
Application of Lipophilic Prodrug Charge Masking Strategy to Obtain Novel, Potential Oxytocin Prodrugs

 ,
,  , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results
2.1. Peptide Synthesis
2.2. PAMPA
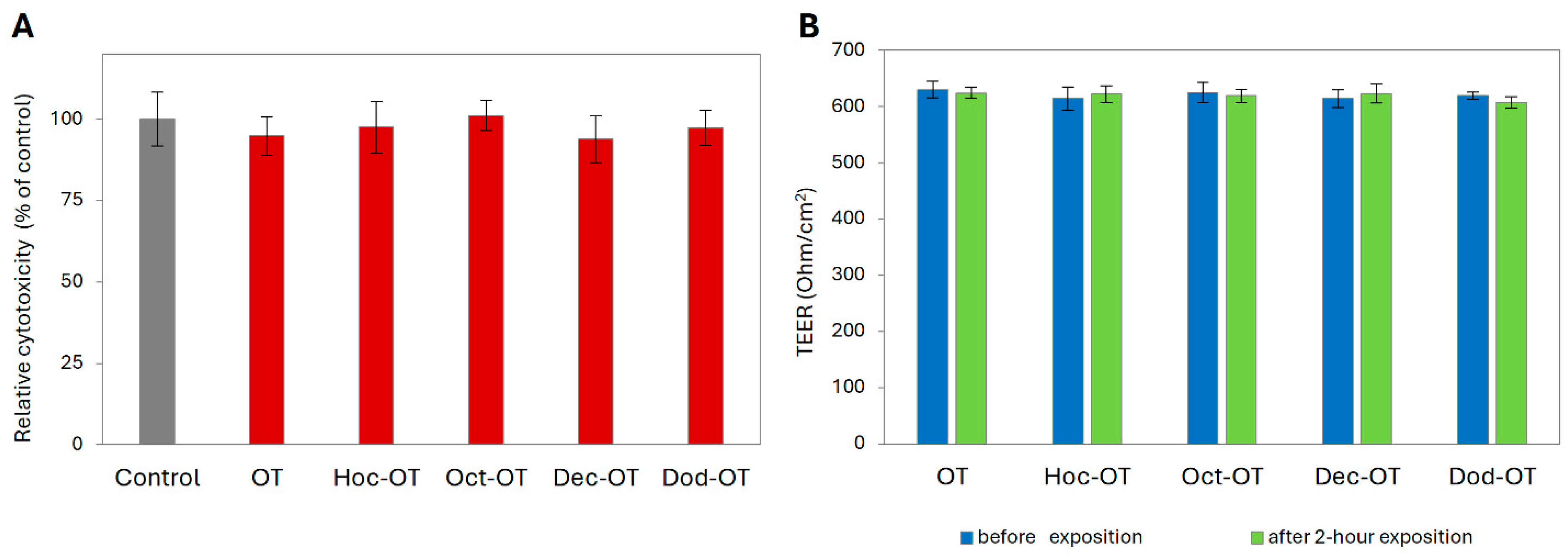
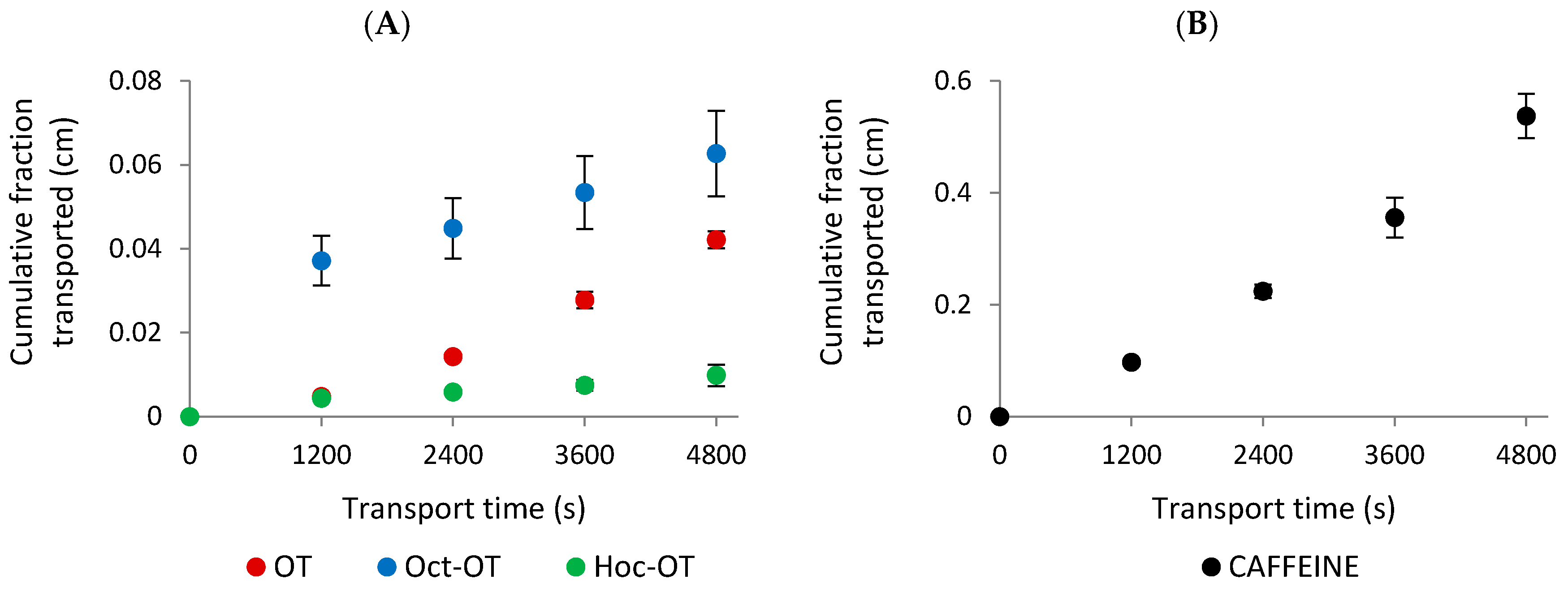
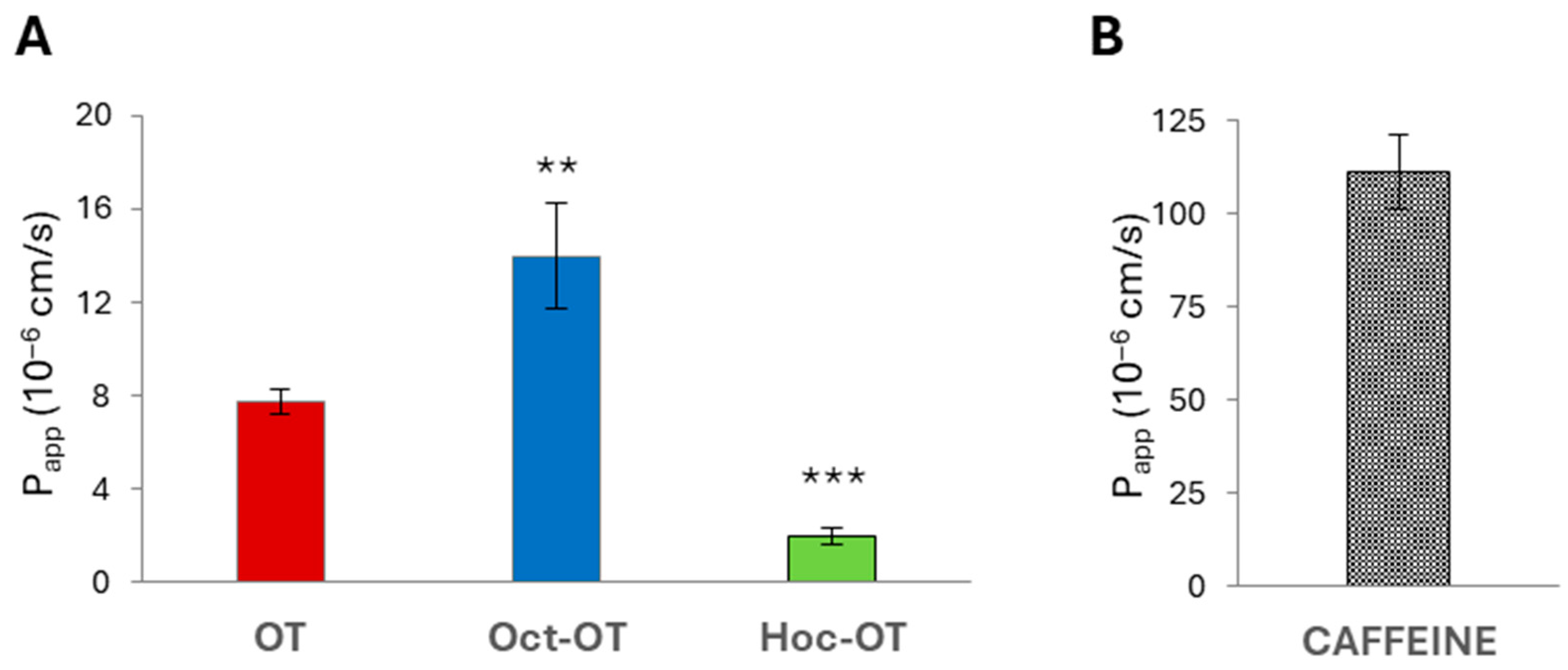
2.3. Permeability Experiments in the Caco-2 Model
3. Discussion
4. Materials and Methods
4.1. Peptide Synthesis
4.2. PAMPA
4.3. Caco-2 Cell Model Assays
4.3.1. Caco-2 Intestine Epithelial Model
4.3.2. Cytotoxicity Test
4.3.3. Transepithelial Transport Experiment
4.4. LC-MS Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bliss, M. The Discovery of Insulin; University of Chicago Press: Chicago, IL, USA, 1984. [Google Scholar]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Pauletti, G.M.; Gangwar, S.; Knipp, G.T.; Nerurkar, M.M.; Okumu, F.W.; Tamura, K.; Siahaan, T.J.; Borchardt, R.T. Structural Requirements for Intestinal Absorption of Peptide Drugs. J. Control. Release 1996, 41, 3–17. [Google Scholar] [CrossRef]
- Adessi, C.; Soto, C. Converting a Peptide into a Drug: Strategies to Improve Stability and Bioavailability. Curr. Med. Chem. 2005, 9, 963–978. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, K.M.; Raunio, H.; Rautio, J. Prodrugs—From Serendipity to Rational Design. Pharmacol. Rev. 2011, 63, 750–771. [Google Scholar] [CrossRef] [PubMed]
- Biron, E.; Chatterjee, J.; Ovadia, O.; Langenegger, D.; Brueggen, J.; Hoyer, D.; Schmid, H.A.; Jelinek, R.; Gilon, C.; Hoffman, A.; et al. Improving Oral Bioavailability of Peptides by Multiple N-Methylation: Somatostatin Analogues. Angew. Chem. Int. Ed. 2008, 47, 2595–2599. [Google Scholar] [CrossRef]
- Modahl, C.; Green, L.; Fein, D.; Morris, M.; Waterhouse, L.; Feinstein, C.; Levin, H. Plasma oxytocin levels in autistic children. Biol. Psychiatry 1998, 43, 270–277. [Google Scholar] [CrossRef]
- Green, J.; Hollander, E. Autism and Oxytocin: New Developments in Translational Approaches to Therapeutics. Neurotherapeutics 2010, 7, 250–257. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Jaeggi, A. Oxytocin levels tend to be lower in autistic children: A meta-analysis of 31 studies. Autism 2021, 25, 2152–2161. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, E.; Soorya, L.; Brian, J.; Dupuis, A.; Mankad, D.; Smile, S.; Jacob, S. Intranasal Oxytocin in the Treatment of Autism Spectrum Disorders: A Review of Literature and Early Safety and Efficacy Data in Youth. Brain Res. 2014, 1580, 188–198. [Google Scholar] [CrossRef] [PubMed]
- McCracken, J.; McGough, J.; Shah, B.; Cronin, P.; Hong, D.; Aman, M.G.; Arnold, L.E.; Lindsay, R.; Nash, P.; Hollway, J.; et al. Research Units on Pediatric Psychopharmacology Autism Network. Risperidone in children with autism and serious behavioral problems. N. Engl. J. Med. 2002, 347, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Guastella, A.J.; Gray, K.M.; Rinehart, N.J.; Alvares, G.A.; Tonge, B.J.; Hickie, I.B.; Keating, C.M.; Cacciotti-Saija, C.; Einfeld, S.L. The Effects of a Course of Intranasal Oxytocin on Social Behaviors in Youth Diagnosed with Autism Spectrum Disorders: A Randomized Controlled Trial. J. Child Psychol. Psychiatry 2015, 56, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Rahim, F.; Qasim, N.; Karlygash, T.; Khozhamkul, F.; Dzhusupovv, K.; Tekmanova, A.; Elmira, K. Intranasal Oxytocin for Patients With Autism Spectrum Disorder: A Comprehensive Meta-Analysis of Preclinical and Clinical Studies. Rev. J. Autism Dev. Disord. 2024. [Google Scholar] [CrossRef]
- Lou, J.; Duan, H.; Qin, Q.; Teng, Z.; Gan, F.; Zhou, X.; Zhou, X. Advances in Oral Drug Delivery Systems: Challenges and Opportunities. Pharmaceutics 2023, 15, 484. [Google Scholar] [CrossRef]
- Liu, F.; Ranmal, S.; Batchelor, H.; Orlu-Gul, M.; Ernest, T.; Thomas, I.; Flangan, T.; Tuleu, C. Patient-Centered Pharmaceutical Design to Improve Acceptability of Medicines: Similarities and Differences in Paediatric and Geriatric Populations. Drugs 2014, 74, 1871–1889. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Q.; Zheng, X.; Yao, S.; Zhao, W.; Becker, B.; Xu, X.; Kendrick, K. Oral Administration of Oxytocin, Like Intranasal Administration, Decreases Top-Down Social Attention. Int. J. Neuropsychopharmacol. 2022, 25, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Choonara, B.F.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; du Toit, L.C.; Pillay, V. A Review of Advanced Oral Drug Delivery Technologies Facilitating the Protection and Absorption of Protein and Peptide Molecules. Biotechnol. Adv. 2014, 32, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Orellana, I. Strategies to Improve Oral Drug Bioavailability. Expert Opin. Drug Deliv. 2005, 2, 419–433. [Google Scholar] [CrossRef]
- Kiptoo, P.; Calcagno, A.M.; Siahaan, T.J. Physiological, Biochemical, and Chemical Barriers to Oral Drug Delivery. In Drug Delivery: Principles and Applications, 2nd ed.; Wiley: Hoboken, NJ, USA, 2016; pp. 19–34. ISBN 9781118833322. [Google Scholar]
- de Groot, A.N.J.A.; Vree, T.B.; Hekster, Y.A.; Pesman, G.J.; Sweep, F.C.G.J.; van Dongen, P.J.W.; van Roosmalen, J. Bioavailability and Pharmacokinetics of Sublingual Oxytocin in Male Volunteers. J. Pharm. Pharmacol. 2011, 47, 571–575. [Google Scholar] [CrossRef]
- Yamasue, H.; Kojima, M.; Kuwabara, H.; Kuroda, M.; Matsumoto, K.; Kanai, C.; Inada, N.; Owada, K.; Ochi, K.; Ono, N.; et al. Effect of a Novel Nasal Oxytocin Spray with Enhanced Bioavailability on Autism: A Randomized Trial. Brain 2022, 145, 490–499. [Google Scholar] [CrossRef]
- Azab, A.E. An Overview of Oxytocin: Chemical Structure, Receptors, Physiological Functions, Measurement Techniques of Oxytocin, and Metabolism. J. Clin. Res. Rep. 2022, 11, 1–11. [Google Scholar] [CrossRef]
- Franke, A.A.; Li, X.; Menden, A.; Lee, M.R.; Lai, J.F. Oxytocin Analysis from Human Serum, Urine, and Saliva by Orbitrap Liquid Chromatography–Mass Spectrometry. Drug Test Anal. 2019, 11, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Stielow, M.; Witczyńska, A.; Kubryń, N.; Fijałkowski, Ł.; Nowaczyk, J.; Nowaczyk, A. The Bioavailability of Drugs—The Current State of Knowledge. Molecules 2023, 28, 8038. [Google Scholar] [CrossRef] [PubMed]
- Fasinu, P.; Pillay, V.; Ndesendo, V.M.K.; Du Toit, L.C.; Choonara, Y.E. Diverse Approaches for the Enhancement of Oral Drug Bioavailability. Biopharm. Drug Dispos. 2011, 32, 185–209. [Google Scholar] [CrossRef] [PubMed]
- Kerns, E.H.; Di, L.; Petusky, S.; Farris, M.; Ley, R.; Jupp, P. Combined Application of Parallel Artificial Membrane Permeability Assay and Caco-2 Permeability Assays in Drug Discovery. J. Pharm. Sci. 2004, 93, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Schumacher-Klinger, A.; Fanous, J.; Merzbach, S.; Weinmüller, M.; Reichart, F.; Räder, A.F.B.; Gitlin-Domagalska, A.; Gilon, C.; Kessler, H.; Hoffman, A. Enhancing Oral Bioavailability of Cyclic RGD Hexa-Peptides by the Lipophilic Prodrug Charge Masking Approach: Redirection of Peptide Intestinal Permeability from a Paracellular to Transcellular Pathway. Mol. Pharm. 2018, 15, 3468–3477. [Google Scholar] [CrossRef] [PubMed]
- Kansy, M.; Senner, F.; Gubernator, K. Physicochemical High Throughput Screening: Parallel Artificial Membrane Permeation Assay in the Description of Passive Absorption Processes. J. Med. Chem. 1998, 41, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Wohnsland, F.; Faller, B. High-Throughput Permeability PH Profile and High-Throughput Alkane/Water Log P with Artificial Membranes. J. Med. Chem. 2001, 44, 923–930. [Google Scholar] [CrossRef]
- Socała, K.; Mogilski, S.; Pieróg, M.; Nieoczym, D.; Abram, M.; Szulczyk, B.; Lubelska, A.; Latacz, G.; Doboszewska, U.; Wlaź, P.; et al. KA-11, a Novel Pyrrolidine-2,5-Dione Derived Broad-Spectrum Anticonvulsant: Its Antiepileptogenic, Antinociceptive Properties and In Vitro Characterization. ACS Chem. Neurosci. 2019, 10, 636–648. [Google Scholar] [CrossRef]
- Zhu, C.; Jiang, L.; Chen, T.M.; Hwang, K.K. A Comparative Study of Artificial Membrane Permeability Assay for High Throughput Profiling of Drug Absorption Potential. Eur. J. Med. Chem. 2002, 37, 399–407. [Google Scholar] [CrossRef]
- Balimane, P.V.; Chong, S.; Morrison, R.A. Current Methodologies Used for Evaluation of Intestinal Permeability and Absorption. J. Pharmacol. Toxicol. Methods 2000, 44, 301–312. [Google Scholar] [CrossRef]
- Masungi, C.; Mensch, J.; Van Dijck, A.; Borremans, C.; Willems, B.; Mackie, C.; Noppe, M.; Brewster, M.E. Parallel Artificial Membrane Permeability Assay (PAMPA) Combined with a 10-Day Multiscreen Caco-2 Cell Culture as a Tool for Assessing New Drug Candidates. Pharmazie 2008, 63, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, M.; Avdeef, A.; Ruiz, A.; Nalda, R.; Ruell, J.A.; Tsinman, O.; González, I.; Fernández, C.; Sánchez, G.; Garrigues, T.M.; et al. PAMPA—A Drug Absorption In Vitro Model: 7. Comparing Rat In Situ, Caco-2, and PAMPA Permeability of Fluoroquinolones. Eur. J. Pharm. Sci. 2004, 21, 429–441. [Google Scholar] [CrossRef]
- Chen, X.; Murawski, A.; Patel, K.; Crespi, C.L.; Balimane, P.V. A Novel Design of Artificial Membrane for Improving the PAMPA Model. Pharm. Res. 2008, 25, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Ruell, J.A.; Avdeef, A. Absorption Screening Using the PAMPA Approach. In Optimization in Drug Discovery; Humana Press: Totowa, NJ, USA, 2004; pp. 37–64. [Google Scholar]
- Pinto, M.; Robine-Leon, S.; Appay, M.; Kedinger, M.; Triadou, N.; Dussaulx, E.; Lacroix, B.; Simon-Assmann, P.; Hafen, K.; Fogh, J.; et al. Enterocyte-like differentiation and Polarization of the human colon carcinoma cell line Caco-2 in culture. Biol. Cell 1983, 47, 323–330. [Google Scholar]
- Shah, P.; Jogani, V.; Bagchi, T.; Misra, A. Role of Caco-2 Cell Monolayers in Prediction of Intestinal Drug Absorption. Biotechnol. Prog. 2006, 22, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Van Breemen, R.B.; Li, Y. Caco-2 Cell Permeability Assays to Measure Drug Absorption. Expert Opin. Drug Metab. Toxicol. 2005, 1, 175–185. [Google Scholar] [CrossRef]
- Sambuy, Y.; De Angelis, I.; Ranaldi, G.; Scarino, M.L.; Stammati, A.; Zucco, F. The Caco-2 Cell Line as a Model of the Intestinal Barrier: Influence of Cell and Culture-Related Factors on Caco-2 Cell Functional Characteristics. Cell Biol. Toxicol. 2005, 21, 1–26. [Google Scholar] [CrossRef]
- ICH M9 on Biopharmaceutics Classification System Based Biowaivers—Scientific Guideline|European Medicines Agency (EMA). Available online: https://www.ema.europa.eu/en/ich-m9-biopharmaceutics-classification-system-based-biowaivers-scientific-guideline (accessed on 20 February 2025).
- Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System—Digital Collections—National Library of Medicine. Available online: https://collections.nlm.nih.gov/catalog/nlm:nlmuid-101720038-pdf (accessed on 20 February 2025).
- Yee, S. In Vitro Permeability across Caco-2 Cells (Colonic) Can Predict In Vivo (Small Intestinal) Absorption in Man—Fact or Myth. Pharm. Res. 1997, 14, 763–766. [Google Scholar] [CrossRef] [PubMed]
- Smetanova, L.; Stetinova, V.; Kholova, D.; Kvetina, J.; Smetana, J.; Svoboda, Z. Caco-2 Cells and Biopharmaceutics Classification System (BCS) for Prediction of Transepithelial Transport of Xenobiotics (Model Drug: Caffeine). Neuroendocrinol. Lett. 2009, 30, 101–105. [Google Scholar]
- Volpe, D.A.; Faustino, P.J.; Ciavarella, A.B.; Asafu-Adjaye, E.B.; Ellison, C.D.; Yu, L.X.; Hussain, A.S. Classification of Drug Permeability with a Caco-2 Cell Monolayer Assay. Clin. Res. Regul. Aff. 2007, 24, 39–47. [Google Scholar] [CrossRef]
- Lin, J.; Yamazaki, M. Role of P-Glycoprotein in Pharmacokinetics. Clin. Pharmacokinet. 2003, 42, 59–98. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jin, H.; Kim, w.; Han, Y.; Kim, D.; Chung, S.; Shim, C. Involvement of P-glycoprotein, Multidrug Resistance Protein 2 and Breast Cancer Resistance Protein in the Transport of Belotecan and Topotecan in Caco-2 and MDCKII Cells. Pharm. Res. 2008, 25, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Choi, S. Quantitative analysis for lipophilic drug transport through a model lipid membrane with membrane retention. Eur. J. Pharm. Biopharm. 2019, 134, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Wils, P.; Warnery, A.; Pbung-Ba, V.; Legrain, S.; Scherman, D. High lipophilicity decreases drug transport across intestinal epithelial cells. J. Pharmacol. Exp. Ther. 1994, 269, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Buckley, S.; Ficher, S.; Fricker, G.; Brandl, M. In vitro models to evaluate the permeability of poorly soluble drug entities: Challenges and perspectives. Eur. J. Pharm. Med. Res. 2012, 45, 235–250. [Google Scholar] [CrossRef]
- Gitlin-Domagalska, A.; Dębowski, D.; Łęgowska, A.; Stirnberg, M.; Okońska, J.; Gütschow, M.; Rolka, K. Design and Chemical Syntheses of Potent Matriptase-2 Inhibitors Based on Trypsin Inhibitor SFTI-1 Isolated from Sunflower Seeds. Biopolymers 2017, 108, e23031. [Google Scholar] [CrossRef]
- Gitlin-Domagalska, A.; Dębowski, D.; Maciejewska, A.; Samsonov, S.; Maszota-Zieleniak, M.; Ptaszyńska, N.; Łęgowska, A.; Rolka, K. Cyclic Peptidic Furin Inhibitors Developed by Combinatorial Chemistry. ACS Med. Chem. Lett. 2023, 14, 458–465. [Google Scholar] [CrossRef]
- Tavelin, S.; Gråsjö, J.; Taipalensuu, J.; Ocklind, G.; Artursson, P. Applications of Epithelial Cell Culture in Studies of Drug Transport. Methods Mol. Biol. 2002, 188, 233–272. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| No | Name a | Structure | Calculated [M+H]+ | Found [M+H]+ | CLogP b |
|---|---|---|---|---|---|
| 1. | OT | Cys(&)-Tyr-Ile-Gln-Asn-Cys(&)-Pro-Leu-Gly-NH2 | 1004.444 | 1007.514 | −0.654 |
| 2. | Et-OT | Et-Cys(&)-Tyr-Ile-Gln-Asn-Cys(&)-Pro-Leu-Gly-NH2 | 1079.466 | 1079.586 | 0.461 |
| 3. | MeOEt-OT | MeOEt-Cys(&)-Tyr-Ile-Gln-Asn-Cys(&)-Pro-Leu-Gly-NH2 | 1109.476 | 1109.520 | 0.129 |
| 4. | Prop-OT | Prop-Cys(&)-Tyr-Ile-Gln-Asn-Cys(&)-Pro-Leu-Gly-NH2 | 1093.481 | 1093.491 | 0.990 |
| 5. | But-OT | But-Cys(&)-Tyr-Ile-Gln-Asn-Cys(&)-Pro-Leu-Gly-NH2 | 1107.497 | 1107.541 | 1.519 |
| 6. | Hoc-OT | Hoc-Cys(&)-Tyr-Ile-Gln-Asn-Cys(&)-Pro-Leu-Gly-NH2 | 1135.528 | 1135.622 | 2.577 |
| 7. | Oct-OT | Oct-Cys(&)-Tyr-Ile-Gln-Asn-Cys(&)-Pro-Leu-Gly-NH2 | 1163.559 | 1163.635 | 3.635 |
| 8. | Dec-OT | Dec-Cys(&)-Tyr-Ile-Gln-Asn-Cys(&)-Pro-Leu-Gly-NH2 | 1191.591 | 1191.611 | 4.693 |
| 9. | Dod-OT | Dod-Cys(&)-Tyr-Ile-Gln-Asn-Cys(&)-Pro-Leu-Gly-NH2 | 1219.622 | 1219.691 | 5.751 |
| Compound | Papp (10−6 cm/s) ± SD | Mass Retention |
|---|---|---|
| Caffeine | 21.99 ± 11.16 | 26% |
| Atenolol | 1.24 ± 1.28 | 12% |
| OT | 2.22 ± 0.21 | 19% |
| Et-OT | 2.78 ± 0.17 | 16% |
| MeOEt-OT | 2.37 ± 0.52 | 15% |
| Prop-OT | 2.97 ± 0.64 | 33% |
| But-OT | 2.20 ± 0.30 | 30% |
| Hoc-OT | 4.67 ± 0.66 | 43% |
| Oct-OT | 5.93 ± 0.86 | 33% |
| Dec-OT | 9.28 ± 6.11 | 32% |
| Dod-OT | 1.08 ± 0.95 | 67% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gitlin-Domagalska, A.; Olejnik, A.; Ruczyński, J.; Starego, D.; Ptaszyńska, N.; Łęgowska, A.; Dębowski, D.; Gilon, C.; Rolka, K. Application of Lipophilic Prodrug Charge Masking Strategy to Obtain Novel, Potential Oxytocin Prodrugs. Int. J. Mol. Sci. 2025, 26, 4772. https://doi.org/10.3390/ijms26104772
Gitlin-Domagalska A, Olejnik A, Ruczyński J, Starego D, Ptaszyńska N, Łęgowska A, Dębowski D, Gilon C, Rolka K. Application of Lipophilic Prodrug Charge Masking Strategy to Obtain Novel, Potential Oxytocin Prodrugs. International Journal of Molecular Sciences. 2025; 26(10):4772. https://doi.org/10.3390/ijms26104772
Chicago/Turabian StyleGitlin-Domagalska, Agata, Anna Olejnik, Jarosław Ruczyński, Dominika Starego, Natalia Ptaszyńska, Anna Łęgowska, Dawid Dębowski, Chaim Gilon, and Krzysztof Rolka. 2025. "Application of Lipophilic Prodrug Charge Masking Strategy to Obtain Novel, Potential Oxytocin Prodrugs" International Journal of Molecular Sciences 26, no. 10: 4772. https://doi.org/10.3390/ijms26104772
APA StyleGitlin-Domagalska, A., Olejnik, A., Ruczyński, J., Starego, D., Ptaszyńska, N., Łęgowska, A., Dębowski, D., Gilon, C., & Rolka, K. (2025). Application of Lipophilic Prodrug Charge Masking Strategy to Obtain Novel, Potential Oxytocin Prodrugs. International Journal of Molecular Sciences, 26(10), 4772. https://doi.org/10.3390/ijms26104772