
Targeting the IL-23 Receptor Gene: A Promising Approach in Inflammatory Bowel Disease Treatment





Abstract

{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Role of IL-23 and IL-23R in IBD
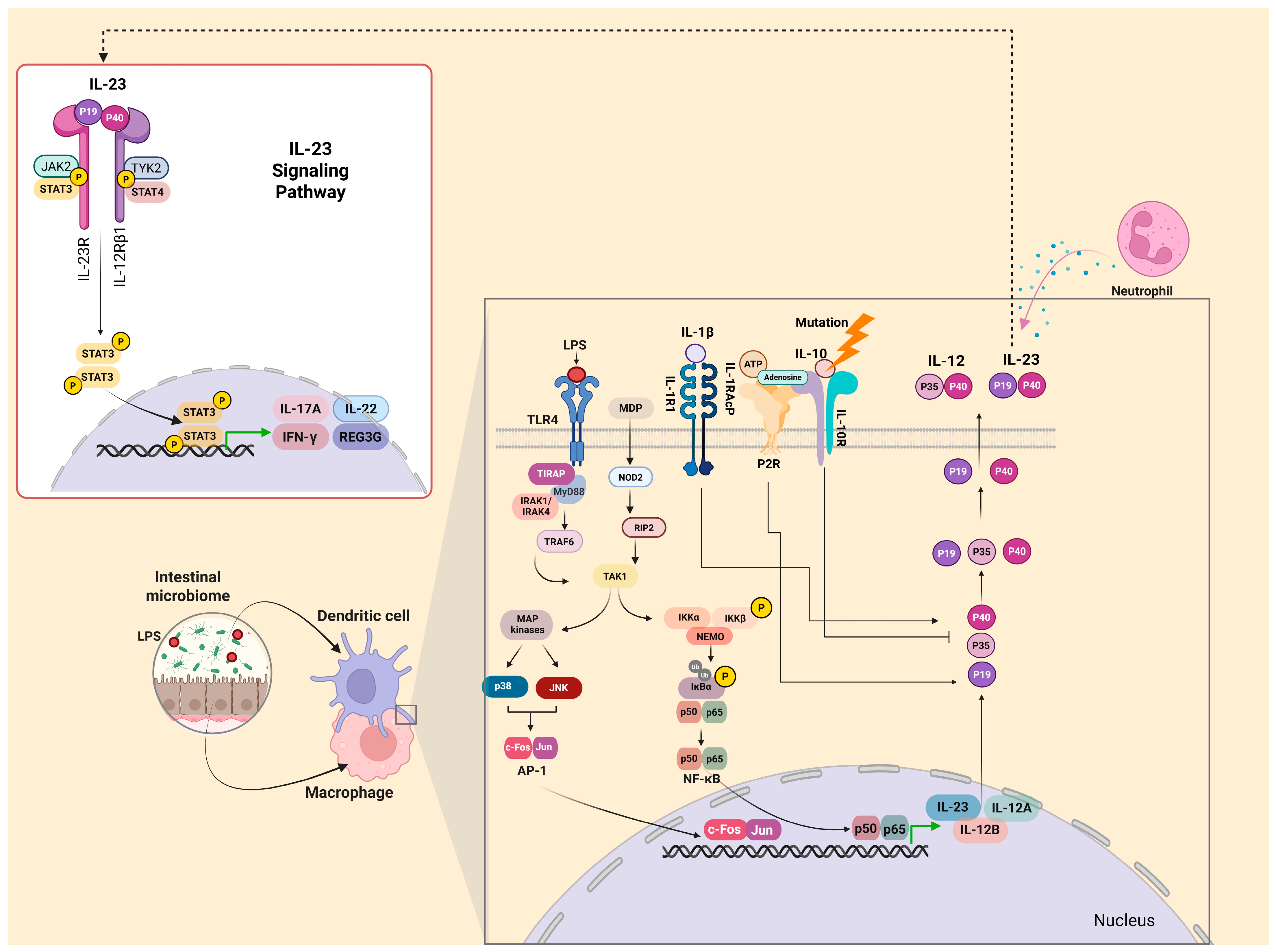
2.1. IL-23 and IL-23R
2.2. IL-23 Production
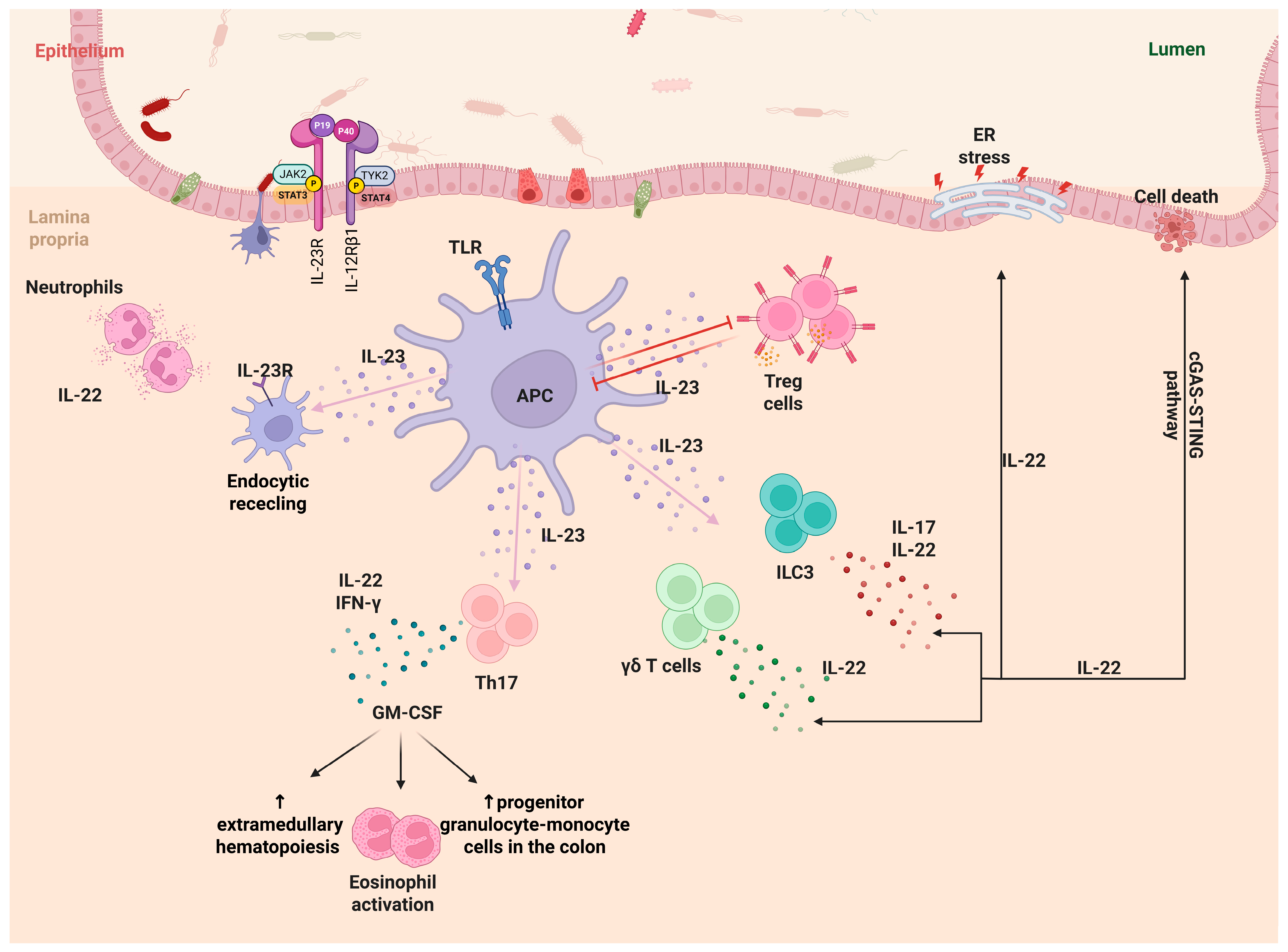
2.3. IL-23 and Its Multifaceted Role in IBD Pathogenesis
3. IL-23R Gene in IBD
3.1. R381Q (Arg381Gln, rs11209026)
3.2. G149R (Gly149Arg, rs76418789)
3.3. V362I (Val362Ile, rs41313262)
3.4. Other IL-23R-Related Gene Variants
4. IL-23R-Based Treatment Options in IBD
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ahluwalia, B.; Magnusson, M.K.; Öhman, L. Mucosal immune system of the gastrointestinal tract: Maintaining balance between the good and the bad. Scand. J. Gastroenterol. 2017, 52, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Le Berre, C.; Ananthakrishnan, A.N.; Danese, S.; Singh, S.; Peyrin-Biroulet, L. Ulcerative Colitis and Crohn’s Disease Have Similar Burden and Goals for Treatment. Clin. Gastroenterol. Hepatol. 2020, 18, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 2019, 7247238. [Google Scholar] [CrossRef]
- Gomollón, F.; Dignass, A.; Annese, V.; Tilg, H.; Van Assche, G.; Lindsay, J.O.; Peyrin-Biroulet, L.; Cullen, G.J.; Daperno, M.; Kucharzik, T.; et al. 3rd European Evidence-based Consensus on the Diagnosis and Management of Crohn’s Disease 2016: Part 1: Diagnosis and Medical Management. J. Crohns Colitis 2017, 11, 3–25. [Google Scholar] [CrossRef]
- Wallace, K.L.; Zheng, L.B.; Kanazawa, Y.; Shih, D.Q. Immunopathology of inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.R.; Liu, C.Q.; Feng, B.S.; Liu, Z.J. Dysregulation of mucosal immune response in pathogenesis of inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 3255–3264. [Google Scholar] [CrossRef]
- de Souza, H.S.P.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Li, Y.Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Kaplan, G.G.; Ng, S.C. Changing Global Epidemiology of Inflammatory Bowel Diseases: Sustaining Health Care Delivery Into the 21st Century. Clin. Gastroenterol. Hepatol. 2020, 18, 1252–1260. [Google Scholar] [CrossRef]
- Park, Y.M.; Ha, E.; Gu, K.N.; Shin, G.Y.; Lee, C.K.; Kim, K.; Kim, H.J. Host Genetic and Gut Microbial Signatures in Familial Inflammatory Bowel Disease. Clin. Transl. Gastroenterol. 2020, 11, e00213. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Duerr, R.H.; Taylor, K.D.; Brant, S.R.; Rioux, J.D.; Silverberg, M.S.; Daly, M.J.; Steinhart, A.H.; Abraham, C.; Regueiro, M.; Griffiths, A.; et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006, 314, 1461–1463. [Google Scholar] [CrossRef] [PubMed]
- Kastelein, R.A.; Hunter, C.A.; Cua, D.J. Discovery and Biology of IL-23 and IL-27: Related but Functionally Distinct Regulators of Inflammation. Annu. Rev. Immunol. 2007, 25, 221–242. [Google Scholar] [CrossRef]
- Jefremow, A.; Neurath, M.F. All are Equal, Some are More Equal: Targeting IL 12 and 23 in IBD—A Clinical Perspective. ImmunoTargets Ther. 2020, 9, 289. [Google Scholar] [CrossRef]
- Lupardus, P.J.; Garcia, K.C. The structure of Interleukin-23 reveals the molecular basis of p40 subunit sharing with IL-12. J. Mol. Biol. 2008, 382, 931. [Google Scholar] [CrossRef]
- Schnurr, M.; Toy, T.; Shin, A.; Wagner, M.; Cebon, J.; Maraskovsky, E. Extracellular nucleotide signaling by P2 receptors inhibits IL-12 and enhances IL-23 expression in human dendritic cells: A novel role for the cAMP pathway. Blood 2005, 105, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A microbial signature for Crohn’s disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Wirtz, S.; Blessing, M.; Pirhonen, J.; Strand, D.; Bechthold, O.; Frick, J.; Galle, P.R.; Autenrieth, I.; Neurath, M.F. Constitutive p40 promoter activation and IL-23 production in the terminal ileum mediated by dendritic cells. J. Clin. Investig. 2003, 112, 693–706. [Google Scholar] [CrossRef]
- Liu, Z.; Yadav, P.K.; Xu, X.; Su, J.; Chen, C.; Tang, M.; Lin, H.; Yu, J.; Qian, J.; Yang, P.C.; et al. The increased expression of IL-23 in inflammatory bowel disease promotes intraepithelial and lamina propria lymphocyte inflammatory responses and cytotoxicity. J. Leukoc. Biol. 2011, 89, 597–606. [Google Scholar] [CrossRef]
- Schmidt, C.; Giese, T.; Ludwig, B.; Mueller-Molaian, I.; Marth, T.; Zeuzem, S.; Meurer, S.C.; Stallmach, A. Expression of interleukin-12-related cytokine transcripts in inflammatory bowel disease: Elevated interleukin-23p19 and interleukin-27p28 in Crohn’s disease but not in ulcerative colitis. Inflamm. Bowel Dis. 2005, 11, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, H.; Billmeier, U.; Dieterich, W.; Rath, T.; Sonnewald, S.; Reid, S.; Hirschmann, S.; Hildner, K.; Waldner, M.J.; Mudter, J.; et al. Expansion of IL-23 receptor bearing TNFR2+ T cells is associated with molecular resistance to anti-TNF therapy in Crohn’s disease. Gut 2019, 68, 814–828. [Google Scholar] [CrossRef]
- Verreck, F.A.W.; de Boer, T.; Langenberg, D.M.L.; Hoeve, M.A.; Kramer, M.; Vaisberg, E.; Kastelein, R.; Kolk, A.; de Waal-Malefyt, R.; Ottenhoff, T.H.M. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc. Natl. Acad. Sci. USA 2004, 101, 4560–4565. [Google Scholar] [CrossRef] [PubMed]
- Re, F.; Strominger, J.L. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J. Biol. Chem. 2001, 276, 37692–37699. [Google Scholar] [CrossRef] [PubMed]
- Veckman, V.; Miettinen, M.; Pirhonen, J.; Sirén, J.; Matikainen, S.; Julkunen, I. Streptococcus pyogenes and Lactobacillus rhamnosus differentially induce maturation and production of Th1-type cytokines and chemokines in human monocyte-derived dendritic cells. J. Leukoc. Biol. 2004, 75, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Hisamatsu, T.; Okamoto, S.; Chinen, H.; Kobayashi, T.; Sato, T.; Sakurba, A.; Kitazume, M.T.; Sugita, A.; Koganei, K.; et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J. Clin. Investig. 2008, 118, 2269–2280. [Google Scholar]
- Brain, O.; Owens, B.M.J.; Pichulik, T.; Allan, P.; Khatamzas, E.; Leslie, A.; Steevels, T.; Sharma, S.; Mayer, A.; Catuneanu, A.M.; et al. The intracellular sensor NOD2 induces microRNA-29 expression in human dendritic cells to limit IL-23 release. Immunity 2013, 39, 521–536. [Google Scholar] [CrossRef]
- Cekic, C.; Linden, J. Purinergic regulation of the immune system. Nat. Rev. Immunol. 2016, 16, 177–192. [Google Scholar] [CrossRef]
- Mogilenko, D.A.; Haas, J.T.; L’homme, L.; Fleury, S.; Quemener, S.; Levavasseur, M.; Becquart, C.; Wartelle, J.; Bogomolova, A.; Pineau, L.; et al. Metabolic and Innate Immune Cues Merge into a Specific Inflammatory Response via the UPR. Cell 2019, 177, 1201–1216.e19. [Google Scholar] [CrossRef]
- Cader, M.Z.; de Almeida Rodrigues, R.P.; West, J.A.; Sewell, G.W.; Md-Ibrahim, M.N.; Reikine, S.; Sirago, G.; Unger, L.W.; Iglesias-Romero, A.B.; Ramshorn, K.; et al. FAMIN Is a Multifunctional Purine Enzyme Enabling the Purine Nucleotide Cycle. Cell 2020, 180, 278–295.e23. [Google Scholar] [CrossRef]
- Aschenbrenner, D.; Quaranta, M.; Banerjee, S.; Ilott, N.; Jansen, J.; Steere, B.; Chen, Y.H.; Ho, S.; Cox, K.; Arancibia-Carcamo, C.V.; et al. Deconvolution of monocyte responses in inflammatory bowel disease reveals an IL-1 cytokine network that regulates IL-23 in genetic and acquired IL-10 resistance. Gut 2021, 70, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Natoli, G. Molecular control of activation and priming in macrophages. Nat. Immunol. 2016, 17, 26–33. [Google Scholar] [CrossRef]
- Russo, R.C.; Garcia, C.C.; Teixeira, M.M.; Amaral, F.A. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert. Rev. Clin. Immunol. 2014, 10, 593–619. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Su, J.; Zhang, X.; Cheng, X.; Zhou, J.; Shi, R.; Zhang, H. Elevated levels of Th17 cells and Th17-related cytokines are associated with disease activity in patients with inflammatory bowel disease. Inflamm. Res. 2014, 63, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef]
- Zhou, L.; Lopes, J.E.; Chong, M.M.W.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 2008, 453, 236–240. [Google Scholar] [CrossRef]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 2000, 13, 715–725. [Google Scholar] [CrossRef]
- El-Behi, M.; Ciric, B.; Dai, H.; Yan, Y.; Cullimore, M.; Safavi, F.; Zhang, G.X.; Dittel, B.N.; Rostami, A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 2011, 12, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Turner, H.; Maynard, C.L.; Oliver, J.R.; Chen, D.; Elson, C.O.; Weaver, C.T. Late developmental plasticity in the T helper 17 lineage. Immunity 2009, 30, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, H.; Neurath, M.F.; Atreya, R. Role of the IL23/IL17 Pathway in Crohn’s Disease. Front. Immunol. 2021, 12, 622934. [Google Scholar] [CrossRef] [PubMed]
- Sewell, G.W.; Kaser, A. Interleukin-23 in the Pathogenesis of Inflammatory Bowel Disease and Implications for Therapeutic Intervention. J. Crohns Colitis. 2022, 16 (Suppl. 2), ii3–ii19. [Google Scholar] [CrossRef] [PubMed]
- Targan, S.R.; Feagan, B.; Vermeire, S.; Panaccione, R.; Melmed, G.Y.; Landers, C.; Li, D.; Russell, C.; Newmark, R.; Zhang, N.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study of Brodalumab in Patients With Moderate-to-Severe Crohn’s Disease. Am. J. Gastroenterol. 2016, 111, 1599–1607. [Google Scholar] [CrossRef]
- Hommes, D.W.; Mikhajlova, T.L.; Stoinov, S.; Stimac, D.; Vucelic, B.; Lonovics, J.; Zakuciova, M.; D’Haens, G.; Van Assche, G.; Ba, S.; et al. Fontolizumab, a humanised anti-interferon gamma antibody, demonstrates safety and clinical activity in patients with moderate to severe Crohn’s disease. Gut 2006, 55, 1131–1137. [Google Scholar] [CrossRef]
- Li, O.; Li, X.; He, J. Knockdown of TOP2A suppresses IL-17 signaling pathway and alleviates the progression of ulcerative colitis. Immun. Inflamm. Dis. 2024, 12, e1207. [Google Scholar] [CrossRef]
- Ahern, P.P.; Schiering, C.; Buonocore, S.; McGeachy, M.J.; Cua, D.J.; Maloy, K.J.; Powrie, F. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity 2010, 33, 279–288. [Google Scholar] [CrossRef]
- Takayama, T.; Kamada, N.; Chinen, H.; Okamoto, S.; Kitazume, M.T.; Chang, J.; Matuzaki, Y.; Suzuki, S.; Sugita, A.; Koganei, K.; et al. Imbalance of NKp44(+)NKp46(-) and NKp44(-)NKp46(+) natural killer cells in the intestinal mucosa of patients with Crohn’s disease. Gastroenterology 2010, 139, 882–892.e3. [Google Scholar] [CrossRef]
- Bauché, D.; Joyce-Shaikh, B.; Jain, R.; Grein, J.; Ku, K.S.; Blumenschein, W.M.; Ganal-Vonarburg, S.C.; Wilson, D.C.; McClanahan, T.K.; de Wall Malefyt, R.; et al. LAG3+ Regulatory T Cells Restrain Interleukin-23-Producing CX3CR1+ Gut-Resident Macrophages during Group 3 Innate Lymphoid Cell-Driven Colitis. Immunity 2018, 49, 342.e5–352.e5. [Google Scholar] [CrossRef] [PubMed]
- Izcue, A.; Coombes, J.L.; Powrie, F. Regulatory lymphocytes and intestinal inflammation. Annu. Rev. Immunol. 2009, 27, 313–338. [Google Scholar] [CrossRef]
- Powrie, F.; Leach, M.W.; Mauze, S.; Caddle, L.B.; Coffman, R.L. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 1993, 5, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Mottet, C.; Uhlig, H.H.; Powrie, F. Cutting edge: Cure of colitis by CD4+CD25+ regulatory T cells. J. Immunol. 2003, 170, 3939–3943. [Google Scholar] [CrossRef] [PubMed]
- Izcue, A.; Hue, S.; Buonocore, S.; Arancibia-Cárcamo, C.V.; Ahern, P.P.; Iwakura, Y.; Maloy, K.J.; Powrie, F. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity 2008, 28, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Jacobse, J.; Brown, R.E.; Li, J.; Pilat, J.M.; Pham, L.; Short, S.P.; Peek, C.T.; Rolong, A.; Washington, M.K.; Martinez-Barricarte, R.; et al. Interleukin-23 receptor signaling impairs the stability and function of colonic regulatory T cells. Cell Rep. 2023, 42, 112128. [Google Scholar] [CrossRef]
- Longman, R.S.; Diehl, G.E.; Victorio, D.A.; Huh, J.R.; Galan, C.; Miraldi, E.R.; Swaminath, A.; Bonneau, R.; Scherl, E.J.; Littman, D.R. CX3CR1+ mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J. Exp. Med. 2014, 211, 1571–1583. [Google Scholar] [CrossRef]
- Bauché, D.; Joyce-Shaikh, B.; Fong, J.; Villarino, A.V.; Ku, K.S.; Jain, R.; Lee, Y.C.; Annamalai, L.; Yearley, J.H.; Cua, D.J. IL-23 and IL-2 activation of STAT5 is required for optimal IL-22 production in ILC3s during colitis. Sci. Immunol. 2020, 5, eaav1080. [Google Scholar] [CrossRef]
- Guo, X.; Qiu, J.; Tu, T.; Yang, X.; Deng, L.; Anders, R.A.; Zhou, L.; Fu, Y.X. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity 2014, 40, 25–39. [Google Scholar] [CrossRef]
- Geremia, A.; Arancibia-Cárcamo, C.V.; Fleming, M.P.P.; Rust, N.; Singh, B.; Mortensen, N.J.; Travis, S.P.L.; Powrie, F. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J. Exp. Med. 2011, 208, 1127–1133. [Google Scholar] [CrossRef]
- Eken, A.; Singh, A.K.; Treuting, P.M.; Oukka, M. IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol. 2014, 7, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.; Yi, T.; Lu, T.; Ghilardi, N. The role of IL-22 in intestinal health and disease. J. Exp. Med. 2020, 217, e20192195. [Google Scholar] [CrossRef] [PubMed]
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Stevens, S.; Flavell, R.A. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 2008, 29, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Valdez, P.A.; Danilenko, D.M.; Hu, Y.; Sa, S.M.; Gong, Q.; Abbas, A.R.; Modrusan, Z.; Ghilardi, N.; de Sauvage, F.J.; et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008, 14, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Powell, N.; Pantazi, E.; Pavlidis, P.; Tsakmaki, A.; Li, K.; Yang, F.; Parker, A.; Pin, C.; Cozzetto, D.; Minns, D.; et al. Interleukin-22 orchestrates a pathological endoplasmic reticulum stress response transcriptional programme in colonic epithelial cells. Gut 2020, 69, 578–590. [Google Scholar] [CrossRef]
- Aden, K.; Tran, F.; Ito, G.; Sheibani-Tezerji, R.; Lipinski, S.; Kuiper, J.W.; Tschurtschnthaler, M.; Saveljeva, S.; Bhattacharyya, J.; Hasler, R.; et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS-STING. J. Exp. Med. 2018, 215, 2868–2886. [Google Scholar] [CrossRef]
- Fournier, B.M.; Parkos, C.A. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012, 5, 354–366. [Google Scholar] [CrossRef]
- Griseri, T.; McKenzie, B.S.; Schiering, C.; Powrie, F. Dysregulated hematopoietic stem and progenitor cell activity promotes interleukin-23-driven chronic intestinal inflammation. Immunity 2012, 37, 1116–1129. [Google Scholar] [CrossRef]
- Griseri, T.; Arnold, I.C.; Pearson, C.; Krausgruber, T.; Schiering, C.; Franchini, F.; Schulthess, J.; McKenzie, B.S.; Crocker, P.R.; Powrie, F. Granulocyte Macrophage Colony-Stimulating Factor-Activated Eosinophils Promote Interleukin-23 Driven Chronic Colitis. Immunity 2015, 43, 187–199. [Google Scholar] [CrossRef]
- Sun, R.; Hedl, M.; Abraham, C. IL23 induces IL23R recycling and amplifies innate receptor-induced signalling and cytokines in human macrophages, and the IBD-protective IL23R R381Q variant modulates these outcomes. Gut 2020, 69, 264–273. [Google Scholar] [CrossRef]
- Chognard, G.; Bellemare, L.; Pelletier, A.N.; Dominguez-Punaro, M.C.; Beauchamp, C.; Guyon, M.J.; Charron, G.; Morin, N.; Sivanesan, D.; Kuchroo, V.; et al. The dichotomous pattern of IL-12r and IL-23R expression elucidates the role of IL-12 and IL-23 in inflammation. PLoS ONE 2014, 9, e89092. [Google Scholar] [CrossRef] [PubMed]
- Parham, C.; Chirica, M.; Timans, J.; Vaisberg, E.; Travis, M.; Cheung, J.; Pflanz, S.; Zhang, R.; Singh, K.P.; Vega, F.; et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J. Immunol. 2002, 168, 5699–5708. [Google Scholar] [CrossRef] [PubMed]
- Budarf, M.L.; Labbé, C.; David, G.; Rioux, J.D. GWA studies: Rewriting the story of IBD. Trends Genet. 2009, 25, 137–146. [Google Scholar] [CrossRef]
- Ouyang, W.; Valdez, P. IL-22 in mucosal immunity. Mucosal Immunol. 2008, 1, 335–338. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Chen, Y.; Tato, C.M.; Laurence, A.; Joyce-Shaikh, B.; Blumenschein, W.M.; McClanahan, T.K.; O’Shea, J.J.; Cua, D.J. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat. Immunol. 2009, 10, 314–324. [Google Scholar] [CrossRef]
- Guo, W.; Luo, C.; Wang, C.; Zhu, Y.; Wang, X.; Gao, X.; Yao, W. Protection against Th17 cells differentiation by an interleukin-23 receptor cytokine-binding homology region. PLoS ONE 2012, 7, e45625. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.; Cho, J. Interleukin-23/Th17 pathways and inflammatory bowel disease. Inflamm. Bowel Dis. 2009, 15, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Amre, D.K.; Mack, D.; Israel, D.; Morgan, K.; Lambrette, P.; Law, L.; Grimard, G.; Deslandres, C.; Krupoves, A.; Bucionis, V.; et al. Association between genetic variants in the IL-23R gene and early-onset Crohn’s disease: Results from a case-control and family-based study among Canadian children. Am. J. Gastroenterol. 2008, 103, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef]
- Sivanesan, D.; Beauchamp, C.; Quinou, C.; Lee, J.; Lesage, S.; Chemtob, S.; Rioux, J.D.; Michnick, S.W. IL23R (Interleukin 23 Receptor) Variants Protective against Inflammatory Bowel Diseases (IBD) Display Loss of Function due to Impaired Protein Stability and Intracellular Trafficking. J. Biol. Chem. 2016, 291, 8673–8685. [Google Scholar] [CrossRef]
- Krause-Kyora, B.; da Silva, N.A.; Kaplan, E.; Kolbe, D.; Archaeological Civilization Disease Consortium; Wohlers, I.; Busch, H.; Ellinghaus, D.; Caliebe, A.; Sezgin, E.; et al. Neolithic introgression of IL23R-related protection against chronic inflammatory bowel diseases in modern Europeans. EBioMedicine 2025, 113, 105591. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Jiang, H.; Chen, Z.; Lu, B.; Li, J.; Shen, X. Genetic association between IL23R rs11209026 and rs10889677 polymorphisms and risk of Crohn’s disease and ulcerative colitis: Evidence from 41 studies. Inflamm. Res. 2020, 69, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.R.; Han, D.Y.; Fraser, A.G.; Huebner, C.; Lam, W.J.; Morgan, A.R. IL23R and IL12B SNPs and Haplotypes Strongly Associate with Crohn’s Disease Risk in a New Zealand Population. Gastroenterol. Res. Pract. 2010, 2010, 539461. [Google Scholar] [CrossRef]
- Lerch-Bader, M.; Lundin, C.; Kim, H.; Nilsson, I.; von Heijne, G. Contribution of positively charged flanking residues to the insertion of transmembrane helices into the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2008, 105, 4127–4132. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.; Rueda, B.; López-Nevot, M.A.; Gómez-García, M.; Martín, J. Replication of an association between IL23R gene polymorphism with inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 2007, 5, 977–981.e2. [Google Scholar] [CrossRef]
- Guan, Q.; Ma, Y.; Aboud, L.; Weiss, C.R.; Qing, G.; Warrington, R.J.; Peng, Z. Targeting IL-23 by employing a p40 peptide-based vaccine ameliorates murine allergic skin and airway inflammation. Clin. Exp. Allergy 2012, 42, 1397–1405. [Google Scholar] [CrossRef]
- Pidasheva, S.; Trifari, S.; Phillips, A.; Hackney, J.A.; Ma, Y.; Smith, A.; Sohn, S.J.; Spits, H.; Little, R.D.; Behrens, T.W.; et al. Functional studies on the IBD susceptibility gene IL23R implicate reduced receptor function in the protective genetic variant R381Q. PLoS ONE 2011, 6, e25038. [Google Scholar] [CrossRef]
- Sarin, R.; Wu, X.; Abraham, C. Inflammatory disease protective R381Q IL23 receptor polymorphism results in decreased primary CD4+ and CD8+ human T-cell functional responses. Proc. Natl. Acad. Sci. USA 2011, 108, 9560–9565. [Google Scholar] [CrossRef]
- Di Meglio, P.; Di Cesare, A.; Laggner, U.; Chu, C.C.; Napolitano, L.; Villanova, F.; Tosi, I.; Capon, F.; Trembath, R.C.; Peris, K.; et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PLoS ONE 2011, 6, e17160. [Google Scholar] [CrossRef]
- Abdollahi, E.; Tavasolian, F.; Ghasemi, N.; Mirghanizadeh, S.A.; Azizi, M.; Ghoryani, M.; Samadi, M. Association between lower frequency of R381Q variant (rs11209026) in IL-23 receptor gene and increased risk of recurrent spontaneous abortion (RSA). J. Immunotoxicol. 2015, 12, 317–321. [Google Scholar] [CrossRef]
- Yu, R.Y.; Brazaitis, J.; Gallagher, G. The human IL-23 receptor rs11209026 A allele promotes the expression of a soluble IL-23R-encoding mRNA species. J. Immunol. 2015, 194, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, D.; Kawai, Y.; Kakuta, Y.; Naito, T.; Torisu, T.; Hirano, A.; Umero, J.; Fuyuno, Y.; Li, D.; Nakano, T.; et al. Genetic analysis of ulcerative colitis in Japanese individuals using population-specific SNP array. Inflamm. Bowel Dis. 2020, 26, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.D.; Choi, H.; Hong, M.; Yun, W.J.; Low, H.Q.; Haritunians, T.; Kim, K.J.; Park, S.H.; Lee, I.; Bang, S.Y.; et al. Identification of Ten Additional Susceptibility Loci for Ulcerative Colitis Through Immunochip Analysis in Koreans. Inflamm. Bowel Dis. 2016, 22, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Bin, C.; Zhirong, Z.; Xiaoqin, W.; Minhu, C.; Mei, L.; Xiang, G.; Baili, C.; Pinjin, H. Contribution of rs11465788 in IL23R gene to Crohn’s disease susceptibility and phenotype in Chinese population. J. Genet. 2009, 88, 191–196. [Google Scholar] [CrossRef]
- Ju, Y.S.; Kim, J.I.; Kim, S.; Hong, D.; Park, H.; Shin, J.Y.; Lee, S.; Lee, W.C.; Kim, S.; Yu, S.B.; et al. Extensive genomic and transcriptional diversity identified through massively parallel DNA and RNA sequencing of eighteen Korean individuals. Nat. Genet. 2011, 43, 745–752. [Google Scholar] [CrossRef]
- Miyake, Y.; Tanaka, K.; Nagata, C.; Furukawa, S.; Andoh, A.; Yokoyama, T.; Yoshimura, N.; Mori, K.; Ninomiya, T.; Yamamoto, Y.; et al. Case-control study of IL23R rs76418789 polymorphism, smoking, and ulcerative colitis in Japan. Cytokine 2024, 183, 156743. [Google Scholar] [CrossRef]
- Momozawa, Y.; Mni, M.; Nakamura, K.; Coppieters, W.; Almer, S.; Amininejad, L.; Cleynen, I.; Colombel, J.F.; de Rijk, P.; Dewit, O.; et al. Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nat. Genet. 2011, 43, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Ali, B.R.; Ben-Rebeh, I.; John, A.; Akawi, N.A.; Milhem, R.M.; Al-Shehhi, N.A.; Al-Ameri, M.M.; Al-Shamisi, S.A.; Al-Gazali, L. Endoplasmic reticulum quality control is involved in the mechanism of endoglin-mediated hereditary haemorrhagic telangiectasia. PLoS ONE 2011, 6, e26206. [Google Scholar] [CrossRef] [PubMed]
- Kizhakkedath, P.; Loregger, A.; John, A.; Bleijlevens, B.; Al-Blooshi, A.S.; Al-Hosani, A.H.; Al-Nuaimi, A.M.; Al-Gazali, L.; Zelcer, N.; Ali, B.R. Impaired trafficking of the very low density lipoprotein receptor caused by missense mutations associated with dysequilibrium syndrome. Biochim. Biophys. Acta 2014, 1843, 2871–2877. [Google Scholar] [CrossRef]
- Spoerri, L.; Vella, L.J.; Pham, C.L.L.; Barnham, K.J.; Cappai, R. The amyloid precursor protein copper binding domain histidine residues 149 and 151 mediate APP stability and metabolism. J. Biol. Chem. 2012, 287, 26840–26853. [Google Scholar] [CrossRef]
- Seidah, N.G.; Sadr, M.S.; Chrétien, M.; Mbikay, M. The multifaceted proprotein convertases: Their unique, redundant, complementary, and opposite functions. J. Biol. Chem. 2013, 288, 21473–21481. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G. The proprotein convertases, 20 years later. Methods Mol. Biol. 2011, 768, 23–57. [Google Scholar] [PubMed]
- Lu, Z.K.; Chen, Z.R.; Zhu, J.Y.; Xu, Y.; Hua, X. Analysis of the association of single nucleotide polymorphisms of interleukin-23 receptor (IL-23R) and inflammatory bowel disease in a Chinese Han cohort. Oncotarget 2016, 7, 67851–67856. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, M.; Goyette, P.; Boucher, G.; Lo, K.S.; Rivas, M.A.; Stevens, C.; Alikashani, A.; Ladouceur, M.; Ellinghaus, D.; Torkvist, L.; et al. Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet. 2013, 9, e1003723. [Google Scholar] [CrossRef] [PubMed]
- Heim, E.N.; Marston, J.L.; Federman, R.S.; Edwards, A.P.B.; Karabadzhak, A.G.; Petti, L.M.; Engelman, D.M.; DiMaio, D. Biologically active LIL proteins built with minimal chemical diversity. Proc. Natl. Acad. Sci. USA 2015, 112, E4717–E4725. [Google Scholar] [CrossRef]
- Zwiers, A.; Kraal, L.; van de Pouw Kraan, T.C.T.M.; Wurdinger, T.; Bouma, G.; Kraal, G. Cutting edge: A variant of the IL-23R gene associated with inflammatory bowel disease induces loss of microRNA regulation and enhanced protein production. J. Immunol. 2012, 188, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Hayatbakhsh, M.M.; Zahedi, M.J.; Shafiepour, M.; Nikpoor, A.R.; Mohammadi, M. IL-23 receptor gene rs7517847 and rs1004819 SNPs in ulcerative colitis. Iran. J. Immunol. 2012, 9, 128–135. [Google Scholar]
- Glas, J.; Seiderer, J.; Wetzke, M.; Konrad, A.; Török, H.P.; Schmechel, S.; Tonenchi, L.; Grassl, C.; Dambacher, J.; Pfennig, S.; et al. rs1004819 is the main disease-associated IL23R variant in German Crohn’s disease patients: Combined analysis of IL23R, CARD15, and OCTN1/2 variants. PLoS ONE 2007, 2, e819. [Google Scholar] [CrossRef]
- Magyari, L.; Varszegi, D.; Sarlos, P.; Jaromi, L.; Melegh, B.I.; Duga, B.; Kisfali, P.; Kovesdi, E.; Matyas, P.; Szabo, A.; et al. Marked differences of haplotype tagging SNP distribution, linkage, and haplotype profile of IL23 receptor gene in Roma and Hungarian population samples. Cytokine 2014, 65, 148–152. [Google Scholar] [CrossRef]
- Cătană, C.S.; Berindan Neagoe, I.; Cozma, V.; Magdaş, C.; Tăbăran, F.; Dumitraşcu, D.L. Contribution of the IL-17/IL-23 axis to the pathogenesis of inflammatory bowel disease. World J. Gastroenterol. 2015, 21, 5823–5830. [Google Scholar] [CrossRef]
- Verstockt, B.; Salas, A.; Sands, B.E.; Abraham, C.; Leibovitzh, H.; Neurath, M.F.; Casteeile, N.V. Alimentiv Translational Research Consortium (ATRC) IL-12 and IL-23 pathway inhibition in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 433–446. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, F.; Peyrin-Biroulet, L.; Danese, S. Ustekinumab in Crohn’s Disease: New Data for Positioning in Treatment Algorithm. J. Crohns Colitis 2022, 16 (Suppl. 2), ii30–ii41. [Google Scholar] [CrossRef] [PubMed]
- Almradi, A.; Hanzel, J.; Sedano, R.; Parker, C.E.; Feagan, B.G.; Ma, C.; Jairath, V. Clinical Trials of IL-12/IL-23 Inhibitors in Inflammatory Bowel Disease. BioDrugs 2020, 34, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; D’Cunha, R.; Winzenborg, I.; Veldman, G.; Pivorunas, V.; Wallace, K. Risankizumab: Mechanism of action, clinical and translational science. Clin. Transl. Sci. 2024, 17, e13706. [Google Scholar] [CrossRef]
- Lusetti, F.; D’Amico, F.; Allocca, M.; Furfaro, F.; Zilli, A.; Fiorino, G. Positioning risankizumab in the treatment algorithm of moderate-to-severe Crohn’s disease. Immunotherapy 2024, 16, 581–595. [Google Scholar] [CrossRef]
- Caron, B.; Habert, A.; Bonsack, O.; Camara, H.; Jeanbert, E.; Parigi, T.L.; Pagini, T.L.; Radice, S.; Peyrin-Biroulet, L.; Danese, S.; et al. Difficult-to-treat inflammatory bowel disease: Effectiveness and safety of 4th and 5th lines of treatment. United Eur. Gastroenterol. J. 2024, 12, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Risankizumab Induction Therapy in Patients With Moderately to Severely Active Ulcerative Colitis: Efficacy and Safety in the Randomized Phase 3 INSPIRE Study. Gastroenterol. Hepatol. 2023, 19 (Suppl. 9), 9–10.
- Bourgonje, A.R.; Ungaro, R.C.; Mehandru, S.; Colombel, J.F. Targeting the Interleukin 23 Pathway in Inflammatory Bowel Disease. Gastroenterology 2025, 168, 29–52.e3. [Google Scholar] [CrossRef]
- D’Haens, G.; Dubinsky, M.; Kobayashi, T.; Irving, P.M.; Howaldt, S.; Pokrotnieks, J.; Krueger, K.; Laskowski, J.; Li, X.; Lissoos, T.; et al. Mirikizumab as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2023, 388, 2444–2455. [Google Scholar] [CrossRef]
- Sands, B.E.; Peyrin-Biroulet, L.; Kierkus, J.; Higgins, P.D.R.; Fischer, M.; Jairath, V.; Hirai, F.; D’Haens, G.; Belin, R.M.; Miller, D.; et al. Efficacy and Safety of Mirikizumab in a Randomized Phase 2 Study of Patients With Crohn’s Disease. Gastroenterology 2022, 162, 495–508. [Google Scholar] [CrossRef]
- Massironi, S.; Furfaro, F.; Bencardino, S.; Allocca, M.; Danese, S. Immunity in digestive diseases: New drugs for inflammatory bowel disease treatment—Insights from Phase II and III trials. J. Gastroenterol. 2024, 59, 761. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Panaccione, R.; Feagan, B.G.; Afzali, A.; Rubin, D.T.; Sands, B.E.; Reinisch, W.; Panes, J.; Sahoo, A.; Terry, N.A.; et al. Efficacy and safety of 48 weeks of guselkumab for patients with Crohn’s disease: Maintenance results from the phase 2, randomised, double-blind GALAXI-1 trial. Lancet Gastroenterol. Hepatol. 2024, 9, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Peyrin-Biroulet, L.; Allegretti, J.R.; Rubin, D.T.; Bressler, B.; Germinaro, M.; Huang, K.H.G.; Shipitofsky, N.; Zhang, H.; Wilson, R.; Han, C.; et al. Guselkumab in Patients With Moderately to Severely Active Ulcerative Colitis: QUASAR Phase 2b Induction Study. Gastroenterology 2023, 165, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Beaton, A.; Duncan, E.A.; Mercier, A.K.; Neisen, J.; Seth, H.; Zetterstrand, S.; Sands, B.E. Long-term safety of brazikumab in the open-label period of a randomized phase 2a study of patients with Crohn’s disease. BMC Gastroenterol. 2023, 23, 451. [Google Scholar] [CrossRef]
- Fourie, A.M.; Cheng, X.; Chang, L.; Greving, C.; Li, X.; Knight, B.; Polidori, D.; Patrick, A.; Bains, T.; Steele, R.; et al. JNJ-77242113, a highly potent, selective peptide targeting the IL-23 receptor, provides robust IL-23 pathway inhibition upon oral dosing in rats and humans. Sci. Rep. 2024, 14, 17515. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.B.; Gasparetto, M. Novel pharmacological developments in the management of paediatric inflammatory bowel disease: Time for guideline update—A narrative review. J. Paediatr. Child. Health 2024, 60, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, E.; Siegmund, B. [Pregnancy and breastfeeding in Crohn’s disease]. Dtsch. Med. Wochenschr. 2024, 149, 46–56. [Google Scholar]
- Honap, S.; Meade, S.; Ibraheim, H.; Irving, P.M.; Jones, M.P.; Samaan, M.A. Effectiveness and Safety of Ustekinumab in Inflammatory Bowel Disease: A Systematic Review and Meta-Analysis. Dig Dis Sci. 2022, 67, 1018–1035. [Google Scholar] [CrossRef]
- Peyrin-Biroulet, L.; Vermeire, S.; D’Heans, G.; Panes, J.; Dignass, A.; Margo, F.; Nazar, M.; Le Bars, M.; Lahaye, M.; Ni, L.; et al. Clinical trial: Clinical and endoscopic outcomes with ustekinumab in patients with Crohn’s disease: Results from the long-term extension period of STARDUST. Aliment. Pharmacol. Ther. 2024, 59, 175–185. [Google Scholar] [CrossRef]
- Sands, B.E.; D’Haens, G.; Clemow, D.B.; Irving, P.M.; Johns, J.T.; Gibble, T.H.; Abreu, M.T.; Lee, S.; Hisamatsu, T.; Kobayashi, T.; et al. Two-Year Efficacy and Safety of Mirikizumab Following 104 Weeks of Continuous Treatment for Ulcerative Colitis: Results From the LUCENT-3 Open-Label Extension Study. Inflamm. Bowel Dis. 2024, 30, 2245–2258. [Google Scholar] [CrossRef]
- Bertani, L.; Antonioli, L.; Fornili, M.; D’Antogiovanni, V.; Ceccarelli, L.; Carmisciano, L.; Benvenuti, L.; Mumolo, M.G.; Bottari, A.; Pardi, V.; et al. Baseline Assessment of Serum Cytokines Predicts Clinical and Endoscopic Response to Ustekinumab in Patients With Crohn’s Disease: A Prospective Pilot Study. Inflamm. Bowel Dis. 2024, 30, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.C.L.; Dulai, P.S.; Marshall, J.K.; Jairath, V.; Reinisch, W.; Narula, N. Improvement in serum eosinophilia is observed in clinical responders to ustekinumab but not adalimumab in inflammatory bowel disease. J. Crohns Colitis 2025, 19, jjaf006. [Google Scholar] [CrossRef] [PubMed]
- Rostami, M.; Haidari, K.; Shahbazi, M. Genetically Engineered Adipose Mesenchymal Stem Cells Using HIV-Based Lentiviral Vectors as Gene Therapy for Autoimmune Diseases. Cell Reprogram. 2018, 20, 337–346. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastras, P.; Aggeletopoulou, I.; Papantoniou, K.; Triantos, C. Targeting the IL-23 Receptor Gene: A Promising Approach in Inflammatory Bowel Disease Treatment. Int. J. Mol. Sci. 2025, 26, 4775. https://doi.org/10.3390/ijms26104775
Pastras P, Aggeletopoulou I, Papantoniou K, Triantos C. Targeting the IL-23 Receptor Gene: A Promising Approach in Inflammatory Bowel Disease Treatment. International Journal of Molecular Sciences. 2025; 26(10):4775. https://doi.org/10.3390/ijms26104775
Chicago/Turabian StylePastras, Ploutarchos, Ioanna Aggeletopoulou, Konstantinos Papantoniou, and Christos Triantos. 2025. "Targeting the IL-23 Receptor Gene: A Promising Approach in Inflammatory Bowel Disease Treatment" International Journal of Molecular Sciences 26, no. 10: 4775. https://doi.org/10.3390/ijms26104775
APA StylePastras, P., Aggeletopoulou, I., Papantoniou, K., & Triantos, C. (2025). Targeting the IL-23 Receptor Gene: A Promising Approach in Inflammatory Bowel Disease Treatment. International Journal of Molecular Sciences, 26(10), 4775. https://doi.org/10.3390/ijms26104775