
An Evaluation of Cation–Chloride Cotransporters NKCC1 and KCC2 in Carbamazepine-Resistant Rats

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
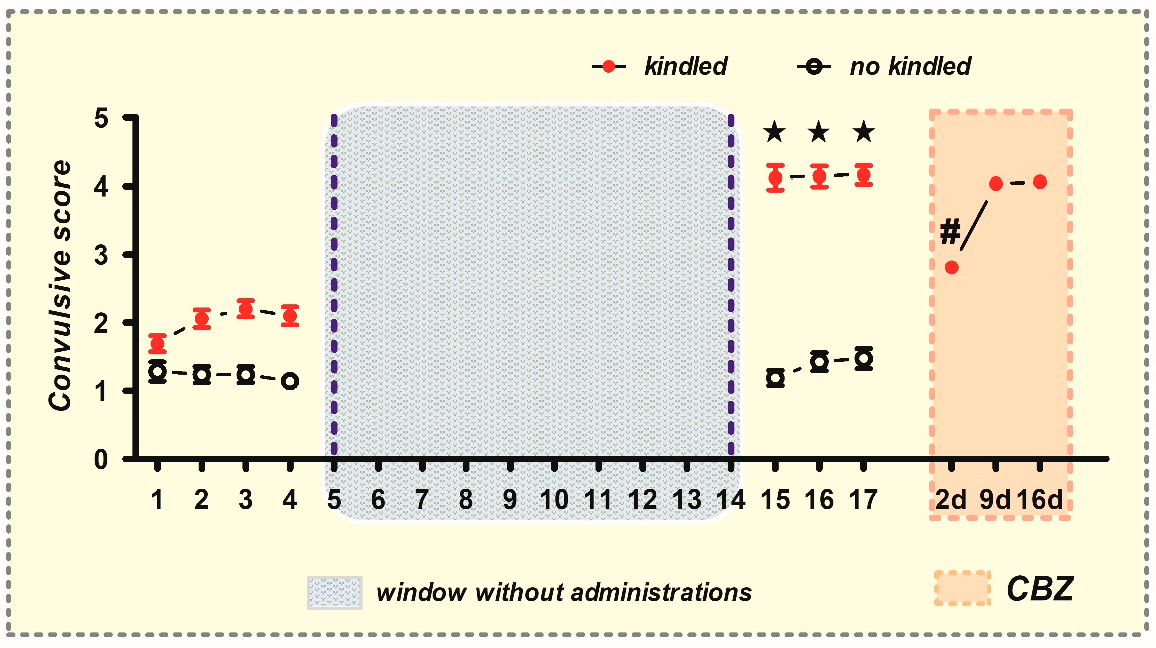
2.1. Generation of CBZ-Resistant Animals
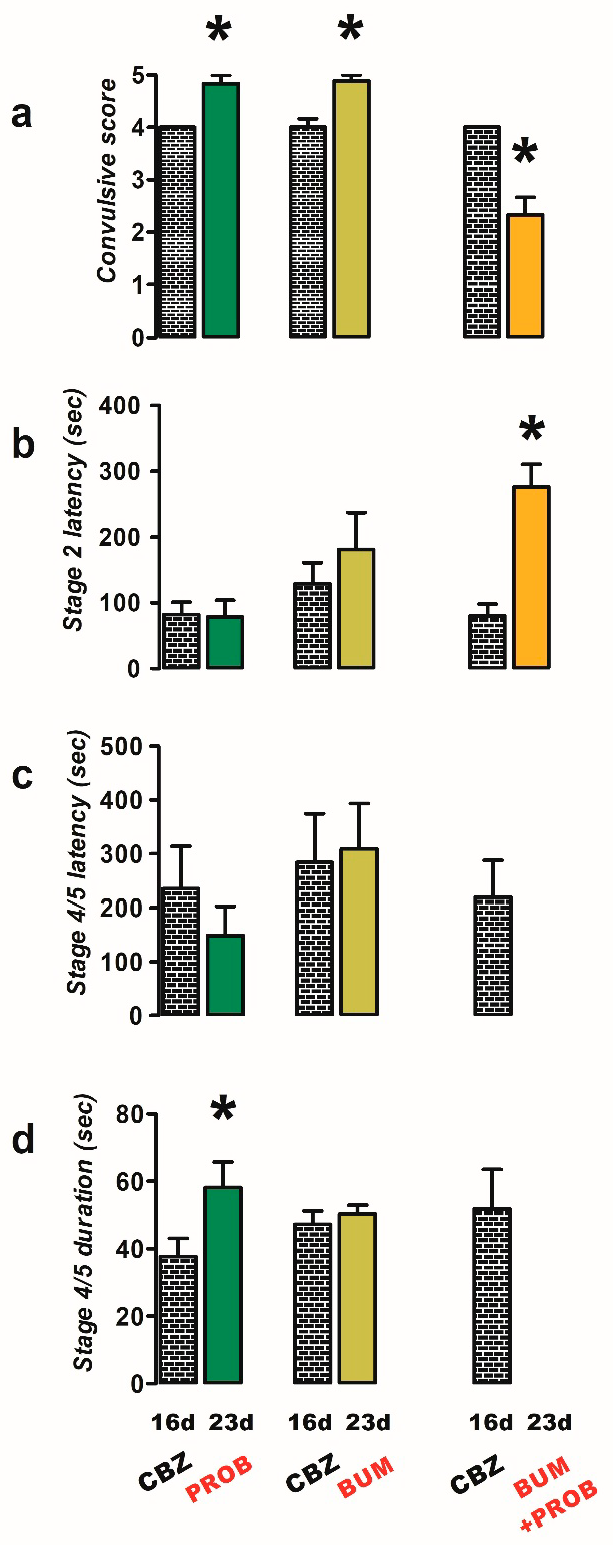
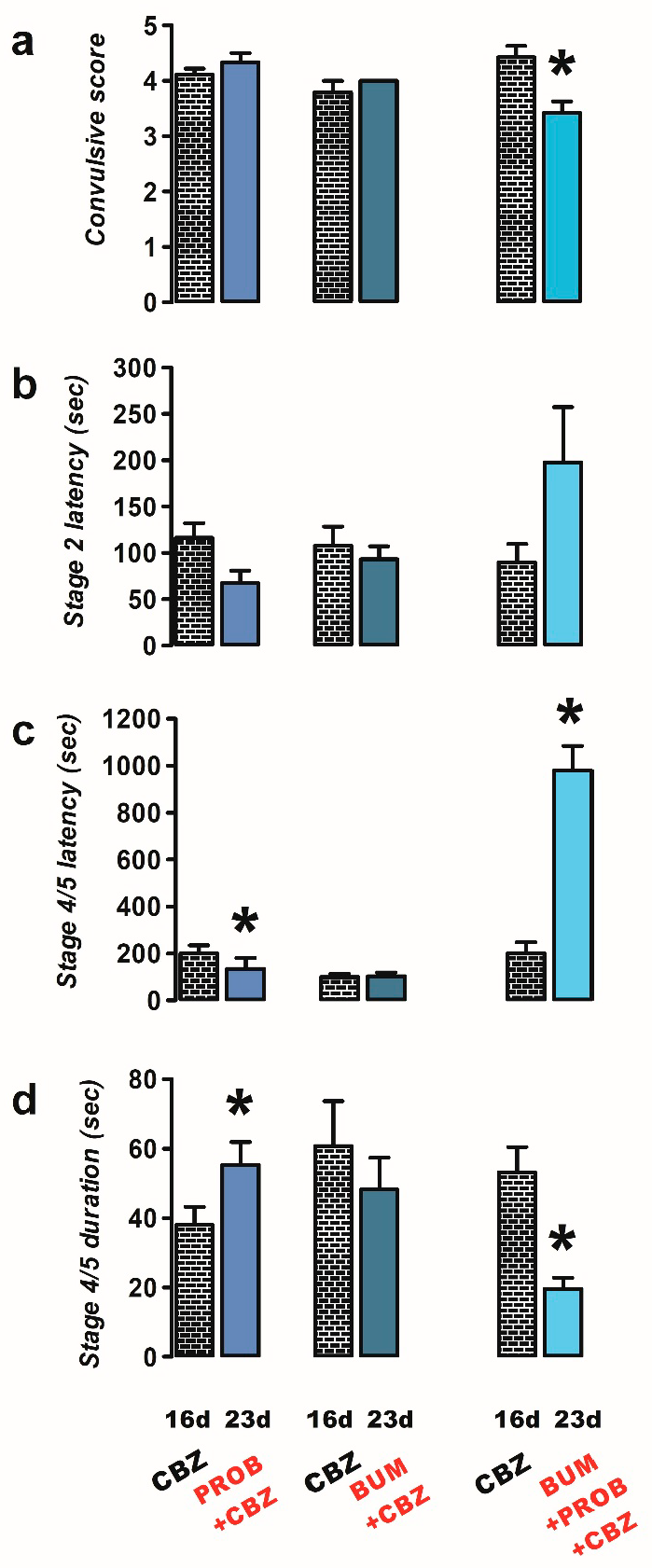
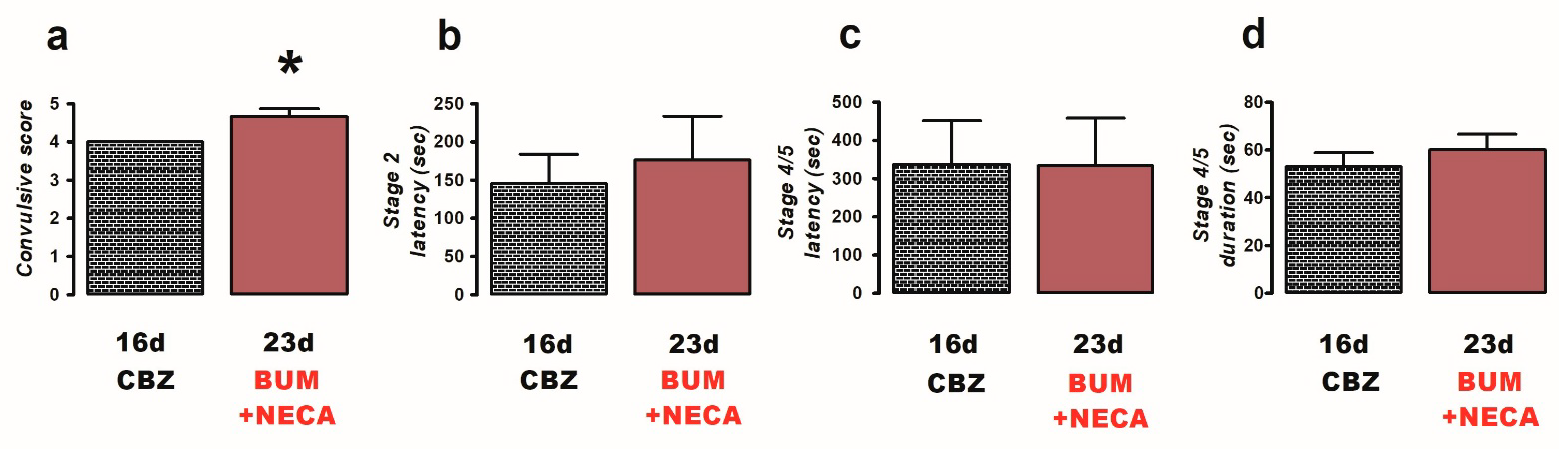
2.2. Effects of Bumetanide, Probenecid, and NECA on PTZ Induced Convulsions in CBZ-Resistant Rats
2.3. Changes in Protein Expression Levels of NKCC1 and KCC2 in Rat Brain Regions
2.3.1. NKCC1 in the Hippocampus
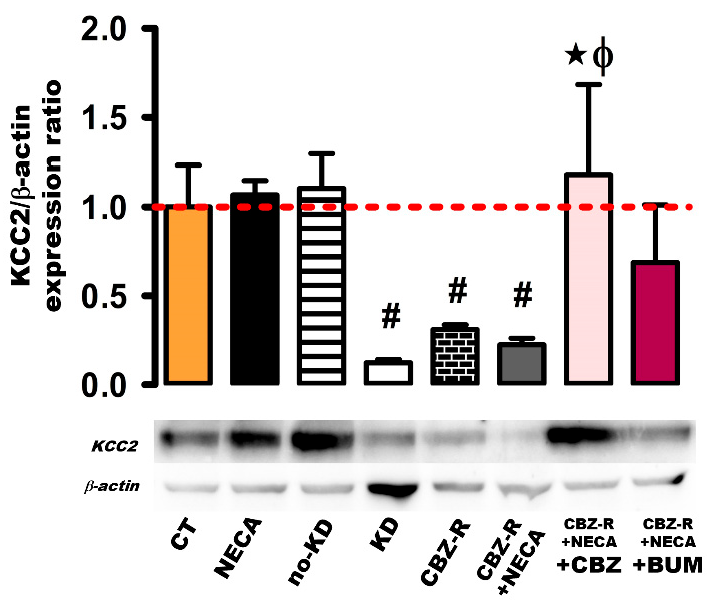
2.3.2. KCC2 in the Hippocampus
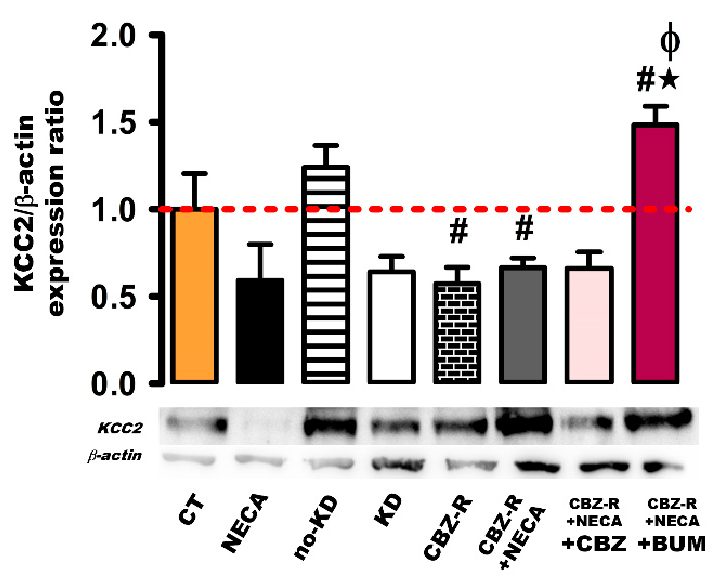
2.3.3. KCC2 in the Cortex
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Drugs and Chemicals
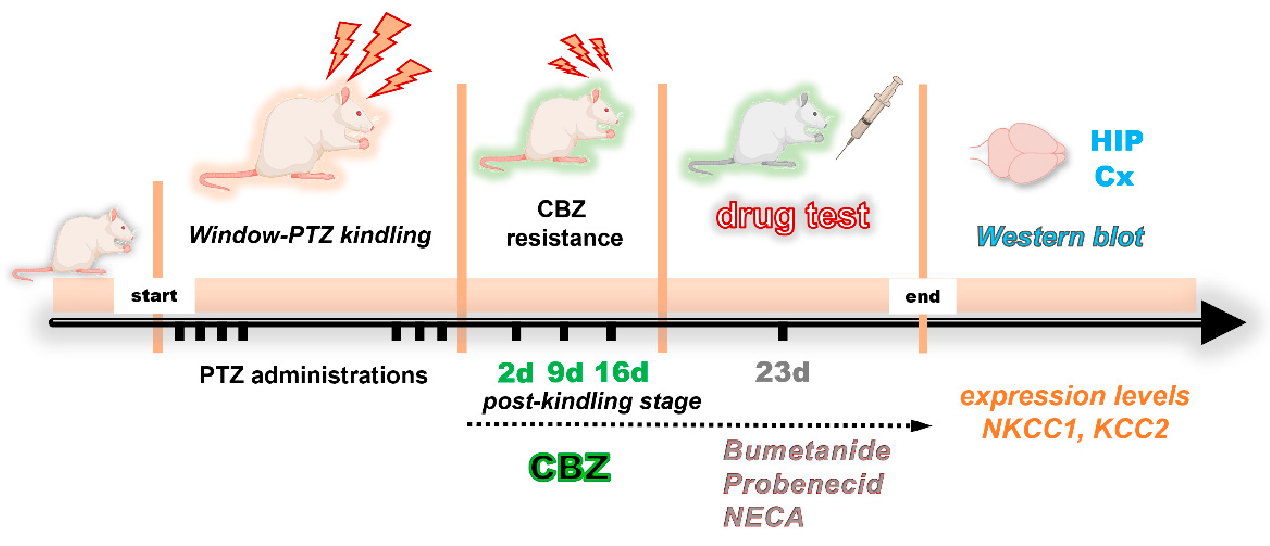
4.3. Development of Chemical Kindling Model and the Induction of Drug Resistance
4.4. Pharmacological Evaluation Design
- Probenecid group (n = 6): one week after the last administration of CBZ (day 23), rats were administered probenecid (50 mg/kg i.p.) 50 min prior to stimulation with PTZ;
- Bumetanide group (n = 9): these animals were treated as above, except that the rats were administered with bumetanide (10 mg/kg i.p.) 20 min prior to stimulation with PTZ;
- Probenecid + bumetanide group (n = 6): rats were administered with both probenecid and bumetanide, as specified in the previous groups;
- Probenecid + CBZ (n = 9): rats were administered probenecid (as specified in group 1) and CBZ (40 mg/kg i.p., 60 min before PTZ), respectively;
- Bumetanide + CBZ (n = 6): rats were treated as previously described, with the exception that they were administered bumetanide (10 mg/kg i.p.) and CBZ (40 mg/kg i.p.), as specified in the preceding groups;
- Bumetanide + probenecid + CBZ (n = 7): rats were treated with bumetanide (10 mg/kg i.p.), probenecid (50 mg/kg i.p.), and CBZ (40 mg/kg i.p.), as specified in the previous groups;
- Bumetanide + NECA (n = 6): rats were administered with both bumetanide (10 mg/kg i.p.) and NECA (1 mg/kg i.p., 8 h prior to stimulation with PTZ).
4.5. Western Blot
- CT: control rats that received a saline solution only;
- NECA: rats that received a single administration of NECA (1 mg/kg, i.p.) and were sacrificed 8 h later;
- no-KD: no kindled animals, animals that did not present three consequent Stage 4 or 5 convulsions during the last three PTZ injections;
- KD: fully kindled rats;
- CBZ-R: carbamazepine resistant rats;
- CBZ-R + NECA: CBZ-resistant rats treated with a single administration of NECA (1 mg/kg, i.p.) and sacrificed 8 h later;
- CBZ-R + NECA + CBZ: CBZ-resistant rats treated with a single administration of NECA (1 mg/kg, i.p.) and CBZ (40 mg/kg, i.p.);
- CBZ-R + NECA + BUM: CBZ-resistant rats treated with a single administration of NECA (1 mg/kg, i.p.) and bumetanide (10 mg/kg, i.p.).
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J.J.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef]
- Kwan, P.; Arzimanoglou, A.; Berg, A.T.; Brodie, M.J.; Allen Hauser, W.; Mathern, G.; Moshé, S.L.; Perucca, E.; Wiebe, S.; French, J. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010, 51, 1069–1077. [Google Scholar] [CrossRef]
- Löscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol. Rev. 2020, 72, 606–638. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, J.; Liang, S.; Zhang, G.; Yang, X. Role of NKCC1 and KCC2 in Epilepsy: From Expression to Function. Front. Neurol. 2020, 10, 1407. [Google Scholar] [CrossRef]
- McMoneagle, E.; Zhou, J.; Zhang, S.; Huang, W.; Josiah, S.S.; Ding, K.; Wang, Y.; Zhang, J. Neuronal K+-Cl− cotransporter KCC2 as a promising drug target for epilepsy treatment. Acta Pharmacol. Sin. 2024, 45, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.D.; Ishibashi, M.; Sinha, A.S.; Watanabe, M.; Kato, D.; Horiuchi, H.; Wake, H.; Fukuda, A. Astrocytic NKCC1 inhibits seizures by buffering Cl− and antagonizing neuronal NKCC1 at GABAergic synapses. Epilepsia 2023, 64, 3389–3403. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.E.C.; Almeida, A.G.; Silva, S.C.B.; Rodrigues, A.M.; Cecílio, S.G.; Scorza, C.A.; Finsterer, J.; Moret, M.; Scorza, F.A. The amygdala lesioning due to status epilepticus-Changes in mechanisms controlling chloride homeostasis. Clinics 2023, 78, 100159. [Google Scholar] [CrossRef]
- Palma, E.; Amici, M.; Sobrero, F.; Spinelli, G.; Di Angelantonio, S.; Ragozzino, D.; Mascia, A.; Scoppetta, C.; Esposito, V.; Miledi, R.; et al. Anomalous levels of Cl- transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc. Natl. Acad. Sci. USA 2006, 103, 8465–8468. [Google Scholar] [CrossRef]
- Sen, A.; Martinian, L.; Nikolic, M.; Walker, M.C.; Thom, M.; Sisodiya, S.M. Increased NKCC1 expression in refractory human epilepsy. Epilepsy Res. 2007, 74, 220–227. [Google Scholar] [CrossRef]
- Savardi, A.; Borgogno, M.; De Vivo, M.; Cancedda, L. Pharmacological tools to target NKCC1 in brain disorders. Trends Pharmacol. Sci. 2021, 42, 1009–1034. [Google Scholar] [CrossRef]
- Löscher, W.; Kaila, K. CNS pharmacology of NKCC1 inhibitors. Neuropharmacology 2022, 205, 108910. [Google Scholar] [CrossRef] [PubMed]
- Töllner, K.; Brandt, C.; Römermann, K.; Löscher, W. The organic anion transport inhibitor probenecid increases brain concentrations of the NKCC1 inhibitor bumetanide. Eur. J. Pharmacol. 2015, 746, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Carman, A.J.; Mills, J.H.; Krenz, A.; Kim, D.G.; Bynoe, M.S. Adenosine receptor signaling modulates permeability of the blood-brain barrier. J. Neurosci. 2011, 31, 13272–13280. [Google Scholar] [CrossRef]
- Kim, D.G.; Bynoe, M.S. A2A adenosine receptor modulates drug efflux transporter P-glycoprotein at the blood-brain barrier. J. Clin. Investig. 2016, 126, 1717–1733. [Google Scholar] [CrossRef]
- Zavala-Tecuapetla, C.; Orozco-Suarez, S.; Manjarrez, J.; Cuellar-Herrera, M.; Vega-Garcia, A.; Buzoianu-Anguiano, V. Activation of adenosine receptors modulates the efflux transporters in brain capillaries and restores the anticonvulsant effect of carbamazepine in carbamazepine resistant rats developed by window-pentylenetetrazole kindling. Brain Res. 2020, 1726, 146516. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Nigro, M.; Travagli, A.; Pasquini, S.; Vincenzi, F.; Varani, K.; Borea, P.A.; Merighi, S.; Gessi, S. Strategies for Drug Delivery into the Brain: A Review on Adenosine Receptors Modulation for Central Nervous System Diseases Therapy. Pharmaceutics 2023, 15, 2441. [Google Scholar] [CrossRef]
- Potschka, H.; Fedrowitz, M.; Löscher, W. Multidrug resistance protein MRP2 contributes to blood-brain barrier function and restricts antiepileptic drug activity. J. Pharmacol. Exp. Ther. 2003, 306, 124–131. [Google Scholar] [CrossRef]
- Yao, D.; Liu, L.; Jin, S.; Li, J.; Liu, X.D. Overexpression of multidrug resistance-associated protein 2 in the brain of pentylenetetrazole-kindled rats. Neuroscience 2012, 227, 283–292. [Google Scholar] [CrossRef]
- García-Rodríguez, C.; Mujica, P.; Illanes-González, J.; López, A.; Vargas, C.; Sáez, J.C.; González-Jamett, A.; Ardiles, Á.O. Probenecid, an Old Drug with Potential New Uses for Central Nervous System Disorders and Neuroinflammation. Biomedicines 2023, 11, 1516. [Google Scholar] [CrossRef]
- González-Guevara, E.; Lara-González, E.; Rendon-Ochoa, E.; Franco-Pérez, J.; Hernández-Cerón, M.; Laville, A.; Pérez-Severiano, F.; Martínez-de Los Santos, C.; Custodio, V.; Bargas, J.; et al. Inhibition of the NMDA Currents by Probenecid in Amygdaloid Kindling Epilepsy Model. Mol. Neurobiol. 2024, 61, 6264–6278. [Google Scholar] [CrossRef]
- Virtanen, M.A.; Uvarov, P.; Hübner, C.A.; Kaila, K. NKCC1, an Elusive Molecular Target in Brain Development: Making Sense of the Existing Data. Cells 2020, 9, 2607. [Google Scholar] [CrossRef]
- Puskarjov, M.; Kahle, K.T.; Ruusuvuori, E.; Kaila, K. Pharmacotherapeutic targeting of cation-chloride cotransporters in neonatal seizures. Epilepsia 2014, 55, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Römermann, K.; Fedrowitz, M.; Hampel, P.; Kaczmarek, E.; Töllner, K.; Erker, T.; Sweet, D.H.; Löscher, W. Multiple blood-brain barrier transport mechanisms limit bumetanide accumulation, and therapeutic potential, in the mammalian brain. Neuropharmacology 2017, 117, 182–194. [Google Scholar] [CrossRef]
- Kharod, S.C.; Kang, S.K.; Kadam, S.D. Off-Label Use of Bumetanide for Brain Disorders: An Overview. Front. Neurosci. 2019, 13, 310. [Google Scholar] [CrossRef] [PubMed]
- Töllner, K.; Brandt, C.; Töpfer, M.; Brunhofer, G.; Erker, T.; Gabriel, M.; Feit, P.W.; Lindfors, J.; Kaila, K.; Löscher, W. A novel prodrug-based strategy to increase effects of bumetanide in epilepsy. Ann. Neurol. 2014, 75, 550–562. [Google Scholar] [CrossRef]
- Töpfer, M.; Töllner, K.; Brandt, C.; Twele, F.; Bröer, S.; Löscher, W. Consequences of inhibition of bumetanide metabolism in rodents on brain penetration and effects of bumetanide in chronic models of epilepsy. Eur. J. Neurosci. 2014, 39, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.; Wielinga, P.; Zelcer, N.; De Haas, M.; Van Deemter, L.; Wijnholds, J.; Balzarini, J.; Borst, P. Characterization of the transport of nucleoside analog drugs by the human multidrug resistance proteins MRP4 and MRP5. Mol. Pharmacol. 2003, 63, 1094–1103. [Google Scholar] [CrossRef]
- VanWert, A.L.; Gionfriddo, M.R.; Sweet, D.H. Organic anion transporters: Discovery, pharmacology, regulation and roles in pathophysiology. Biopharm. Drug Dispos. 2010, 31, 1–71. [Google Scholar] [CrossRef]
- Donovan, M.D.; O’Brien, F.E.; Boylan, G.B.; Cryan, J.F.; Griffin, B.T. The effect of organic anion transporter 3 inhibitor probenecid on bumetanide levels in the brain: An integrated in vivo microdialysis study in the rat. J. Pharm. Pharmacol. 2015, 67, 501–510. [Google Scholar] [CrossRef]
- Zhang, G.; Franklin, P.H.; Murray, T.F. Anticonvulsant effect of N-ethylcarboxamidoadenosine against kainic acid-induced behavioral seizures in the rat prepiriform cortex. Neurosci. Lett. 1990, 114, 345–350. [Google Scholar] [CrossRef]
- Adami, M.; Bertorelli, R.; Ferri, N.; Foddi, M.C.; Ongini, E. Effects of repeated administration of selective adenosine A1 and A2A receptor agonists on pentylenetetrazole-induced convulsions in the rat. Eur. J. Pharmacol. 1995, 294, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Yang, L.; Zhou, J.; Zhu, D.; Guo, Q.; Chen, Z.; Chen, S.; Zhou, L. Anomalous expression of chloride transporters in the sclerosed hippocampus of mesial temporal lobe epilepsy patients. Neural Regen. Res. 2013, 8, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Gharaylou, Z.; Tafakhori, A.; Agah, E.; Aghamollaii, V.; Kebriaeezadeh, A.; Hadjighassem, M. A Preliminary Study Evaluating the Safety and Efficacy of Bumetanide, an NKCC1 Inhibitor, in Patients with Drug-Resistant Epilepsy. CNS Drugs 2019, 33, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, J.; Chen, Z.; Chen, S.; Zhu, F.; Zhou, L. Long-term expressional changes of Na+-K+-Cl− co-transporter 1 (NKCC1) and K+-Cl− co-transporter 2 (KCC2) in CA1 region of hippocampus following lithium-pilocarpine induced status epilepticus (PISE). Brain Res. 2008, 1221, 141–146. [Google Scholar] [CrossRef]
- Sivakumaran, S.; Maguire, J. Bumetanide reduces seizure progression and the development of pharmacoresistant status epilepticus. Epilepsia 2016, 57, 222–232. [Google Scholar] [CrossRef]
- Kourdougli, N.; Pellegrino, C.; Renko, J.M.; Khirug, S.; Chazal, G.; Kukko-Lukjanov, T.K.; Lauri, S.E.; Gaiarsa, J.L.; Zhou, L.; Peret, A.; et al. Depolarizing γ-aminobutyric acid contributes to glutamatergic network rewiring in epilepsy. Ann. Neurol. 2017, 81, 251–265. [Google Scholar] [CrossRef]
- Bonet-Fernández, J.M.; Tranque, P.; Aroca-Aguilar, J.D.; Muñoz, L.J.; López, D.E.; Escribano, J.; de Cabo, C. Seizures regulate the cation-Cl− cotransporter NKCC1 in a hamster model of epilepsy: Implications for GABA neurotransmission. Front. Neurol. 2023, 14, 1207616. [Google Scholar] [CrossRef]
- Othman, M.Z.; Mohd Nasir, M.H.; Wan Ahmad, W.A.N.; Abdullah, J.M.; Che Has, A.T. Differential regulation of KCC2 and NKCC1 expression by zolpidem in CA1 and CA3 hippocampal subregions of the lithium-pilocarpine status epilepticus rat model. Exp. Anim. 2025, 74, 286–299. [Google Scholar] [CrossRef]
- Muñoz, A.; Méndez, P.; DeFelipe, J.; Alvarez-Leefmans, F.J. Cation-chloride cotransporters and GABA-ergic innervation in the human epileptic hippocampus. Epilepsia 2007, 48, 663–673. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, S.; Jiang, Y.; Yang, Y.; Zhang, M.; Guo, Y.; Wang, S.; Ding, M.P. Fructose-1,6-diphosphate protects against epileptogenesis by modifying cation-chloride co-transporters in a model of amygdaloid-kindling temporal epilepticus. Brain Res. 2013, 1539, 87–94. [Google Scholar] [CrossRef]
- Eftekhari, S.; Mehrabi, S.; Soleimani, M.; Hassanzadeh, G.; Shahrokhi, A.; Mostafavi, H.; Hayat, P.; Barati, M.; Mehdizadeh, H.; Rahmanzadeh, R.; et al. BDNF modifies hippocampal KCC2 and NKCC1 expression in a temporal lobe epilepsy model. Acta Neurobiol. Exp. 2014, 74, 276–287. [Google Scholar] [CrossRef] [PubMed]
- González, M.I. Regulation of the cell surface expression of chloride transporters during epileptogenesis. Neurosci. Lett. 2016, 628, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wan, L.; Wu, Z.; Ren, W.; Huang, Y.; Qian, B.; Wang, Y. KCC2 downregulation facilitates epileptic seizures. Sci. Rep. 2017, 7, 156. [Google Scholar] [CrossRef]
- Hui, K.K.; Chater, T.E.; Goda, Y.; Tanaka, M. How Staying Negative Is Good for the (Adult) Brain: Maintaining Chloride Homeostasis and the GABA-Shift in Neurological Disorders. Front. Mol. Neurosci. 2022, 15, 893111. [Google Scholar] [CrossRef]
- Sica, D.A. Diuretic-related side effects: Development and treatment. J. Clin. Hypertens. 2004, 6, 532–540. [Google Scholar] [CrossRef]
- Ding, D.; Liu, H.; Qi, W.; Jiang, H.; Li, Y.; Wu, X.; Sun, H.; Gross, K.; Salvi, R. Ototoxic effects and mechanisms of loop diuretics. J. Otol. 2016, 11, 145–156. [Google Scholar] [CrossRef]
- Cheung, D.L.; Cooke, M.J.; Goulton, C.S.; Chaichim, C.; Cheung, L.F.; Khoshaba, A.; Nabekura, J.; Moorhouse, A.J. Global transgenic upregulation of KCC2 confers enhanced diazepam efficacy in treating sustained seizures. Epilepsia 2022, 63, e15–e22. [Google Scholar] [CrossRef]
- Jarvis, R.; Josephine Ng, S.F.; Nathanson, A.J.; Cardarelli, R.A.; Abiraman, K.; Wade, F.; Evans-Strong, A.; Fernandez-Campa, M.P.; Deeb, T.Z.; Smalley, J.L.; et al. Direct activation of KCC2 arrests benzodiazepine refractory status epilepticus and limits the subsequent neuronal injury in mice. Cell Rep. Med. 2023, 4, 100957. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Xin, H.; Shao, Y.; Dai, S.; Tan, N.; Li, Z.; Fei, F.; Wu, D.; Wang, Y.; Ping, Y.; et al. CRISPR-Based KCC2 Upregulation Attenuates Drug-Resistant Seizure in Mouse Models of Epilepsy. Ann. Neurol. 2023, 94, 91–105. [Google Scholar] [CrossRef]
- Zavala-Tecuapetla, C.; Manjarrez-Marmolejo, J.; Ramírez-Jarquín, J.O.; Rivera-Cerecedo, C.V. Eslicarbazepine, but Not Lamotrigine or Ranolazine, Shows Anticonvulsant Efficacy in Carbamazepine-Resistant Rats Developed by Window-Pentylenetetrazole Kindling. Brain Sci. 2022, 12, 629. [Google Scholar] [CrossRef]
- Hubrecht, R.C.; Carter, E. The 3Rs and Humane Experimental Technique: Implementing Change. Animals 2019, 9, 754. [Google Scholar] [CrossRef] [PubMed]
- Davoudi, M.; Shojaei, A.; Palizvan, M.R.; Javan, M.; Mirnajafi-Zadeh, J. Comparison between standard protocol and a novel window protocol for induction of pentylenetetrazol kindled seizures in the rat. Epilepsy Res. 2013, 106, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Racine, R.J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Corda, M.G.; Giorgi, O.; Longoni, B.; Orlandi, M.; Biggio, G. Decrease in the function of the gamma-aminobutyric acid-coupled chloride channel produced by the repeated administration of pentylenetetrazol to rats. J. Neurochem. 1990, 55, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Alex, A.B.; Wilcox, K.S.; White, H.S. Rapid loss of efficacy to the antiseizure drugs lamotrigine and carbamazepine: A novel experimental model of pharmacoresistant epilepsy. Epilepsia 2013, 54, 1186–1194. [Google Scholar] [CrossRef]
- Brandt, C.; Nozadze, M.; Heuchert, N.; Rattka, M.; Löscher, W. Disease-modifying effects of phenobarbital and the NKCC1 inhibitor bumetanide in the pilocarpine model of temporal lobe epilepsy. J. Neurosci. 2010, 30, 8602–8612. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zavala-Tecuapetla, C.; Orozco-Suárez, S.; Vega-García, A.; Manjarrez-Marmolejo, J. An Evaluation of Cation–Chloride Cotransporters NKCC1 and KCC2 in Carbamazepine-Resistant Rats. Int. J. Mol. Sci. 2025, 26, 4764. https://doi.org/10.3390/ijms26104764
Zavala-Tecuapetla C, Orozco-Suárez S, Vega-García A, Manjarrez-Marmolejo J. An Evaluation of Cation–Chloride Cotransporters NKCC1 and KCC2 in Carbamazepine-Resistant Rats. International Journal of Molecular Sciences. 2025; 26(10):4764. https://doi.org/10.3390/ijms26104764
Chicago/Turabian StyleZavala-Tecuapetla, Cecilia, Sandra Orozco-Suárez, Angélica Vega-García, and Joaquín Manjarrez-Marmolejo. 2025. "An Evaluation of Cation–Chloride Cotransporters NKCC1 and KCC2 in Carbamazepine-Resistant Rats" International Journal of Molecular Sciences 26, no. 10: 4764. https://doi.org/10.3390/ijms26104764
APA StyleZavala-Tecuapetla, C., Orozco-Suárez, S., Vega-García, A., & Manjarrez-Marmolejo, J. (2025). An Evaluation of Cation–Chloride Cotransporters NKCC1 and KCC2 in Carbamazepine-Resistant Rats. International Journal of Molecular Sciences, 26(10), 4764. https://doi.org/10.3390/ijms26104764