestinal Microbiome by the Bile Acid Derivative Obeticholic Acid. Gastroenterology 2018, 155, 1741–1752.e5. [Google Scholar] [CrossRef] [PubMed]
- Santos-Marcos, J.A.; Mora-Ortiz, M.; Tena-Sempere, M.; Lopez-Miranda, J.; Camargo, A. Interaction between gut microbiota and sex hormones and their relation to sexual dimorphism in metabolic diseases. Biol. Sex. Differ. 2023, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Bolnick, D.I.; Snowberg, L.K.; Hirsch, P.E.; Lauber, C.L.; Org, E.; Parks, B.; Lusis, A.J.; Knight, R.; Caporaso, J.G.; Svanbäck, R. Individual diet has sex-dependent effects on vertebrate gut microbiota. Nat. Commun. 2014, 5, 4500. [Google Scholar] [CrossRef] [PubMed]
- Org, E.; Mehrabian, M.; Parks, B.W.; Shipkova, P.; Liu, X.; Drake, T.A.; Lusis, A.J. Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes 2016, 7, 313–322. [Google Scholar] [CrossRef]
- Fransen, F.; van Beek, A.A.; Borghuis, T.; Meijer, B.; Hugenholtz, F.; van der Gaast-de Jongh, C.; Savelkoul, H.F.; de Jonge, M.I.; Faas, M.M.; Boekschoten, M.V.; et al. The Impact of Gut Microbiota on Gender-Specific Differences in Immunity. Front. Immunol. 2017, 8, 754. [Google Scholar] [CrossRef]
- Elderman, M.; Hugenholtz, F.; Belzer, C.; Boekschoten, M.; van Beek, A.; de Haan, B.; Savelkoul, H.; de Vos, P.; Faas, M. Sex and strain dependent differences in mucosal immunology and microbiota composition in mice. Biol. Sex. Differ. 2018, 9, 26. [Google Scholar] [CrossRef]
- Sinha, T.; Vich Vila, A.; Garmaeva, S.; Jankipersadsing, S.A.; Imhann, F.; Collij, V.; Bonder, M.J.; Jiang, X.; Gurry, T.; Alm, E.J.; et al. Analysis of 1135 gut metagenomes identifies sex-specific resistome profiles. Gut Microbes 2019, 10, 358–366. [Google Scholar] [CrossRef]
- Liu, D.S.; Wang, X.S.; Zhong, X.H.; Cao, H.; Zhang, F. Sexual dimorphism in the gut microbiota and sexual dimorphism in chronic diseases: Association or causation? J. Steroid Biochem. Mol. Biol. 2024, 237, 106451. [Google Scholar] [CrossRef]
- Ma, Z.S.; Li, W. How and Why Men and Women Differ in Their Microbiomes: Medical Ecology and Network Analyses of the Microgenderome. Adv. Sci. 2019, 6, 1902054. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Schloss, P.D. Dynamics and associations of microbial community types across the human body. Nature 2014, 509, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Xu, Y.; Yi, C.X.; Tong, Q.; Cai, D. The hypothalamus for whole-body physiology: From metabolism to aging. Protein Cell 2022, 13, 394–421. [Google Scholar] [CrossRef] [PubMed]
- Chowen, J.A.; Argente-Arizón, P.; Freire-Regatillo, A.; Argente, J. Sex differences in the neuroendocrine control of metabolism and the implication of astrocytes. Front. Neuroendocrinol. 2018, 48, 3–12. [Google Scholar] [CrossRef]
- Tang, C.; Wang, Y.; Xu, Z.; Chen, D.; Xu, J.; Yang, D.; Zhang, L.; Liu, J.; Kan, J. The relationships between high-fat diet and metabolic syndrome: Potential mechanisms. Food Biosci. 2024, 59, 104261. [Google Scholar] [CrossRef]
- Mann, E.R.; Lam, Y.K.; Uhlig, H.H. Short-chain fatty acids: Linking diet, the microbiome and immunity. Nat. Rev. Immunol. 2024, 24, 577–595. [Google Scholar] [CrossRef]
- Kwon, Y.J.; Lee, H.S.; Lee, J.W. Association of carbohydrate and fat intake with metabolic syndrome. Clin. Nutr. 2018, 37, 746–751. [Google Scholar] [CrossRef]
- MacGregor, K.; Ellefsen, S.; Pillon, N.J.; Hammarström, D.; Krook, A. Sex differences in skeletal muscle metabolism in exercise and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2025, 21, 166–179. [Google Scholar] [CrossRef]
- Tomé, D.; Benoit, S.; Azzout-Marniche, D. Protein metabolism and related body function: Mechanistic approaches and health consequences. Proc. Nutr. Soc. 2021, 80, 243–251. [Google Scholar] [CrossRef]
- Bailey, R.L.; Dog, T.L.; Smith-Ryan, A.E.; Das, S.K.; Baker, F.C.; Madak-Erdogan, Z.; Hammond, B.R.; Sesso, H.D.; Eapen, A.; Mitmesser, S.H.; et al. Sex Differences Across the Life Course: A Focus On Unique Nutritional and Health Considerations among Women. J. Nutr. 2022, 152, 1597–1610. [Google Scholar] [CrossRef]
- DiNicolantonio, J.J.; O’Keefe, J.H. Monounsaturated Fat vs Saturated Fat: Effects on Cardio-Metabolic Health and Obesity. Mo. Med. 2022, 119, 69–73. [Google Scholar] [PubMed]
- Delgado-Lista, J.; Alcala-Diaz, J.F.; Torres-Peña, J.D.; Quintana-Navarro, G.M.; Fuentes, F.; Garcia-Rios, A.; Ortiz-Morales, A.M.; Gonzalez-Requero, A.I.; Perez-Caballero, A.I.; Yubero-Serrano, E.M.; et al. Long-term secondary prevention of cardiovascular disease with a Mediterranean diet and a low-fat diet (CORDIOPREV): A randomised controlled trial. Lancet 2022, 399, 1876–1885. [Google Scholar] [CrossRef]
- Güzey Akansel, M.; Baş, M.; Gençalp, C.; Kahrıman, M.; Şahin, E.; Öztürk, H.; Gür, G.; Gür, C. Effects of the Ketogenic Diet on Microbiota Composition and Short-Chain Fatty Acids in Women with Overweight/Obesity. Nutrients 2024, 16, 4374. [Google Scholar] [CrossRef] [PubMed]
- Didier, A.J.; Stiene, J.; Fang, L.; Watkins, D.; Dworkin, L.D.; Creeden, J.F. Antioxidant and Anti-Tumor Effects of Dietary Vitamins A, C, and E. Antioxidants 2023, 12, 632. [Google Scholar] [CrossRef]
- Di Berardino, C.; Barceviciute, U.; Camerano Spelta Rapini, C.; Peserico, A.; Capacchietti, G.; Bernabò, N.; Russo, V.; Gatta, V.; Konstantinidou, F.; Donato, M.; et al. High-fat diet-negative impact on female fertility: From mechanisms to protective actions of antioxidant matrices. Front. Nutr. 2024, 11, 1415455. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.H.; Sholzberg, M. Iron deficiency anemia among women: An issue of health equity. Blood Rev. 2024, 64, 101159. [Google Scholar] [CrossRef]
- Zucker, I.; Prendergast, B.J. Sex differences in pharmacokinetics predict adverse drug reactions in women. Biol. Sex. Differ. 2020, 11, 32. [Google Scholar] [CrossRef]
- Kautzky-Willer, A.; Leutner, M.; Harreiter, J. Sex differences in type 2 diabetes. Diabetologia 2023, 66, 986–1002. [Google Scholar] [CrossRef]
- Wilding, J.P.; Overgaard, R.V.; Jacobsen, L.V.; Jensen, C.B.; le Roux, C.W. Exposure-response analyses of liraglutide 3.0 mg for weight management. Diabetes Obes. Metab. 2016, 18, 491–499. [Google Scholar] [CrossRef]
- Moyer, A.M.; Matey, E.T.; Miller, V.M. Individualized medicine: Sex, hormones, genetics, and adverse drug reactions. Pharmacol. Res. Perspect. 2019, 7, e00541. [Google Scholar] [CrossRef]
- Aroda, V.R.; Christophi, C.A.; Edelstein, S.L.; Zhang, P.; Herman, W.H.; Barrett-Connor, E.; Delahanty, L.M.; Montez, M.G.; Ackermann, R.T.; Zhuo, X.; et al. The effect of lifestyle intervention and metformin on preventing or delaying diabetes among women with and without gestational diabetes: The Diabetes Prevention Program outcomes study 10-year follow-up. J. Clin. Endocrinol. Metab. 2015, 100, 1646–1653. [Google Scholar] [CrossRef] [PubMed]
- Raparelli, V.; Elharram, M.; Moura, C.S.; Abrahamowicz, M.; Bernatsky, S.; Behlouli, H.; Pilote, L. Sex Differences in Cardiovascular Effectiveness of Newer Glucose-Lowering Drugs Added to Metformin in Type 2 Diabetes Mellitus. J. Am. Heart Assoc. 2020, 9, e012940. [Google Scholar] [CrossRef] [PubMed]
- Funck, K.L.; Bjerg, L.; Isaksen, A.A.; Sandbæk, A.; Grove, E.L. Gender disparities in time-to-initiation of cardioprotective glucose-lowering drugs in patients with type 2 diabetes and cardiovascular disease: A Danish nationwide cohort study. Cardiovasc. Diabetol. 2022, 21, 279. [Google Scholar] [CrossRef]
- Christakis, M.K.; Hasan, H.; De Souza, L.R.; Shirreff, L. The effect of menopause on metabolic syndrome: Cross-sectional results from the Canadian Longitudinal Study on Aging. Menopause 2020, 27, 999–1009. [Google Scholar] [CrossRef]
- Tishova, Y.; Kalinchenko, S.; Mskhalaya, G.; Hackett, G.; Livingston, M.; König, C.; Strange, R.; Zitzmann, M.; Mann, A.; Maarouf, A.; et al. Testosterone therapy reduces insulin resistance in men with adult-onset testosterone deficiency and metabolic syndrome. Results from the Moscow Study, a randomized controlled trial with an open-label phase. Diabetes Obes. Metab. 2024, 26, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Kanakis, G.A.; Pofi, R.; Goulis, D.G.; Isidori, A.M.; Armeni, E.; Erel, C.T.; Fistonić, I.; Hillard, T.; Hirschberg, A.L.; Meczekalski, B.; et al. EMAS position statement: Testosterone replacement therapy in older men. Maturitas 2023, 178, 107854. [Google Scholar] [CrossRef]
- Maseroli, E.; Comeglio, P.; Corno, C.; Cellai, I.; Filippi, S.; Mello, T.; Galli, A.; Rapizzi, E.; Presenti, L.; Truglia, M.C.; et al. Testosterone treatment is associated with reduced adipose tissue dysfunction and nonalcoholic fatty liver disease in obese hypogonadal men. J. Endocrinol. Investig. 2021, 44, 819–842. [Google Scholar] [CrossRef]
- Archacki, D.; Zieliński, J.; Ciekot-Sołtysiak, M.; Zarębska, E.A.; Kusy, K. Sex Differences in the Energy System Contribution during Sprint Exercise in Speed-Power and Endurance Athletes. J. Clin. Med. 2024, 13, 4812. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The role of estrogens in control of energy balance and glucose homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef]
- Landen, S.; Hiam, D.; Voisin, S.; Jacques, M.; Lamon, S.; Eynon, N. Physiological and molecular sex differences in human skeletal muscle in response to exercise training. J. Physiol. 2023, 601, 419–434. [Google Scholar] [CrossRef]
- Tarnopolsky, M.A. Sex differences in exercise metabolism and the role of 17-beta estradiol. Med. Sci. Sports Exerc. 2008, 40, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.K.; Angadi, S.S.; Bhargava, A.; Harper, J.; Hirschberg, A.L.; Levine, B.D.; Moreau, K.L.; Nokoff, N.J.; Stachenfeld, N.S.; Bermon, S. The Biological Basis of Sex Differences in Athletic Performance: Consensus Statement for the American College of Sports Medicine. Med. Sci. Sports Exerc. 2023, 55, 2328–2360. [Google Scholar] [CrossRef]
- Brown, K.D.; Waggy, E.D.; Nair, S.; Robinson, T.J.; Schmitt, E.E.; Bruns, D.R.; Thomas, D.P. Sex Differences in Cardiac AMP-Activated Protein Kinase Following Exhaustive Exercise. Sports Med. Int. Open 2020, 4, E13–E18. [Google Scholar] [CrossRef] [PubMed]
- Mauvais-Jarvis, F. Sex differences in energy metabolism: Natural selection, mechanisms and consequences. Nat. Rev. Nephrol. 2024, 20, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Ansdell, P.; Thomas, K.; Hicks, K.M.; Hunter, S.K.; Howatson, G.; Goodall, S. Physiological sex differences affect the integrative response to exercise: Acute and chronic implications. Exp. Physiol. 2020, 105, 2007–2021. [Google Scholar] [CrossRef]
- Blüher, S.; Käpplinger, J.; Herget, S.; Reichardt, S.; Böttcher, Y.; Grimm, A.; Kratzsch, J.; Petroff, D. Cardiometabolic risk markers, adipocyte fatty acid binding protein (aFABP) and the impact of high-intensity interval training (HIIT) in obese adolescents. Metabolism 2017, 68, 77–87. [Google Scholar] [CrossRef]
- Ji, H.; Gulati, M.; Huang, T.Y.; Kwan, A.C.; Ouyang, D.; Ebinger, J.E.; Casaletto, K.; Moreau, K.L.; Skali, H.; Cheng, S. Sex Differences in Association of Physical Activity With All-Cause and Cardiovascular Mortality. J. Am. Coll. Cardiol. 2024, 83, 783–793. [Google Scholar] [CrossRef]
- Geng, J.; Zhang, X.; Guo, Y.; Wen, H.; Guo, D.; Liang, Q.; Pu, S.; Wang, Y.; Liu, M.; Li, Z.; et al. Moderate-intensity interval exercise exacerbates cardiac lipotoxicity in high-fat, high-calories diet-fed mice. Nat. Commun. 2025, 16, 613. [Google Scholar] [CrossRef]
- Kusuyama, J.; Alves-Wagner, A.B.; Makarewicz, N.S.; Goodyear, L.J. Effects of maternal and paternal exercise on offspring metabolism. Nat. Metab. 2020, 2, 858–872. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mo, Q.; Deng, X.; Zhou, Z.; Yin, L. High-Fat Diet and Metabolic Diseases: A Comparative Analysis of Sex-Dependent Responses and Mechanisms. Int. J. Mol. Sci. 2025, 26, 4777. https://doi.org/10.3390/ijms26104777
Mo Q, Deng X, Zhou Z, Yin L. High-Fat Diet and Metabolic Diseases: A Comparative Analysis of Sex-Dependent Responses and Mechanisms. International Journal of Molecular Sciences. 2025; 26(10):4777. https://doi.org/10.3390/ijms26104777
Chicago/Turabian StyleMo, Qiaoling, Xinquan Deng, Ziyu Zhou, and Lijun Yin. 2025. "High-Fat Diet and Metabolic Diseases: A Comparative Analysis of Sex-Dependent Responses and Mechanisms" International Journal of Molecular Sciences 26, no. 10: 4777. https://doi.org/10.3390/ijms26104777
APA StyleMo, Q., Deng, X., Zhou, Z., & Yin, L. (2025). High-Fat Diet and Metabolic Diseases: A Comparative Analysis of Sex-Dependent Responses and Mechanisms. International Journal of Molecular Sciences, 26(10), 4777. https://doi.org/10.3390/ijms26104777