


Current Methods in Synovial Fluid Microbiota Characterization: A Systematic Review
 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Materials and Methods
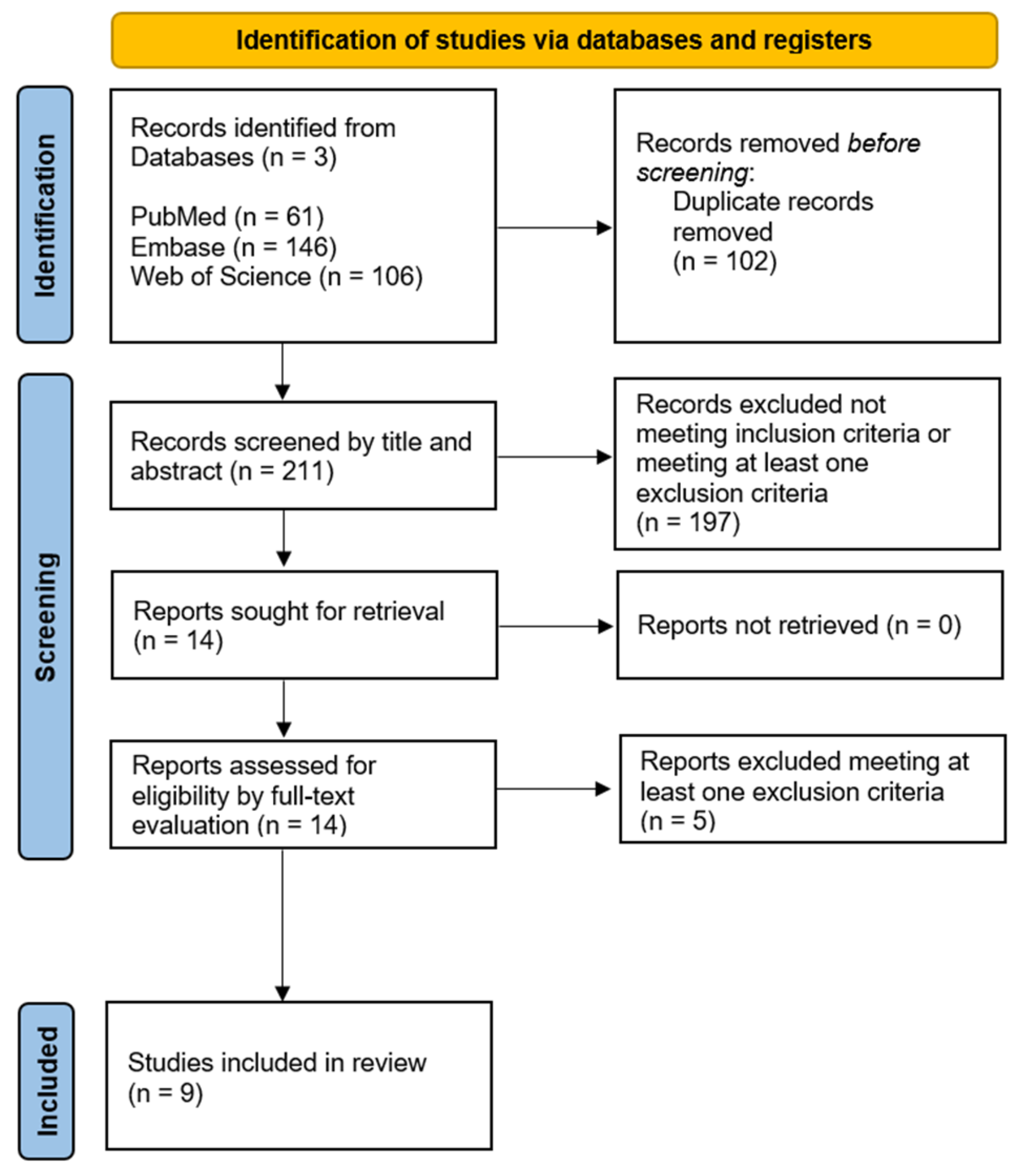
2.1. Database Search
2.2. Study Selection Process
2.3. Data Extraction and Synthesis
- sample collection, handling, and processing;
- DNA extraction method;
- library preparation (targeted regions, amplification, and contaminant control);
- sequencing technology;
- bioinformatics pipeline (quality filtering and denoising algorithms, clustering and classification, and quality control);
- downstream analyses (i.e., microbial diversity or others).
3. Results
3.1. Database Search and Study Selection
3.2. Data Extraction
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ASV | Amplicon sequence variant |
| GBD | Global Burden of Disease |
| IHMS | International Human Microbiome Standards |
| LPS | Lipopolysaccharides |
| NGS | Next-generation sequencing |
| OA | Osteoarthritis |
| OTU | Operational taxonomic unit |
| PCoA | Principal coordinate analysis |
| PCR | Polymerase chain reaction |
| PJI | Periprosthetic joint infection |
| PRISMA | Preferred Reporting Items for Systematic Reviews and Meta-analyses |
| RA | Rheumatoid arthritis |
| SF | Synovial fluid |
| SpA | Spondyloarthritis |
| STORMS | Strengthening The Organization and Reporting of Microbiome Studies |
References
- Steinmetz, J.D.; Culbreth, G.T.; Haile, L.M.; Rafferty, Q.; Lo, J.; Fukutaki, K.G.; Cruz, J.A.; Smith, A.E.; Vollset, S.E.; Brooks, P.M.; et al. Global, Regional, and National Burden of Osteoarthritis, 1990–2020 and Projections to 2050: A Systematic Analysis for the Global Burden of Disease Study 2021. Lancet Rheumatol. 2023, 5, e508–e522. [Google Scholar] [CrossRef] [PubMed]
- Losina, E.; Walensky, R.P.; Reichmann, W.M.; Holt, H.L.; Gerlovin, H.; Solomon, D.H.; Jordan, J.M.; Hunter, D.J.; Suter, L.G.; Weinstein, A.M.; et al. Impact of Obesity and Knee Osteoarthritis on Morbidity and Mortality in Older Americans. Ann. Intern. Med. 2011, 154, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Puig-Junoy, J.; Ruiz Zamora, A. Socio-Economic Costs of Osteoarthritis: A Systematic Review of Cost-of-Illness Studies. Semin. Arthritis Rheum. 2015, 44, 531–541. [Google Scholar] [CrossRef]
- Hunter, D.J.; Schofield, D.; Callander, E. The Individual and Socioeconomic Impact of Osteoarthritis. Nat. Rev. Rheumatol. 2014, 10, 437–441. [Google Scholar] [CrossRef]
- Ayral, X.; Pickering, E.H.; Woodworth, T.G.; Mackillop, N.; Dougados, M. Synovitis: A Potential Predictive Factor of Structural Progression of Medial Tibiofemoral Knee Osteoarthritis—Results of a 1 Year Longitudinal Arthroscopic Study in 422 Patients. Osteoarthr. Cartil. 2005, 13, 361–367. [Google Scholar] [CrossRef]
- De Roover, A.; Escribano-Núñez, A.; Monteagudo, S.; Lories, R. Fundamentals of Osteoarthritis: Inflammatory Mediators in Osteoarthritis. Osteoarthr. Cartil. 2023, 31, 1303–1311. [Google Scholar] [CrossRef]
- Dahaghin, S.; Bierma-Zeinstra, S.M.A.; Koes, B.W.; Hazes, J.M.W.; Pols, H.A.P. Do Metabolic Factors Add to the Effect of Overweight on Hand Osteoarthritis? The Rotterdam Study. Ann. Rheum. Dis. 2007, 66, 916–920. [Google Scholar] [CrossRef]
- Courties, A.; Berenbaum, F.; Sellam, J. The Phenotypic Approach to Osteoarthritis: A Look at Metabolic Syndrome-Associated Osteoarthritis. Jt. Bone Spine 2019, 86, 725–730. [Google Scholar] [CrossRef]
- Liu, S.; Li, G.; Xu, H.; Wang, Q.; Wei, Y.; Yang, Q.; Xiong, A.; Yu, F.; Weng, J.; Zeng, H. “Cross-Talk” between Gut Microbiome Dysbiosis and Osteoarthritis Progression: A Systematic Review. Front. Immunol. 2023, 14, 1150572. [Google Scholar]
- Collins, K.H.; Paul, H.A.; Reimer, R.A.; Seerattan, R.A.; Hart, D.A.; Herzog, W. Relationship between Inflammation, the Gut Microbiota, and Metabolic Osteoarthritis Development: Studies in a Rat Model. Osteoarthr. Cartil. 2015, 23, 1989–1998. [Google Scholar] [CrossRef]
- Sun, C.; Zhou, X.; Guo, T.; Meng, J. The Immune Role of the Intestinal Microbiome in Knee Osteoarthritis: A Review of the Possible Mechanisms and Therapies. Front. Immunol. 2023, 14, 1168818. [Google Scholar] [CrossRef] [PubMed]
- Guido, G.; Ausenda, G.; Iascone, V.; Chisari, E. Gut Permeability and Osteoarthritis, towards a Mechanistic Understanding of the Pathogenesis: A Systematic Review. Ann. Med. 2021, 53, 2380–2390. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.Y.; Stabler, T.; Pei, F.X.; Kraus, V.B. Both Systemic and Local Lipopolysaccharide (LPS) Burden Are Associated with Knee OA Severity and Inflammation. Osteoarthr. Cartil. 2016, 24, 1769–1775. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, B.; Li, S.; Yang, L.; Zhu, D.; Wang, Y.; Wang, H.; Wang, T.; Shi, B.; Gai, Z.; et al. Detection and Characterization of Bacterial Nucleic Acids in Culture-Negative Synovial Tissue and Fluid Samples from Rheumatoid Arthritis or Osteoarthritis Patients. Sci. Rep. 2018, 8, 14305. [Google Scholar] [CrossRef]
- Fernández-Rodríguez, D.; Baker, C.M.; Tarabichi, S.; Johnson, E.E.; Ciccotti, M.G.; Parvizi, J. Mark Coventry Award: Human Knee Has a Distinct Microbiome: Implications for Periprosthetic Joint Infection. J. Arthroplast. 2023, 38, S2–S6. [Google Scholar] [CrossRef]
- Borsinger, T.; Torchia, M.; Malskis, B.; Levy, B.A.; Werth, P.M.; Moschetti, W.E. Characterizing the Native Microbiome Using Next-Generation Sequencing of Bilateral ‘Aseptic’ Knees. J. Arthroplast. 2024, 39, 1317–1322. [Google Scholar] [CrossRef]
- Goswami, K.; Clarkson, S.; Tipton, C.; Phillips, C.D.; Dennis, D.A.; Klatt, B.A.; O’Malley, M.; Smith, E.L.; Gililland, J.; Pelt, C.E.; et al. The Microbiome of Osteoarthritic Hip and Knee Joints: A Prospective Multicenter Investigation. J. Bone Jt. Surg. 2023, 105, 821–829. [Google Scholar] [CrossRef]
- Torchia, M.T.; Amakiri, I.; Werth, P.; Moschetti, W. Characterization of Native Knee Microorganisms Using Next-Generation Sequencing in Patients Undergoing Primary Total Knee Arthroplasty. Knee 2020, 27, 1113–1119. [Google Scholar] [CrossRef]
- Tsai, J.C.; Casteneda, G.; Lee, A.; Dereschuk, K.; Li, W.T.; Chakladar, J.; Lombardi, A.F.; Ongkeko, W.M.; Chang, E.Y. Identification and Characterization of the Intra-Articular Microbiome in the Osteoarthritic Knee. Int. J. Mol. Sci. 2020, 21, 8618. [Google Scholar] [CrossRef]
- Bonanzinga, T.; Conte, P.; Anzillotti, G.; Longobardi, V.; Kon, E.; Rescigno, M.; Marcacci, M. Native Intra-Articular Knee Microbiome Is a Matter of Facts: A Systematic Review of Clinical Evidence. EFORT Open Rev. 2024, 9, 969–979. [Google Scholar] [CrossRef]
- Gilat, R.; Yazdi, A.A.; Weissman, A.C.; Joyce, K.M.; Bouftas, F.A.; Muth, S.A.; Chisari, E.; Shohat, N.; Cole, B.J. The Gut Microbiome and Joint Microbiome Show Alterations in Patients with Knee Osteoarthritis Versus Controls: A Systematic Review. Arthrosc. J. Arthrosc. Relat. Surg. 2024, 41, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, M.G.; Núñez-Carro, C.; Blanco-Blanco, M.; Blanco, F.J.; de Andrés, M.C. Inflammaging Contributes to Osteoarthritis Development and Human Microbiota Variations and Vice Versa: A Systematic Review. Osteoarthr. Cartil. 2024, 33, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 Statement: An Updated Guideline for Reporting Systematic Reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Elsawy, N.A.; Ibrahiem, A.H.; Younis, G.A.; Meheissen, M.A.; Abdel-Fattah, Y.H. Microbiome and Femoral Cartilage Thickness in Knee Osteoarthritis: Is There a Link? Cartilage 2024, 19476035241276852. [Google Scholar] [CrossRef]
- Cheng, M.; Zhao, Y.; Cui, Y.; Zhong, C.; Zha, Y.; Li, S.; Cao, G.; Li, M.; Zhang, L.; Ning, K.; et al. Stage-Specific Roles of Microbial Dysbiosis and Metabolic Disorders in Rheumatoid Arthritis. Ann. Rheum. Dis. 2022, 81, 1669–1677. [Google Scholar] [CrossRef]
- Hammad, D.B.M.; Liyanapathirana, V.; Tonge, D.P. Molecular Characterisation of the Synovial Fluid Microbiome in Rheumatoid Arthritis Patients and Healthy Control Subjects. PLoS ONE 2019, 14, e0225110. [Google Scholar] [CrossRef]
- Cristea, D.; Trandafir, M.; Bojinca, V.; Ciontea, A.; Andrei, M.; Popa, A.; Lixandru, B.; Militaru, C.; Nascutiu, A.; Predeteanu, D.; et al. Usefulness of Complex Bacteriological and Serological Analysis in Patients with Spondyloarthritis. Exp. Ther. Med. 2019, 17, 3465–3476. [Google Scholar] [CrossRef]
- Wensel, C.R.; Pluznick, J.L.; Salzberg, S.L.; Sears, C.L. Next-Generation Sequencing: Insights to Advance Clinical Investigations of the Microbiome. J. Clin. Investig. 2022, 132, e154944. [Google Scholar] [CrossRef]
- Ghyselinck, J.; Pfeiffer, S.; Heylen, K.; Sessitsch, A.; De Vos, P. The Effect of Primer Choice and Short Read Sequences on the Outcome of 16S RRNA Gene Based Diversity Studies. PLoS ONE 2013, 8, e71360. [Google Scholar] [CrossRef]
- Abellan-Schneyder, I.; Matchado, M.S.; Reitmeier, S.; Sommer, A.; Sewald, Z.; Baumbach, J.; List, M.; Neuhaus, K. Primer, Pipelines, Parameters: Issues in 16S RRNA Gene Sequencing. mSphere 2021, 6, e01202-20. [Google Scholar] [CrossRef]
- Mirzayi, C.; Renson, A.; Furlanello, C.; Sansone, S.A.; Zohra, F.; Elsafoury, S.; Geistlinger, L.; Kasselman, L.J.; Eckenrode, K.; van de Wijgert, J.; et al. Reporting Guidelines for Human Microbiome Research: The STORMS Checklist. Nat. Med. 2021, 27, 1885–1892. [Google Scholar] [CrossRef] [PubMed]
- Panek, M.; Čipčić Paljetak, H.; Barešić, A.; Perić, M.; Matijašić, M.; Lojkić, I.; Bender, D.V.; Krznarić, Ž.; Verbanac, D. Methodology Challenges in Studying Human Gut Microbiota-Effects of Collection, Storage, DNA Extraction and next Generation Sequencing Technologies. Sci. Rep. 2018, 8, 5143. [Google Scholar] [CrossRef] [PubMed]
- Nearing, J.T.; Comeau, A.M.; Langille, M.G.I. Identifying Biases and Their Potential Solutions in Human Microbiome Studies. Microbiome 2021, 9, 113. [Google Scholar] [CrossRef] [PubMed]
- Chiarello, M.; McCauley, M.; Villéger, S.; Jackson, C.R. Ranking the Biases: The Choice of OTUs vs. ASVs in 16S RRNA Amplicon Data Analysis Has Stronger Effects on Diversity Measures than Rarefaction and OTU Identity Threshold. PLoS ONE 2022, 17, e0264443. [Google Scholar] [CrossRef]
- Kers, J.G.; Saccenti, E. The Power of Microbiome Studies: Some Considerations on Which Alpha and Beta Metrics to Use and How to Report Results. Front. Microbiol. 2022, 12, 796025. [Google Scholar] [CrossRef]

{kind=link}
| Authors, Year | Aim | Population | Analyses | Results |
|---|---|---|---|---|
| Elsawy, 2024 [24] | Assess the relation between microbiome and LPS in the blood and SF with FCT knee OA patients | 40 patients with knee OA (F: 72.5%)—Egypt | • PCR for microbiomes and ELISA for LPS in serum and SF • US for FCT | • 100% OA patients had bacterial DNA in SF • Firmicutes was the most abundant (63.59%), followed by Actinobacteria (24.14%), Proteobacteria (11.51%), and Bacteroidetes. |
| Fernandez-Rodríguez, 2023 [15] | Investigate presence of the microbiome in human knee and compare the profile in different knee conditions | 15 healthy volunteers, 14 patients with OA undergoing TKA, 12 septic revision surgery, 10 contralateral knee non-OA, 14 aseptic revision surgery (F: 50.9%)—United States | Culture and NGS analysis of SF | • 13.8% positive SF cultures • Highest number of species in native OA knees • Cutibacterium, Staphylococcus, and Paracoccus dominant in non-OA knees • Proteobacteria dominant in OA knees • Similar trend in composition for contralateral and aseptic revision knees • NGS of PJI SF confirmed culture results |
| Goswami, 2023 [17] | • Explore the microbial composition within joints of OA patients during joint arthroplasty • Quantify impact of factors associated with microbiome activity and their source | 117 patients with hip or knee OA undergoing arthroplasty from 13 institutions—United States | NGS of SF, ST, and swab specimens | • 87% of SF samples positive to NGS, only 38% ≥ 800 reads • Escherichia, Cutibacterium, Staphylococcus, Acinetobacter, and Pseudomonas were the most abundant genera • Hospital explained a portion (18.5%) of the variance in composition • Corticosteroid injection associated with high abundance of several lineages |
| Borsinger, 2024 [16] | Evaluate the presence of microorganisms in bilateral native knees with OA | 40 patients undergoing primary UKA (n = 30) or bilateral (n = 10) TKA (F: 54%)—United States | • NGS of SF, ST and swabs • SF biomarkers: CRP, WBC count, and %PMNs. | • Positive NGS in 3 of 80 samples (3.8%), two below threshold • Two multiple microorganisms identified (1 knee with 4 microorganisms; 1 knee with 2 microorganisms) • Cutibacterium acnes was the most common (2 out of 3 samples) species • No association between patient characteristic and positive NGS |
| Cheng, 2022 [25] | • Investigatestage-specific roles of microbial dysbiosis and metabolic disorders in RA • Comparison healthy vs. OA vs. RA | 76 with RA (stages I–IV), 19 with OA and 27 healthy for fecal-plasma study, and 271 with RA (stages I–IV) for knee SF study (F: 77.9%)—China | • Stage-based profiles of faecal metagenome and plasma metabolome • SF NGS analysis, bacterial isolation and scanning electron microscopy | • Not enough bacteria DNA in stages I-III • Most of the microbes in SF were Proteobacteria and Firmicutes |
| Torchia, 2020 [18] | Characterize the native knee microorganism profile in patients undergoing TKA | 40 patients with OA undergoing TKA (F: 52.5%)—Unites States | NGS for SF bacterial detection | • 30% of TKA patients with organisms identified by NGS • An average of 4.6 organisms found among patients with positive NGS results, and 48 unique organisms identified by NGS from all samples |
| Hammad, 2019 [26] | • Investigate SF for presence of bacterial and fungal DNA • Investigate association between bacterial and fungi community composition to synovial inflammation markers | 16 patients with RA and 9 healthy volunteers (F: 48%)—United Kingdom | • NGS for SF bacterial and fungal detection • ELISA for inflammatory markers levels (IL-6, IL-17A, IL-22, and IL-23) | • Bacterial DNA detected in the SF of 87.5% patients with RA, and in 100% healthy controls • Fungal DNA detected in 75% RA samples, and 89% healthy controls • SF predominated by Proteobacteria (Control = 83.5%, RA = 79.3%) and Firmicutes (Control = 16.1%, RA = 20.3%) and Actinobacteria (Control = 0.2%, RA = 0.3%) and Bacteroidetes (Control = 0.1%, RA = 0.1%) • SF predominated by members of the Basidiomycota (Control = 53.9%, RA = 46.9%) and Ascomycota (Control = 35.1%, RA = 50.8%) |
| Cristea, 2019 [27] | Identify which parameters best explain the correlation between SpA activity/severity andmicrobiological results/immune status against intestinal and/or urogenital pathogens. | 27 patients with SpA and 26 healthy volunteers (F: 32.1%)—Romania | • Microbiological investigation (culture, nucleic acid-based assays) of stool, urine, SF and serum. • Screening of anti-thyroid antibodies and peroxidase • Detection of HLA-B27 by PCR and the determination of CRP biomarker | • SF from 3 SpA patients negative on culture and PCR analysis |
| Zhao, 2018 [14] | Characterize potential SF bacterial nucleic acids | 125 patients with RA (5 ST and 110 SF samples), and 58 with OA (16 ST and 42 SF samples) (F: 83.1%)—China | NGS for bacterial detection | • Abundant diversity of bacterial DNA, including Porphyromonas and Bacteroides in 100% samples • Veillonella dispar, Haemophilus parainfluenzae, Prevotella copri and Treponema amylovorum more abundant in SF of RA • Bacteroides caccae more abundant in SF of OA |
| Author, Year | Sample Collection | DNA Extraction | Library Preparation | Sequencing | Bioinformatics Pipeline | Downstream Analyses | ||
|---|---|---|---|---|---|---|---|---|
| Size, collection, sterility | Method—Kit | Targeted regions and amplification | Contaminants | Platform | Filtering/Denoising | Clustering and classification | ||
| NGS | ||||||||
| Fernàndez-Rodrìguez, 2023 [15] | • knee • 3 mL min • aseptic technique • in the operating room • before arthrotomy/at the start of the elective procedure | • mechanical lysis with Qiagen TissueLyser • extraction with KingFisher Flex Purification System | • 16S rRNA V2-V3 •PCR on LightCycler 480 II (Roche Life Science) | negative extraction controls included | Illumina MiSeq | N/A | • clustering not directly reported • removal of commonly recognized contaminants, ‘No hit’ OTUs, OTUs below 2% of relative abundance | • rarefaction • relative abundance |
| Goswami, 2023 [17] | • hip and knee • 2 mL • aseptic technique • in the operating room • before arthrotomy • using 18-gauge needle | not directly reported but similar to methods described in previous studies | • 16S rRNA V1-V2 • PCR not directly reported but similar to methods described in previous studies | negative laboratory procedures controls included | IonTorrent PGM4 and Illumina Miseq26 | not directly reported but similar to processing described in previous studies | • clustering not directly reported but similar to processing described in previous studies • removal of contaminants identified by controls with no PCR template, OTUs detected in <1% of samples | • relative abundance • α-diversity: Hill1 (exponent of the Shannon diversity) • β-diversity: Bray–Curtis dissimilarity PCoA |
| Borsinger, 2024 [16] | • knee • 1 cm 3 min • aseptic technique • in the operating room • before arthrotomy after skin incision (for the operative knee) • aseptic aspiration (for the non-operative knee) • transport in sterile containers | extraction with Zymo Research kit | • 16S rRNA V1-V2 and ITS • PCR on Applied Biosystems • products combined based on qualitative band strength • size selection with Agencourt AMPure XP beads and Qiagen Minelute Kit • library quantified with Qubit 4.0 fluorometer | 3 negative extraction controls run every 92 samples | Ilumina MiSeq | • de novo chimera removal in UCHIME • removal of any read below the quality score, quality metric, or length • removal of any sequence that did not contain a valid barcode (demultiplexing with internally developed algorithm) | • OTU global alignment using USEARCH against a database of high-quality sequences • species below 2% reported as number of reads | • percent dominance |
| Cheng, 2022 [25] | • knee • 5 mL • aseptic technique in sterile atmosphere • during therapeutic aspiration • placed in sterile tubes on ice and homogenized within 5 min of collection, immediately frozen and kept w/o heparin or hyaluronidase at −135 °C | extraction with TIANGEN bacterial DNA kit | • 16S rRNA V1-V2 • PCR not directly reported | environmental control (PBS tube left open during the procedure) run in parallel | Illumina HiSeq 2500 | • reads trimmed for adapter and primer sequences, splitted according to the barcodes in QIIME • Reads with average quality less than 25 were removed • sequences joined with FLASH method • de novo chimera removal with USEARCH | • open reference OTU picking protocol QIIME at 97% sequence similarity • RDP classifier vs. GreenGene database with 0.8 confidence. | • relative abundance |
| Torchia, 2020 [18] | • knee • 1 cm3 • in the operating room • before arthrotomy after skin incision • tube placed in sterile container • shipped at ambient temperature | N/A | • 16S rRNA and ITS • emulsion PCR | environmental control (four operative field sterile water collected at different time points) run in parallel | IonTorrent PGM4 | N/A | cross-reference of the sequences against the NIH/Gen-bank database with 90% agreement | • relative quantity (DNA load) |
| Hammad, 2019 [26] | • knee • aseptic technique • aspiration (either therapeutic or for donation) • transferred to sterile micro-centrifuge tube at −80 °C | no extraction to avoid contaminants | • 16S rRNA V4 and ITS2 • direct PCR: two rounds to add sequencing adapters • purification with AMPure XP magnetic beads • DNA quantification using Qubit 3.0 hsDNA kit | one negative reaction control (UV-irradiated nuclease-free water) run in parallel during both reactions | Ilumina MiSeq | Nephele 16S/ITS paired-end QIIME pipeline | open reference OTU clustering against SILVA database for bacteria and ITS for fungi at 99% sequence identity in QIIME | • rarefaction • relative abundance • PCoA based on Bray–Curtis distance matrix |
| Zhao, 2018 [14] | • knee • aseptic technique in sterile environment • during therapeutic aspiration • placed in sterile tubes on ice and homogenized within five minutes of collection, immediately frozen and kept w/o heparin or hyaluronidase at –135 °C | • extraction with DNeasy Blood and Tissue Kit • DNA quantitation with Nanodrop 2000 | • 16S rRNA V1-V2 • PCR • amplicons purified using QIAquick PCR Purification Kit | sample collection controls, reaction mixture controls and environmental control (PBS tube left open during the procedure) run in parallel | Illumina HiSeq | • sequences joined and quality filtered with FLASH method • de novo chimera removal with USEARCH | • first OTU clustering against 2013 GreenGenes database at 97% similarity • second de novo OTU clustering at 97% similarity with UCLUST • QIIME RDP classifier against the GreenGenes data set • UPGMA hierarchical clustering (average linkage) on the distance matrix of OTU abundance • Newick formatted tree with the QIIME package | • rarefaction• relative abundance • α-diversity: Shannon index using OTU tables rarefied to the lowest number of reads obtained for any of the samples analyzed. • PCoA based on unweighted and weighted UniFrac distances using evenly sampled OTU abundances • Prediction of functional composition with PICRUSt using KEGG pathways |
| Real-time PCR | ||||||||
| Elsawy, 2024 [24] | • knee • aspiration in the operating room • aseptic technique | extraction with QIAamp DNA Mini kit | • 16S rRNA (as denominator) • Firmicutes • Bacteroidetes • Proteobacteria (delta and gamma) • Actinobacteria • Rotor Gene Q real-time PCR machine | negative extraction and reaction controls (sterile distilled water) included in each PCR reaction | N/A | N/A | N/A | • relative abundance • α-diversity: Shannon index |
| Broad range PCR | ||||||||
| Cristea, 2019 [27] | N/A | extraction with PureLink Genome DNA Mini kit | • 16S rRNA • Broad range PCR | N/A | N/A | N/A | N/A | N/A because there were no samples positive to PCR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bardi, E.; D’Arrigo, D.; Pozzi, C.; Gatti, A.; Bertolino, L.; Favaro, A.; Rescigno, M.; Bonanzinga, T. Current Methods in Synovial Fluid Microbiota Characterization: A Systematic Review. Int. J. Mol. Sci. 2025, 26, 4690. https://doi.org/10.3390/ijms26104690
Bardi E, D’Arrigo D, Pozzi C, Gatti A, Bertolino L, Favaro A, Rescigno M, Bonanzinga T. Current Methods in Synovial Fluid Microbiota Characterization: A Systematic Review. International Journal of Molecular Sciences. 2025; 26(10):4690. https://doi.org/10.3390/ijms26104690
Chicago/Turabian StyleBardi, Elena, Daniele D’Arrigo, Chiara Pozzi, Andrea Gatti, Luca Bertolino, Alberto Favaro, Maria Rescigno, and Tommaso Bonanzinga. 2025. "Current Methods in Synovial Fluid Microbiota Characterization: A Systematic Review" International Journal of Molecular Sciences 26, no. 10: 4690. https://doi.org/10.3390/ijms26104690
APA StyleBardi, E., D’Arrigo, D., Pozzi, C., Gatti, A., Bertolino, L., Favaro, A., Rescigno, M., & Bonanzinga, T. (2025). Current Methods in Synovial Fluid Microbiota Characterization: A Systematic Review. International Journal of Molecular Sciences, 26(10), 4690. https://doi.org/10.3390/ijms26104690