
A Web of Challenges: The Therapeutic Struggle to Target NETs in Disease



Abstract
1. Introduction
2. NETs in Immunity and Pathology
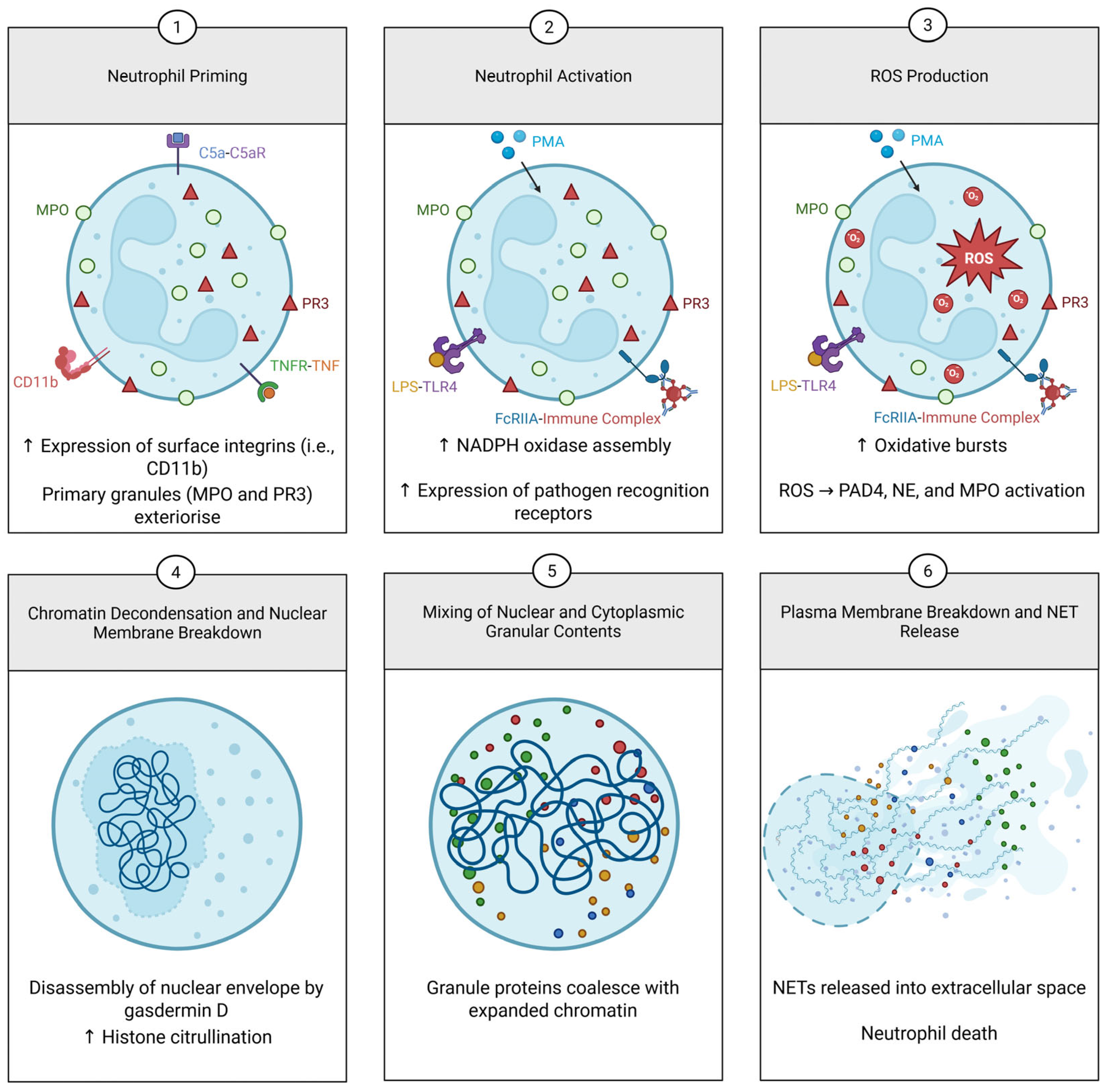
3. Mechanisms of NET Formation
3.1. Types of NETs
3.1.1. Lytic NETosis
3.1.2. Vital NETs
3.1.3. Mitochondrial NETs
3.2. Signalling Pathways of NET Formation by Stimulants
3.2.1. Phorbol 12-Myristate 13-Acetate (PMA) Induction
3.2.2. Lipopolysaccharide (LPS) Induction
3.2.3. Complement Induction
3.2.4. Calcium Induction
3.2.5. Antigen-Antibody Complex Induction
3.2.6. Osteopontin Induction
3.2.7. Hepoxilin A3 Induction
4. Biochemical Modifications to NETs
5. NET-Targeted Therapies
5.1. Repurposing for NET Inhibition
5.1.1. Early-Stage Inhibitors
Eculizumab
Tocilizumab
Metformin
Fostamatinib
SkQ1
5.1.2. Late-Stage Inhibitors
Colchicine
BB-Cl-Amidine
GSK484
BAY 85-8501
AZD9668
CIT-013
Disulfiram
NINJ1 Monoclonal Antibodies
5.2. Enhancing NET Clearance—DNase
5.3. Other Potential Targets for NET Inhibition
5.3.1. Pentraxin 3
5.3.2. Hepoxilin A3
6. Challenges in Translation
6.1. Limitations of NET Inhibitor Laboratory Experiments
6.1.1. Animal Models
6.1.2. In Vitro Experiments
6.2. NETs Are a Biological Conundrum
6.3. Bioavailability
6.4. Study Design Issues
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NETs | Neutrophil extracellular traps |
| TNF | Tumour necrosis factor |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| IL | Interleukin |
| PMA | Phorbol 12-myristate 13-acetate |
| dsDNA | Double-stranded DNA |
| NE | Neutrophil elastase |
| MPO | Myeloperoxidase |
| PR3 | Proteinase 3 |
| SLE | Systemic lupus erythematosus |
| RA | Rheumatoid arthritis |
| SVV | Small vessel vasculitis |
| ANCA | Anti-neutrophil cytoplasmic antibodies |
| AAV | ANCA-associated vasculitis |
| C5aR | C5a receptor |
| ROS | Reactive oxygen species |
| NOX2 | NADPH |
| LPS | Lipopolysaccharide |
| RNP ICs | Ribonucleoprotein immune complexes |
| PKC | Protein kinase C |
| TLR | Toll-like receptor |
| JNK | c-Jun-N-terminal kinase |
| Syk | Spleen tyrosine kinase |
| mPTP | Mitochondrial permeability transition pore |
| FcγRIIa | Fc gamma receptor IIa |
| HXA3 | Hepoxilin A3 |
| PNH | Paroxysmal nocturnal haemoglobulinuria |
| pDCs | Plasmacytoid dendritic cells |
| CRC | Colorectal cancer |
| COPD | Chronic obstructive pulmonary disease |
| NINJ1 | Ninjurin-1 |
| I/R | Ischemia reperfusion |
| IV | Intravenous |
| 12S-LOX | 12S-lupoxygenase |
| cfDNA | Cell-free DNA |
References
- Basu, S.; Hodgson, G.; Katz, M.; Dunn, A.R. Evaluation of role of G-CSF in the production, survival, and release of neutrophils from bone marrow into circulation. Blood 2002, 100, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Borregaard, N.; Wynn, T.A. Phenotypic and functional plasticity of cells of innate immunity: Macrophages, mast cells and neutrophils. Nat. Immunol. 2011, 12, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hossain, M.; Thanabalasuriar, A.; Gunzer, M.; Meininger, C.; Kubes, P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science 2017, 358, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Noseykina, E.M.; Schepetkin, I.A.; Atochin, D.N. Molecular Mechanisms for Regulation of Neutrophil Apoptosis under Normal and Pathological Conditions. J. Evol. Biochem. Physiol. 2021, 57, 429–450. [Google Scholar] [CrossRef]
- Schultze, M. Ein heizbarer Objecttisch und seine Verwendung bei Untersuchungen des Blutes. Arch. Für Mikrosk. Anatomie 1865, 1, 1–42. [Google Scholar] [CrossRef]
- Takeda, Y.; Watanabe, H.; Yonehara, S.; Yamashita, T.; Saito, S.; Sendo, F. Rapid acceleration of neutrophil apoptosis by tumor necrosis factor-alpha. Int. Immunol. 1993, 5, 691–694. [Google Scholar] [CrossRef]
- Takei, H.; Araki, A.; Watanabe, H.; Ichinose, A.; Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Saitoh, T.; Komano, J.; Saitoh, Y.; Misawa, T.; Takahama, M.; Kozaki, T.; Uehata, T.; Iwasaki, H.; Omori, H.; Yamaoka, S.; et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 2012, 12, 109–116. [Google Scholar] [CrossRef]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef]
- Guimarães-Costa, A.B.; Nascimento, M.T.; Froment, G.S.; Soares, R.P.; Morgado, F.N.; Conceição-Silva, F.; Saraiva, E.M. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc. Natl. Acad. Sci. USA 2009, 106, 6748–6753. [Google Scholar] [CrossRef] [PubMed]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Tsourouktsoglou, T.D.; Branzk, N.; Wang, Q.; Reincke, S.; Herbst, S.; Gutierrez, M.; Papayannopoulos, V. Reactive Oxygen Species Localization Programs Inflammation to Clear Microbes of Different Size. Immunity 2017, 46, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Biermann, M.H.; Brauner, J.M.; Liu, Y.; Zhao, Y.; Herrmann, M. New Insights into Neutrophil Extracellular Traps: Mechanisms of Formation and Role in Inflammation. Front. Immunol. 2016, 7, 302. [Google Scholar] [CrossRef]
- Miller, B.F.; Abrams, R.; Dorfman, A.; Klein, M. Antibacterial Properties of Protamine and Histone. Science 1942, 96, 428–430. [Google Scholar] [CrossRef]
- Hirsch, J.G. Bactericidal action of histone. J. Exp. Med. 1958, 108, 925–944. [Google Scholar] [CrossRef]
- Doolin, T.; Amir, H.M.; Duong, L.; Rosenzweig, R.; Urban, L.A.; Bosch, M.; Pol, A.; Gross, S.P.; Siryaporn, A. Mammalian histones facilitate antimicrobial synergy by disrupting the bacterial proton gradient and chromosome organization. Nat. Commun. 2020, 11, 3888. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Monestier, M.; Esmon, N.L.; Esmon, C.T. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J. Immunol. 2011, 187, 2626–2631. [Google Scholar] [CrossRef]
- Schultz, J.; Kaminker, K. Myeloperoxidase of the leucocyte of normal human blood. I. Content and localization. Arch. Biochem. Biophys. 1962, 96, 465–467. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J. Myeloperoxidase: Friend and foe. J. Leukoc. Biol. 2005, 77, 598–625. [Google Scholar] [CrossRef] [PubMed]
- Belaaouaj, A.; Kim, K.S.; Shapiro, S.D. Degradation of outer membrane protein A in Escherichia coli killing by neutrophil elastase. Science 2000, 289, 1185–1188. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Boado, Y.S.; Espinola, M.; Bahr, S.; Belaaouaj, A. Neutrophil serine proteinases cleave bacterial flagellin, abrogating its host response-inducing activity. J. Immunol. 2004, 172, 509–515. [Google Scholar] [CrossRef]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef]
- Wirthmueller, U.; Dewald, B.; Thelen, M.; Schafer, M.K.; Stover, C.; Whaley, K.; North, J.; Eggleton, P.; Reid, K.B.; Schwaeble, W.J. Properdin, a positive regulator of complement activation, is released from secondary granules of stimulated peripheral blood neutrophils. J. Immunol. 1997, 158, 4444–4451. [Google Scholar] [CrossRef]
- Wang, H.; Wang, C.; Zhao, M.H.; Chen, M. Neutrophil extracellular traps can activate alternative complement pathways. Clin. Exp. Immunol. 2015, 181, 518–527. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Dassler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Arita, K.; Hashimoto, H.; Shimizu, T.; Nakashima, K.; Yamada, M.; Sato, M. Structural basis for Ca(2+)-induced activation of human PAD4. Nat. Struct. Mol. Biol. 2004, 11, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Leshner, M.; Wang, S.; Lewis, C.; Zheng, H.; Chen, X.A.; Santy, L.; Wang, Y. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front. Immunol. 2012, 3, 307. [Google Scholar] [CrossRef] [PubMed]
- Kambara, H.; Liu, F.; Zhang, X.; Liu, P.; Bajrami, B.; Teng, Y.; Zhao, L.; Zhou, S.; Yu, H.; Zhou, W.; et al. Gasdermin D Exerts Anti-inflammatory Effects by Promoting Neutrophil Death. Cell Rep. 2018, 22, 2924–2936. [Google Scholar] [CrossRef] [PubMed]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [PubMed]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef]
- Malawista, S.E.; Van Blaricom, G.; Breitenstein, M.G. Cryopreservable neutrophil surrogates. Stored cytoplasts from human polymorphonuclear leukocytes retain chemotactic, phagocytic, and microbicidal function. J. Clin. Investig. 1989, 83, 728–732. [Google Scholar] [CrossRef]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef]
- Remijsen, Q.; Vanden Berghe, T.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; De Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef]
- Wolfson, M.; McPhail, L.C.; Nasrallah, V.N.; Snyderman, R. Phorbol myristate acetate mediates redistribution of protein kinase C in human neutrophils: Potential role in the activation of the respiratory burst enzyme. J. Immunol. 1985, 135, 2057–2062. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Farahvash, A.; Douda, D.N.; Licht, J.C.; Grasemann, H.; Sweezey, N.; Palaniyar, N. JNK Activation Turns on LPS- and Gram-Negative Bacteria-Induced NADPH Oxidase-Dependent Suicidal NETosis. Sci. Rep. 2017, 7, 3409. [Google Scholar] [CrossRef] [PubMed]
- Sogkas, G.; Vogtle, T.; Rau, E.; Gewecke, B.; Stegner, D.; Schmidt, R.E.; Nieswandt, B.; Gessner, J.E. Orai1 controls C5a-induced neutrophil recruitment in inflammation. Eur. J. Immunol. 2015, 45, 2143–2153. [Google Scholar] [CrossRef] [PubMed]
- Mocsai, A.; Zhang, H.; Jakus, Z.; Kitaura, J.; Kawakami, T.; Lowell, C.A. G-protein-coupled receptor signaling in Syk-deficient neutrophils and mast cells. Blood 2003, 101, 4155–4163. [Google Scholar] [CrossRef]
- Dedkova, E.N.; Sigova, A.A.; Zinchenko, V.P. Mechanism of action of calcium ionophores on intact cells: Ionophore-resistant cells. Membr. Cell Biol. 2000, 13, 357–368. [Google Scholar]
- Vorobjeva, N.; Galkin, I.; Pletjushkina, O.; Golyshev, S.; Zinovkin, R.; Prikhodko, A.; Pinegin, V.; Kondratenko, I.; Pinegin, B.; Chernyak, B. Mitochondrial permeability transition pore is involved in oxidative burst and NETosis of human neutrophils. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165664. [Google Scholar] [CrossRef]
- Kent, A.C.; El Baradie, K.B.Y.; Hamrick, M.W. Targeting the Mitochondrial Permeability Transition Pore to Prevent Age-Associated Cell Damage and Neurodegeneration. Oxidative Med. Cell Longev. 2021, 2021, 6626484. [Google Scholar] [CrossRef]
- Junker, F.; Gordon, J.; Qureshi, O. Fc Gamma Receptors and Their Role in Antigen Uptake, Presentation, and T Cell Activation. Front. Immunol. 2020, 11, 1393. [Google Scholar] [CrossRef]
- Park, J.W.; Hoyal, C.R.; Benna, J.E.; Babior, B.M. Kinase-dependent activation of the leukocyte NADPH oxidase in a cell-free system. Phosphorylation of membranes and p47(PHOX) during oxidase activation. J. Biol. Chem. 1997, 272, 11035–11043. [Google Scholar] [CrossRef]
- Hoyal, C.R.; Gutierrez, A.; Young, B.M.; Catz, S.D.; Lin, J.H.; Tsichlis, P.N.; Babior, B.M. Modulation of p47PHOX activity by site-specific phosphorylation: Akt-dependent activation of the NADPH oxidase. Proc. Natl. Acad. Sci. USA 2003, 100, 5130–5135. [Google Scholar] [CrossRef]
- Akhiani, A.A.; Hallner, A.; Kiffin, R.; Aydin, E.; Werlenius, O.; Aurelius, J.; Martner, A.; Thoren, F.B.; Hellstrand, K. Idelalisib Rescues Natural Killer Cells from Monocyte-Induced Immunosuppression by Inhibiting NOX2-Derived Reactive Oxygen Species. Cancer Immunol. Res. 2020, 8, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Tatsiy, O.; McDonald, P.P. Physiological Stimuli Induce PAD4-Dependent, ROS-Independent NETosis, With Early and Late Events Controlled by Discrete Signaling Pathways. Front. Immunol. 2018, 9, 2036. [Google Scholar] [CrossRef] [PubMed]
- Nishimichi, N.; Higashikawa, F.; Kinoh, H.H.; Tateishi, Y.; Matsuda, H.; Yokosaki, Y. Polymeric osteopontin employs integrin alpha9beta1 as a receptor and attracts neutrophils by presenting a de novo binding site. J. Biol. Chem. 2009, 284, 14769–14776. [Google Scholar] [CrossRef] [PubMed]
- Bertolotto, M.; Verzola, D.; Contini, P.; de Totero, D.; Tirandi, A.; Ramoni, D.; Ministrini, S.; Giacobbe, D.R.; Bonaventura, A.; Vecchie, A.; et al. Osteopontin is associated with neutrophil extracellular trap formation in elderly patients with severe sepsis. Eur. J. Clin. Investig. 2024, 54, e14159. [Google Scholar] [CrossRef]
- Hurley, B.P.; Siccardi, D.; Mrsny, R.J.; McCormick, B.A. Polymorphonuclear cell transmigration induced by Pseudomonas aeruginosa requires the eicosanoid hepoxilin A3. J. Immunol. 2004, 173, 5712–5720. [Google Scholar] [CrossRef]
- Dho, S.; Grinstein, S.; Corey, E.J.; Su, W.G.; Pace-Asciak, C.R. Hepoxilin A3 induces changes in cytosolic calcium, intracellular pH and membrane potential in human neutrophils. Biochem. J. 1990, 266, 63–68. [Google Scholar] [CrossRef]
- Douda, D.N.; Grasemann, H.; Pace-Asciak, C.; Palaniyar, N. A lipid mediator hepoxilin A3 is a natural inducer of neutrophil extracellular traps in human neutrophils. Mediat. Inflamm. 2015, 2015, 520871. [Google Scholar] [CrossRef]
- Gehrke, N.; Mertens, C.; Zillinger, T.; Wenzel, J.; Bald, T.; Zahn, S.; Tuting, T.; Hartmann, G.; Barchet, W. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 2013, 39, 482–495. [Google Scholar] [CrossRef]
- Yan, M.; Gu, Y.; Sun, H.; Ge, Q. Neutrophil extracellular traps in tumor progression and immunotherapy. Front. Immunol. 2023, 14, 1135086. [Google Scholar] [CrossRef]
- Lazzaretto, B.; Fadeel, B. Intra- and Extracellular Degradation of Neutrophil Extracellular Traps by Macrophages and Dendritic Cells. J. Immunol. 2019, 203, 2276–2290. [Google Scholar] [CrossRef]
- Pastorek, M.; Konecna, B.; Janko, J.; Janovicova, L.; Podracka, L.; Zahumensky, J.; Stenova, E.; Dubrava, M.; Hodosy, J.; Vlkova, B.; et al. Mitochondria-induced formation of neutrophil extracellular traps is enhanced in the elderly via Toll-like receptor 9. J. Leukoc. Biol. 2023, 114, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Tangsombatvisit, S.; Rosenberg, J.M.; Mandelbaum, G.; Gillespie, E.C.; Gozani, O.P.; Alizadeh, A.A.; Utz, P.J. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res. Ther. 2012, 14, R25. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, E.; Hofstra, J.; Berden, J.; Herrmann, M.; Dieker, J.; van der Vlag, J. Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clin. Exp. Immunol. 2015, 179, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Zeerleder, S.; van Bijnen, S.; Wouters, D.; van Mierlo, G.J.; Muus, P. Neutrophil Extracellular Trap Formation In PNH Patients With and Without a History Of Thrombosis—Effects Of Eculizumab. Blood 2013, 122, 1235. [Google Scholar] [CrossRef]
- Strich, J.R.; Ramos-Benitez, M.J.; Randazzo, D.; Stein, S.R.; Babyak, A.; Davey, R.T.; Suffredini, A.F.; Childs, R.W.; Chertow, D.S. Fostamatinib Inhibits Neutrophils Extracellular Traps Induced by COVID-19 Patient Plasma: A Potential Therapeutic. J. Infect. Dis. 2021, 223, 981–984. [Google Scholar] [CrossRef]
- Li, Y.W.; Chen, S.X.; Yang, Y.; Zhang, Z.H.; Zhou, W.B.; Huang, Y.N.; Huang, Z.Q.; He, J.Q.; Chen, T.F.; Wang, J.F.; et al. Colchicine Inhibits NETs and Alleviates Cardiac Remodeling after Acute Myocardial Infarction. Cardiovasc. Drugs Ther. 2024, 38, 31–41. [Google Scholar] [CrossRef]
- Knight, J.S.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Smith, C.K.; Hodgin, J.B.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann. Rheum. Dis. 2015, 74, 2199–2206. [Google Scholar] [CrossRef]
- Lewis, H.D.; Liddle, J.; Coote, J.E.; Atkinson, S.J.; Barker, M.D.; Bax, B.D.; Bicker, K.L.; Bingham, R.P.; Campbell, M.; Chen, Y.H.; et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 2015, 11, 189–191. [Google Scholar] [CrossRef]
- Chirivi, R.G.S.; van Rosmalen, J.W.G.; van der Linden, M.; Euler, M.; Schmets, G.; Bogatkevich, G.; Kambas, K.; Hahn, J.; Braster, Q.; Soehnlein, O.; et al. Therapeutic ACPA inhibits NET formation: A potential therapy for neutrophil-mediated inflammatory diseases. Cell Mol. Immunol. 2021, 18, 1528–1544. [Google Scholar] [CrossRef]
- Adrover, J.M.; Carrau, L.; Dassler-Plenker, J.; Bram, Y.; Chandar, V.; Houghton, S.; Redmond, D.; Merrill, J.R.; Shevik, M.; tenOever, B.R.; et al. Disulfiram inhibits neutrophil extracellular trap formation and protects rodents from acute lung injury and SARS-CoV-2 infection. JCI Insight 2022, 7, e157342. [Google Scholar] [CrossRef]
- Lin, Y.; Yuan, Y.; Ye, K.; Chen, Z.; Wang, Y.; Li, G.; Song, Y.; Chen, H.; Ma, H.; Xu, Y. NINJ1-mediated Macrophage Plasma Membrane Rupture and Neutrophil Extracellular Trap Formation Contribute to Oxalate Nephropathy. Nephrol. Dial. Transplant. 2024, 40, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xie, T.; Sun, S.; Wang, K.; Liu, B.; Wu, X.; Ding, W. DNase-1 Treatment Exerts Protective Effects in a Rat Model of Intestinal Ischemia-Reperfusion Injury. Sci. Rep. 2018, 8, 17788. [Google Scholar] [CrossRef] [PubMed]
- Kindberg, K.M.; Broch, K.; Andersen, G.O.; Anstensrud, A.K.; Akra, S.; Woxholt, S.; Tollefsen, I.M.; Ueland, T.; Amundsen, B.H.; Klow, N.E.; et al. Neutrophil Extracellular Traps in ST-Segment Elevation Myocardial Infarction: Reduced by Tocilizumab and Associated With Infarct Size. JACC Adv. 2024, 3, 101193. [Google Scholar] [CrossRef] [PubMed]
- Menegazzo, L.; Scattolini, V.; Cappellari, R.; Bonora, B.M.; Albiero, M.; Bortolozzi, M.; Romanato, F.; Ceolotto, G.; Vigili de Kreutzeberg, S.; Avogaro, A.; et al. The antidiabetic drug metformin blunts NETosis in vitro and reduces circulating NETosis biomarkers in vivo. Acta Diabetol. 2018, 55, 593–601. [Google Scholar] [CrossRef]
- Stockley, R.; De Soyza, A.; Gunawardena, K.; Perrett, J.; Forsman-Semb, K.; Entwistle, N.; Snell, N. Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir. Med. 2013, 107, 524–533. [Google Scholar] [CrossRef]
- Elborn, J.S.; Perrett, J.; Forsman-Semb, K.; Marks-Konczalik, J.; Gunawardena, K.; Entwistle, N. Efficacy, safety and effect on biomarkers of AZD9668 in cystic fibrosis. Eur. Respir. J. 2012, 40, 969–976. [Google Scholar] [CrossRef]
- Vogelmeier, C.; Aquino, T.O.; O’Brien, C.D.; Perrett, J.; Gunawardena, K.A. A randomised, placebo-controlled, dose-finding study of AZD9668, an oral inhibitor of neutrophil elastase, in patients with chronic obstructive pulmonary disease treated with tiotropium. COPD 2012, 9, 111–120. [Google Scholar] [CrossRef]
- Watz, H.; Nagelschmitz, J.; Kirsten, A.; Pedersen, F.; van der Mey, D.; Schwers, S.; Bandel, T.J.; Rabe, K.F. Safety and efficacy of the human neutrophil elastase inhibitor BAY 85-8501 for the treatment of non-cystic fibrosis bronchiectasis: A randomized controlled trial. Pulm. Pharmacol. Ther. 2019, 56, 86–93. [Google Scholar] [CrossRef]
- Porter, J.C.; Inshaw, J.; Solis, V.J.; Denneny, E.; Evans, R.; Temkin, M.I.; De Vasconcelos, N.; Aramburu, I.V.; Hoving, D.; Basire, D.; et al. Anti-inflammatory therapy with nebulized dornase alfa for severe COVID-19 pneumonia: A randomized unblinded trial. eLife 2024, 12, RP87030. [Google Scholar] [CrossRef]
- Cofiell, R.; Kukreja, A.; Bedard, K.; Yan, Y.; Mickle, A.P.; Ogawa, M.; Bedrosian, C.L.; Faas, S.J. Eculizumab reduces complement activation, inflammation, endothelial damage, thrombosis, and renal injury markers in aHUS. Blood 2015, 125, 3253–3262. [Google Scholar] [CrossRef]
- Joshi, M.B.; Lad, A.; Bharath Prasad, A.S.; Balakrishnan, A.; Ramachandra, L.; Satyamoorthy, K. High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Lett. 2013, 587, 2241–2246. [Google Scholar] [CrossRef] [PubMed]
- Finzel, S.; Kraus, S.; Figueiredo, C.P.; Regensburger, A.; Kocijan, R.; Rech, J.; Schett, G. Comparison of the effects of tocilizumab monotherapy and adalimumab in combination with methotrexate on bone erosion repair in rheumatoid arthritis. Ann. Rheum. Dis. 2019, 78, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Klearman, M.; Collinson, N. Trial of Tocilizumab in Giant-Cell Arteritis. N. Engl. J. Med. 2017, 377, 1494–1495. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, K.A.; Valentine, R.J.; Ruderman, N.B.; Saha, A.K. AMPK activation: A therapeutic target for type 2 diabetes? Diabetes Metab. Syndr. Obes. 2014, 7, 241–253. [Google Scholar] [CrossRef]
- Cesta, M.C.; Zippoli, M.; Marsiglia, C.; Gavioli, E.M.; Cremonesi, G.; Khan, A.; Mantelli, F.; Allegretti, M.; Balk, R. Neutrophil activation and neutrophil extracellular traps (NETs) in COVID-19 ARDS and immunothrombosis. Eur. J. Immunol. 2023, 53, e2250010. [Google Scholar] [CrossRef]
- Ackermann, M.; Anders, H.J.; Bilyy, R.; Bowlin, G.L.; Daniel, C.; De Lorenzo, R.; Egeblad, M.; Henneck, T.; Hidalgo, A.; Hoffmann, M.; et al. Patients with COVID-19: In the dark-NETs of neutrophils. Cell Death Differ. 2021, 28, 3125–3139. [Google Scholar] [CrossRef]
- Reis, G.; Dos Santos Moreira Silva, E.A.; Medeiros Silva, D.C.; Thabane, L.; Cruz Milagres, A.; Ferreira, T.S.; Quirino Dos Santos, C.V.; de Figueiredo Neto, A.D.; Diniz Callegari, E.; Monteiro Savassi, L.C.; et al. Effect of early treatment with metformin on risk of emergency care and hospitalization among patients with COVID-19: The TOGETHER randomized platform clinical trial. Lancet Reg. Health Am. 2022, 6, 100142. [Google Scholar] [CrossRef]
- Kow, C.S.; Hasan, S.S. Mortality risk with preadmission metformin use in patients with COVID-19 and diabetes: A meta-analysis. J. Med. Virol. 2021, 93, 695–697. [Google Scholar] [CrossRef]
- Carmona-Rivera, C.; Kaplan, M.J. Detection of SLE antigens in neutrophil extracellular traps (NETs). Methods Mol. Biol. 2014, 1134, 151–161. [Google Scholar] [CrossRef]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra19. [Google Scholar] [CrossRef]
- Wang, H.; Li, T.; Chen, S.; Gu, Y.; Ye, S. Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-of-Concept Trial of Metformin. Arthritis Rheumatol. 2015, 67, 3190–3200. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.P.; Shotwell, M.S.; Strich, J.R.; Gibbs, K.W.; de Wit, M.; Files, D.C.; Harkins, M.; Hudock, K.; Merck, L.H.; Moskowitz, A.; et al. Fostamatinib for Hospitalized Adults with COVID-19 and Hypoxemia: A Randomized Clinical Trial. JAMA Netw. Open 2024, 7, e2448215. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.; Perekhvatova, N.; Skulachev, M.; Stein, L.; Ousler, G. SkQ1 Ophthalmic Solution for Dry Eye Treatment: Results of a Phase 2 Safety and Efficacy Clinical Study in the Environment and During Challenge in the Controlled Adverse Environment Model. Adv. Ther. 2016, 33, 96–115. [Google Scholar] [CrossRef] [PubMed]
- Friedhoff, L.T.; Petrov, A.; Ousler, G.W.; Watson, M.; Xue, Q.; Ngiam, M. Safety and Efficacy of First-in-Class mtROS scavenger SkQ1 for the Treatment of Dry Eye Disease: A Phase 3 Clinical Trial. Investig. Ophthalmol. Vis. Sci. 2019, 60, 6750. [Google Scholar]
- Li, F.; Zhao, P.; Zhao, L.; Bai, L.; Su, Q.; Feng, Y.; Ma, W.; Zhu, J.; Yang, J.; Zhang, S. Colchicine Alleviates Interstitial Lung Disease in an Experimental Autoimmune Myositis Murine Model by Inhibiting the Formation of Neutrophil Extracellular Traps. Inflammation 2024. [Google Scholar] [CrossRef]
- Vaidya, K.; Tucker, B.; Kurup, R.; Khandkar, C.; Pandzic, E.; Barraclough, J.; Machet, J.; Misra, A.; Kavurma, M.; Martinez, G.; et al. Colchicine Inhibits Neutrophil Extracellular Trap Formation in Patients With Acute Coronary Syndrome After Percutaneous Coronary Intervention. J. Am. Heart Assoc. 2021, 10, e018993. [Google Scholar] [CrossRef]
- Lopes, M.I.; Bonjorno, L.P.; Giannini, M.C.; Amaral, N.B.; Menezes, P.I.; Dib, S.M.; Gigante, S.L.; Benatti, M.N.; Rezek, U.C.; Emrich-Filho, L.L.; et al. Beneficial effects of colchicine for moderate to severe COVID-19: A randomised, double-blinded, placebo-controlled clinical trial. RMD Open 2021, 7, e001455. [Google Scholar] [CrossRef]
- Meher, A.K.; Spinosa, M.; Davis, J.P.; Pope, N.; Laubach, V.E.; Su, G.; Serbulea, V.; Leitinger, N.; Ailawadi, G.; Upchurch, G.R., Jr. Novel Role of IL (Interleukin)-1beta in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 843–853. [Google Scholar] [CrossRef]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 2013, 190, 1217–1226. [Google Scholar] [CrossRef]
- Dinallo, V.; Marafini, I.; Di Fusco, D.; Laudisi, F.; Franze, E.; Di Grazia, A.; Figliuzzi, M.M.; Caprioli, F.; Stolfi, C.; Monteleone, I.; et al. Neutrophil Extracellular Traps Sustain Inflammatory Signals in Ulcerative Colitis. J. Crohns Colitis 2019, 13, 772–784. [Google Scholar] [CrossRef]
- Elkon, K.B.; Wiedeman, A. Type I IFN system in the development and manifestations of SLE. Curr. Opin. Rheumatol. 2012, 24, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Yuzhalin, A.E.; Gordon-Weeks, A.N.; Tognoli, M.L.; Jones, K.; Markelc, B.; Konietzny, R.; Fischer, R.; Muth, A.; O’Neill, E.; Thompson, P.R.; et al. Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat. Commun. 2018, 9, 4783. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Su, X.; Zhang, B.; Pan, S. GSK484, an inhibitor of peptidyl arginine deiminase 4, increases the radiosensitivity of colorectal cancer and inhibits neutrophil extracellular traps. J. Gene Med. 2023, 25, e3530. [Google Scholar] [CrossRef] [PubMed]
- Rayes, R.F.; Mouhanna, J.G.; Nicolau, I.; Bourdeau, F.; Giannias, B.; Rousseau, S.; Quail, D.; Walsh, L.; Sangwan, V.; Bertos, N.; et al. Primary tumors induce neutrophil extracellular traps with targetable metastasis promoting effects. JCI Insight 2019, 5, e128008. [Google Scholar] [CrossRef]
- Chen, M.B.; Hajal, C.; Benjamin, D.C.; Yu, C.; Azizgolshani, H.; Hynes, R.O.; Kamm, R.D. Inflamed neutrophils sequestered at entrapped tumor cells via chemotactic confinement promote tumor cell extravasation. Proc. Natl. Acad. Sci. USA 2018, 115, 7022–7027. [Google Scholar] [CrossRef]
- Hemmers, S.; Teijaro, J.R.; Arandjelovic, S.; Mowen, K.A. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS ONE 2011, 6, e22043. [Google Scholar] [CrossRef]
- Guiducci, E.; Lemberg, C.; Kung, N.; Schraner, E.; Theocharides, A.P.A.; LeibundGut-Landmann, S. Candida albicans-Induced NETosis Is Independent of Peptidylarginine Deiminase 4. Front. Immunol. 2018, 9, 1573. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef]
- Leonie Middelink, R.C.; van Es, H.; Meldrum, E.; van Zandvoort, P.; Bruurmijn, T.; Moerland, M.; Round, P. Early Clinical Development of CIT-013, a First in Class NETosis Inhibitor, in a Randomized Phase I Dose Escalation Study in Healthy Volunteers Demonstrating Potent Inhibition of LPS Induced Neutrophil Extracellular Trap Formation. In Proceedings of the ACR Convergence 2023, San Diego, CA, USA, 10–15 November 2023. [Google Scholar]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- Mota, A.C.; Sant’Ana Filho, V.C.; Hendrickson, C.M.; DeVay, R.M.; Donne, M.; Nguyen, A.M.; Junqueira, C.; Marino, M.; Mehra, M.; Somaratne, R.; et al. Safety and Efficacy of Disulfiram in Hospitalized Patients with Moderate COVID-19: A Randomized, Double-Blind, Placebo-Controlled Trial. medRxiv 2023. [Google Scholar] [CrossRef]
- Degen, M.; Santos, J.C.; Pluhackova, K.; Cebrero, G.; Ramos, S.; Jankevicius, G.; Hartenian, E.; Guillerm, U.; Mari, S.A.; Kohl, B.; et al. Structural basis of NINJ1-mediated plasma membrane rupture in cell death. Nature 2023, 618, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Chen, C.; Lin, G.; Wu, C.; Xie, J.; Lin, K.; Dai, X.; Chen, Z.; Ye, K.; Yuan, Y.; et al. Caspase-11/GSDMD contributes to the progression of hyperuricemic nephropathy by promoting NETs formation. Cell Mol. Life Sci. 2024, 81, 114. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Alegre, K.; Deshpande, I.; Wu, S.; Lin, Z.; Kornfeld, O.S.; Lee, B.L.; Zhang, J.; Liu, J.; et al. Inhibiting membrane rupture with NINJ1 antibodies limits tissue injury. Nature 2023, 618, 1072–1077. [Google Scholar] [CrossRef]
- Shak, S.; Capon, D.J.; Hellmiss, R.; Marsters, S.A.; Baker, C.L. Recombinant human DNase I reduces the viscosity of cystic fibrosis sputum. Proc. Natl. Acad. Sci. USA 1990, 87, 9188–9192. [Google Scholar] [CrossRef]
- Fuchs, H.J.; Borowitz, D.S.; Christiansen, D.H.; Morris, E.M.; Nash, M.L.; Ramsey, B.W.; Rosenstein, B.J.; Smith, A.L.; Wohl, M.E. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N. Engl. J. Med. 1994, 331, 637–642. [Google Scholar] [CrossRef]
- Lee, B.-H.; Hsu, W.-H.; Lin, C.-H. The Anti-bacterial and Anti-adherent Effects of Pentraxin-3 on Porcine Kidney Epithelial PK15 Cells Against Staphylococcus aureus Infection. Int. J. Pept. Res. Ther. 2019, 25, 645–652. [Google Scholar] [CrossRef]
- Moalli, F.; Doni, A.; Deban, L.; Zelante, T.; Zagarella, S.; Bottazzi, B.; Romani, L.; Mantovani, A.; Garlanda, C. Role of complement and Fcgamma receptors in the protective activity of the long pentraxin PTX3 against Aspergillus fumigatus. Blood 2010, 116, 5170–5180. [Google Scholar] [CrossRef]
- Rovere, P.; Peri, G.; Fazzini, F.; Bottazzi, B.; Doni, A.; Bondanza, A.; Zimmermann, V.S.; Garlanda, C.; Fascio, U.; Sabbadini, M.G.; et al. The long pentraxin PTX3 binds to apoptotic cells and regulates their clearance by antigen-presenting dendritic cells. Blood 2000, 96, 4300–4306. [Google Scholar] [CrossRef]
- Allam, R.; Kumar, S.V.; Darisipudi, M.N.; Anders, H.J. Extracellular histones in tissue injury and inflammation. J. Mol. Med. 2014, 92, 465–472. [Google Scholar] [CrossRef]
- Nigam, S.; Patabhiraman, S.; Ciccoli, R.; Ishdorj, G.; Schwarz, K.; Petrucev, B.; Kuhn, H.; Haeggstrom, J.Z. The rat leukocyte-type 12-lipoxygenase exhibits an intrinsic hepoxilin A3 synthase activity. J. Biol. Chem. 2004, 279, 29023–29030. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, K.; Okuda, H. Selective inhibition of platelet lipoxygenase by baicalein. Biochem. Biophys. Res. Commun. 1982, 105, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Wang, Z.Y.; Bik, D.P.; Mukhtar, H. Nordihydroguaiaretic acid, an inhibitor of lipoxygenase, also inhibits cytochrome P-450-mediated monooxygenase activity in rat epidermal and hepatic microsomes. Drug Metab. Dispos. 1991, 19, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Shochet, L.; Holdsworth, S.; Kitching, A.R. Animal Models of ANCA Associated Vasculitis. Front. Immunol. 2020, 11, 525. [Google Scholar] [CrossRef]
- Li, G.; Tokuno, S.; Tahep ld, P.; Vaage, J.; Lowbeer, C.; Valen, G. Preconditioning protects the severely atherosclerotic mouse heart. Ann. Thorac. Surg. 2001, 71, 1296–1303; discussion 1303–1294. [Google Scholar] [CrossRef]
- Insull, W., Jr. The pathology of atherosclerosis: Plaque development and plaque responses to medical treatment. Am. J. Med. 2009, 122, S3–S14. [Google Scholar] [CrossRef]
- Sjoland, H.; Eitzman, D.T.; Gordon, D.; Westrick, R.; Nabel, E.G.; Ginsburg, D. Atherosclerosis progression in LDL receptor-deficient and apolipoprotein E-deficient mice is independent of genetic alterations in plasminogen activator inhibitor-1. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 846–852. [Google Scholar] [CrossRef]
- O’Neil, L.J.; Alpizar-Rodriguez, D.; Deane, K.D. Rheumatoid Arthritis: The Continuum of Disease and Strategies for Prediction, Early Intervention, and Prevention. J. Rheumatol. 2024, 51, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Kouskoff, V.; Korganow, A.S.; Duchatelle, V.; Degott, C.; Benoist, C.; Mathis, D. Organ-specific disease provoked by systemic autoimmunity. Cell 1996, 87, 811–822. [Google Scholar] [CrossRef]
- Doeing, D.C.; Borowicz, J.L.; Crockett, E.T. Gender dimorphism in differential peripheral blood leukocyte counts in mice using cardiac, tail, foot, and saphenous vein puncture methods. BMC Clin. Pathol. 2003, 3, 3. [Google Scholar] [CrossRef]
- Ermert, D.; Urban, C.F.; Laube, B.; Goosmann, C.; Zychlinsky, A.; Brinkmann, V. Mouse neutrophil extracellular traps in microbial infections. J. Innate Immun. 2009, 1, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Loder, S.J.; Cholok, D.; Li, J.; Bian, G.; Yalavarthi, S.; Li, S.; Carson, W.F.; Hwang, C.; Marini, S.; et al. Disruption of Neutrophil Extracellular Traps (NETs) Links Mechanical Strain to Post-traumatic Inflammation. Front. Immunol. 2019, 10, 2148. [Google Scholar] [CrossRef] [PubMed]
- Khanmohammadi, M.; Danish, H.; Sekar, N.C.; Suarez, S.A.; Chheang, C.; Peter, K.; Khoshmanesh, K.; Baratchi, S. Cyclic stretch enhances neutrophil extracellular trap formation. BMC Biol. 2024, 22, 209. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, M.C.; Fingerhut, L.; Alfonso-Castro, A.; Mergani, A.; Schwennen, C.; von Kockritz-Blickwede, M.; de Buhr, N. How Long Does a Neutrophil Live?-The Effect of 24 h Whole Blood Storage on Neutrophil Functions in Pigs. Biomedicines 2020, 8, 278. [Google Scholar] [CrossRef]
- Blanter, M.; Gouwy, M.; Struyf, S. Studying Neutrophil Function in vitro: Cell Models and Environmental Factors. J. Inflamm. Res. 2021, 14, 141–162. [Google Scholar] [CrossRef]
- Schreiber, A.; Xiao, H.; Jennette, J.C.; Schneider, W.; Luft, F.C.; Kettritz, R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 289–298. [Google Scholar] [CrossRef]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef]
- Parker, H.; Dragunow, M.; Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J. Leukoc. Biol. 2012, 92, 841–849. [Google Scholar] [CrossRef]
- van der Linden, M.; Westerlaken, G.H.A.; van der Vlist, M.; van Montfrans, J.; Meyaard, L. Differential Signalling and Kinetics of Neutrophil Extracellular Trap Release Revealed by Quantitative Live Imaging. Sci. Rep. 2017, 7, 6529. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Lisman, T. Platelet-neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res. 2018, 371, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Czaikoski, P.G.; Mota, J.M.; Nascimento, D.C.; Sonego, F.; Castanheira, F.V.; Melo, P.H.; Scortegagna, G.T.; Silva, R.L.; Barroso-Sousa, R.; Souto, F.O.; et al. Neutrophil Extracellular Traps Induce Organ Damage during Experimental and Clinical Sepsis. PLoS ONE 2016, 11, e0148142. [Google Scholar] [CrossRef] [PubMed]
- D’Cruz, A.A.; Speir, M.; Bliss-Moreau, M.; Dietrich, S.; Wang, S.; Chen, A.A.; Gavillet, M.; Al-Obeidi, A.; Lawlor, K.E.; Vince, J.E.; et al. The pseudokinase MLKL activates PAD4-dependent NET formation in necroptotic neutrophils. Sci. Signal 2018, 11, eaao1716. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Li, H.; Li, Y.; Dai, M.; Zhang, L.; Liu, S.; Tan, H.; Deng, P.; Liu, J.; Mao, Z.; et al. NETs promote ALI/ARDS inflammation by regulating alveolar macrophage polarization. Exp. Cell Res. 2019, 382, 111486. [Google Scholar] [CrossRef]
- Hudock, K.M.; Collins, M.S.; Imbrogno, M.; Snowball, J.; Kramer, E.L.; Brewington, J.J.; Gollomp, K.; McCarthy, C.; Ostmann, A.J.; Kopras, E.J.; et al. Neutrophil extracellular traps activate IL-8 and IL-1 expression in human bronchial epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 319, L137–L147. [Google Scholar] [CrossRef]
- Tillack, K.; Breiden, P.; Martin, R.; Sospedra, M. T lymphocyte priming by neutrophil extracellular traps links innate and adaptive immune responses. J. Immunol. 2012, 188, 3150–3159. [Google Scholar] [CrossRef]
- Dube, D.K.; Seal, G.; Loeb, L.A. Differential heat sensitivity of mammalian DNA polymerases. Biochem. Biophys. Res. Commun. 1976, 76, 483–487. [Google Scholar] [CrossRef]
- Morrison, J.M.; Keir, H.M. A new DNA-exonuclease in cells infected with herpes virus: Partial purification and properties of the enzyme. J. Gen. Virol. 1968, 3, 337–347. [Google Scholar] [CrossRef]
- Kovaliov, M.; Cohen-Karni, D.; Burridge, K.A.; Mambelli, D.; Sloane, S.; Daman, N.; Xu, C.; Guth, J.; Kenneth Wickiser, J.; Tomycz, N.; et al. Grafting strategies for the synthesis of active DNase I polymer biohybrids. Eur. Polym. J. 2018, 107, 15–24. [Google Scholar] [CrossRef]
- Chhabra, D.; Bao, S.; dos Remedios, C.G. The distribution of cofilin and DNase I in vivo. Cell Res. 2002, 12, 207–214. [Google Scholar] [CrossRef]
- Prince, W.S.; Baker, D.L.; Dodge, A.H.; Ahmed, A.E.; Chestnut, R.W.; Sinicropi, D.V. Pharmacodynamics of recombinant human DNase I in serum. Clin. Exp. Immunol. 1998, 113, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Alcazar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renne, C.; Renne, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Englert, H.; Gobel, J.; Khong, D.; Omidi, M.; Wolska, N.; Konrath, S.; Frye, M.; Mailer, R.K.; Beerens, M.; Gerwers, J.C.; et al. Targeting NETs using dual-active DNase1 variants. Front. Immunol. 2023, 14, 1181761. [Google Scholar] [CrossRef] [PubMed]
- Baron, W.F.; Pan, C.Q.; Spencer, S.A.; Ryan, A.M.; Lazarus, R.A.; Baker, K.P. Cloning and characterization of an actin-resistant DNase I-like endonuclease secreted by macrophages. Gene 1998, 215, 291–301. [Google Scholar] [CrossRef]
- Daya, S.; Berns, K.I. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef]
- Anker, P.; Stroun, M.; Maurice, P.A. Spontaneous release of DNA by human blood lymphocytes as shown in an in vitro system. Cancer Res. 1975, 35, 2375–2382. [Google Scholar]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Hayden, H.; Ibrahim, N.; Klopf, J.; Zagrapan, B.; Mauracher, L.M.; Hell, L.; Hofbauer, T.M.; Ondracek, A.S.; Schoergenhofer, C.; Jilma, B.; et al. ELISA detection of MPO-DNA complexes in human plasma is error-prone and yields limited information on neutrophil extracellular traps formed in vivo. PLoS ONE 2021, 16, e0250265. [Google Scholar] [CrossRef]
- Mauracher, L.M.; Posch, F.; Martinod, K.; Grilz, E.; Daullary, T.; Hell, L.; Brostjan, C.; Zielinski, C.; Ay, C.; Wagner, D.D.; et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J. Thromb. Haemost. 2018, 16, 508–518. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Drug | Mechanism/Target | Experimental Model/Disease Context | Key Findings | Reference |
|---|---|---|---|---|
| Eculizumab | Binds to the complement component C5 and inhibits its cleavage | In vitro, plasma from paroxysmal nocturnal haemoglonuria patients with or without a history of thrombosis | Reduced nucleosome levels in the thrombosis group, suggesting reduced NET formation. | [64] |
| Fostamatinib | Inhibits Syk, reducing FcR-mediated NET formation | In vitro, plasma from COVID-19 patients | Reduced NET release by healthy neutrophils stimulated with plasma from COVID-19 patients | [65] |
| SKQ1 | Scavenges excess mitochondrial reactive oxygen species | In vitro, isolated neutrophils from healthy patients or those with CGD | Inhibited calcium-induced NETs but not PMA-induced NETs | [46] |
| Colchicine | Disrupts microtubule mobilisation. Inhibits ROS and calcium influx | In vivo, C57Bl/6 mice subjected to ligation of the left anterior descending coronary artery | Improved survival and cardiac function, and inhibited NET formation | [66] |
| BB-Cl-Amidine | Inhibits PAD enzymes, limiting histone citrullination | In vivo, lupus-prone MRL/lpr mice | Improved endothelial function and downregulated type I IFN-regulated genes while decreasing NET formation | [67] |
| GSK484 | Inhibits PAD4 enzymes, limiting histone citrullination | In vitro, isolated murine neutrophils | H3Cit+ cells and NETs were reduced significantly | [68] |
| CIT-013 | Binds to citrullinated histones, preventing release of NETs | In vivo, the CAIA mouse model, the DSS-induced colitis mouse model, and the LPS-induced sepsis mouse model | For all disease models, severity was reduced, and NET release diminished | [69] |
| Disulfiram | Inhibits gasdermin D, blocking plasma membrane rupture | In vivo, the transfusion-related acute lung injury mouse model and the COVID-19 hamster model | NET formation was blocked in both models. Survival increased in ALI mice, and lung histology improved in both models. | [70] |
| NINJ1 Monoclonal Antibodies | Blocks NINJ1—a plasma membrane protein required for rupture | In vivo, the acute oxalate nephropathy mouse model | Deletion of NIN1 reduced NETs, inflammation, and renal damage | [71] |
| DNase I | Degrades the extracellular DNA backbone of NETs, promoting clearance | In vivo, the ischemia–reperfusion injury rat model | Inflammation and epithelial damage improved due to NET degradation | [72] |
| Drug | Mechanism/Target | Trial Design | Outcomes | Reference |
|---|---|---|---|---|
| Tocilizumab | Binds to IL-6 receptor to prevent IL-6 signalling | Phase II trial assessing tocilizumab (280 mg, intravenous, single dose) vs. placebo during percutaneous coronary intervention in patients with STEMI | All NET markers were reduced compared to the placebo. Myocardial salvage improved | [73] |
| Metformin | Activates AMPK and inhibits mitochondrial complex 1, reducing mtROS | Sub-study from a phase III trial assessing metformin (1500 mg/day for 2 months) vs. placebo in pre-diabetic patients | After 2 months of therapy, metformin significantly reduced NET markers compared to placebo | [74] |
| AZD9668 | Inhibits neutrophil elastase, limiting gasdermin D activation and histone modifications | Phase II trial assessing oral AZD9668 (60 mg, bid, for 28 days) vs. placebo in patients with idiopathic or post-infective bronchiectasis | No significant improvements in sputum neutrophil counts or lung function. AZD9668 was well-tolerated without serious adverse events. | [75] |
| AZD9668 | - | Phase II trial assessing oral AZD9668 (60 mg, bid, for 28 days) with standard care vs. placebo with standard care in cystic fibrosis patients | No significant improvements in sputum neutrophil counts or lung function. AZD9668 was well-tolerated without serious adverse events. | [76] |
| AZD9668 | - | Phase I/II trial assessing oral AZD9668 (5, 20, and 60 mg, bid, for 12 weeks) vs. placebo in COPD patients receiving tiotropium treatment | No difference in post-bronchodilator FEV1 compared to placebo. No difference in adverse events relative to placebo | [77] |
| BAY 85-8501 | Inhibits neutrophil elastase, limiting gasdermin D activation and histone modifications | Phase II trial assessing BAY 85-8501 (1 mg/day for 28 days) vs. placebo in patients with non-CF BE | There was no improvement in the disease. There was no statistical difference between the metformin and placebo groups regarding adverse effects | [78] |
| DNase I | Degrades the extracellular DNA backbone of NETs, promoting clearance | Phase II trial assessing nebulised DNase I (2.5 mg, days 1–7) with best available care (BAC) vs. BAC alone in hospitalised COVID-19 patients | CRP and D-dimer were reduced, indicating reduced inflammation through NET degradation | [79] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espiritu, A.; O’Sullivan, K.M. A Web of Challenges: The Therapeutic Struggle to Target NETs in Disease. Int. J. Mol. Sci. 2025, 26, 4773. https://doi.org/10.3390/ijms26104773
Espiritu A, O’Sullivan KM. A Web of Challenges: The Therapeutic Struggle to Target NETs in Disease. International Journal of Molecular Sciences. 2025; 26(10):4773. https://doi.org/10.3390/ijms26104773
Chicago/Turabian StyleEspiritu, Andre, and Kim Maree O’Sullivan. 2025. "A Web of Challenges: The Therapeutic Struggle to Target NETs in Disease" International Journal of Molecular Sciences 26, no. 10: 4773. https://doi.org/10.3390/ijms26104773
APA StyleEspiritu, A., & O’Sullivan, K. M. (2025). A Web of Challenges: The Therapeutic Struggle to Target NETs in Disease. International Journal of Molecular Sciences, 26(10), 4773. https://doi.org/10.3390/ijms26104773