
In Vitro and In Silico Evaluation of the Potential Anti-Prostate Cancer Activity of Rosmarinus officinalis L. Leaf Extracts

 , , ,
, , , 
Abstract
1. Introduction
2. Results
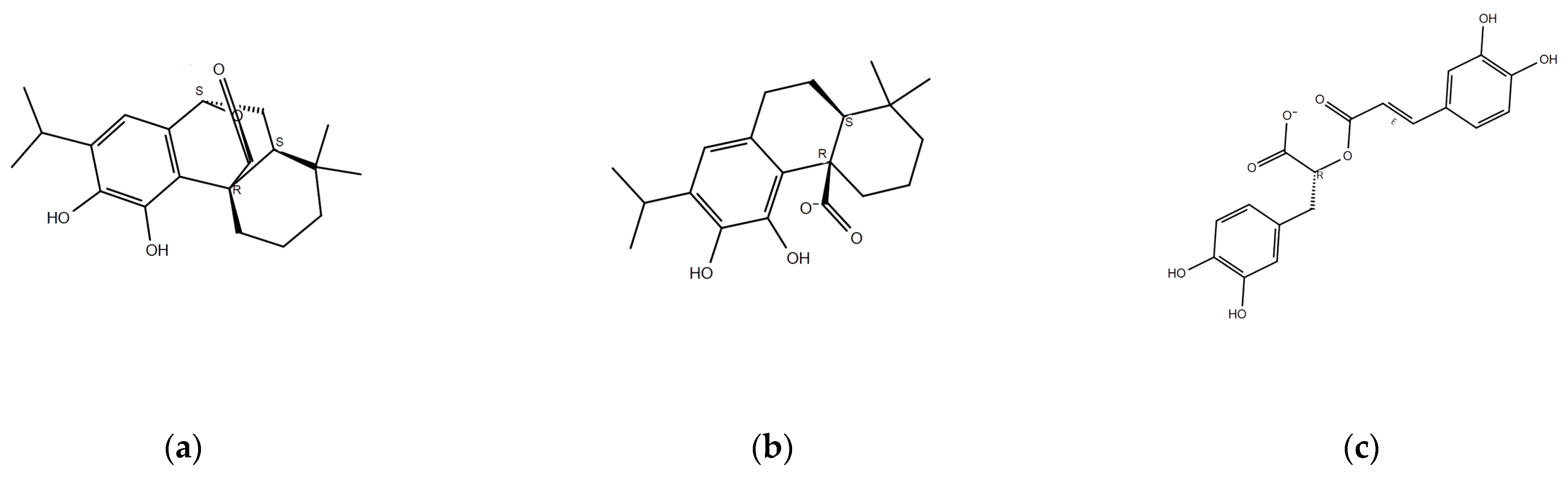
2.1. Identification of Major Components of Rosemary
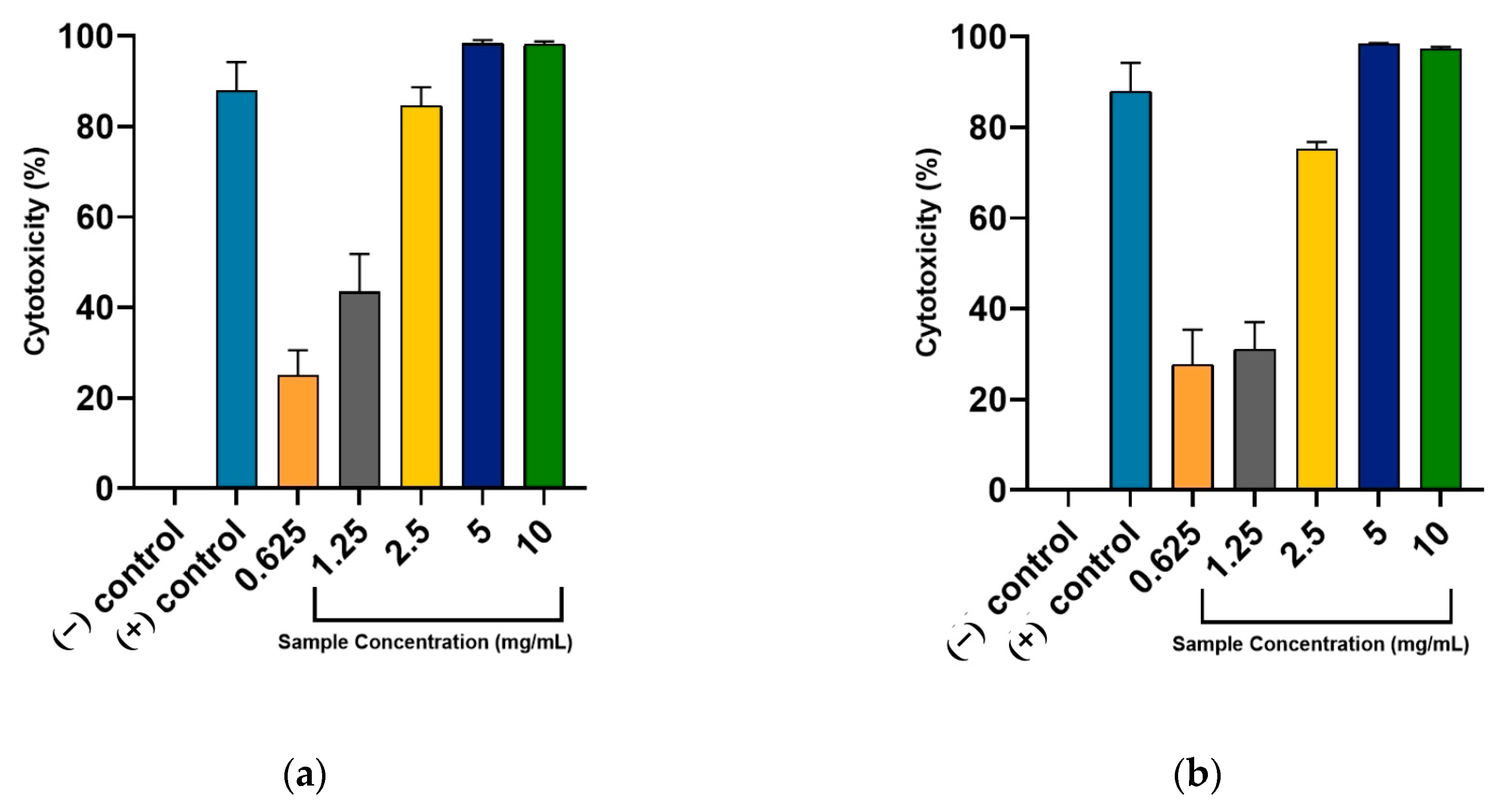
2.2. Anti-Prostate Cancer Activity of Rosemary Extracts
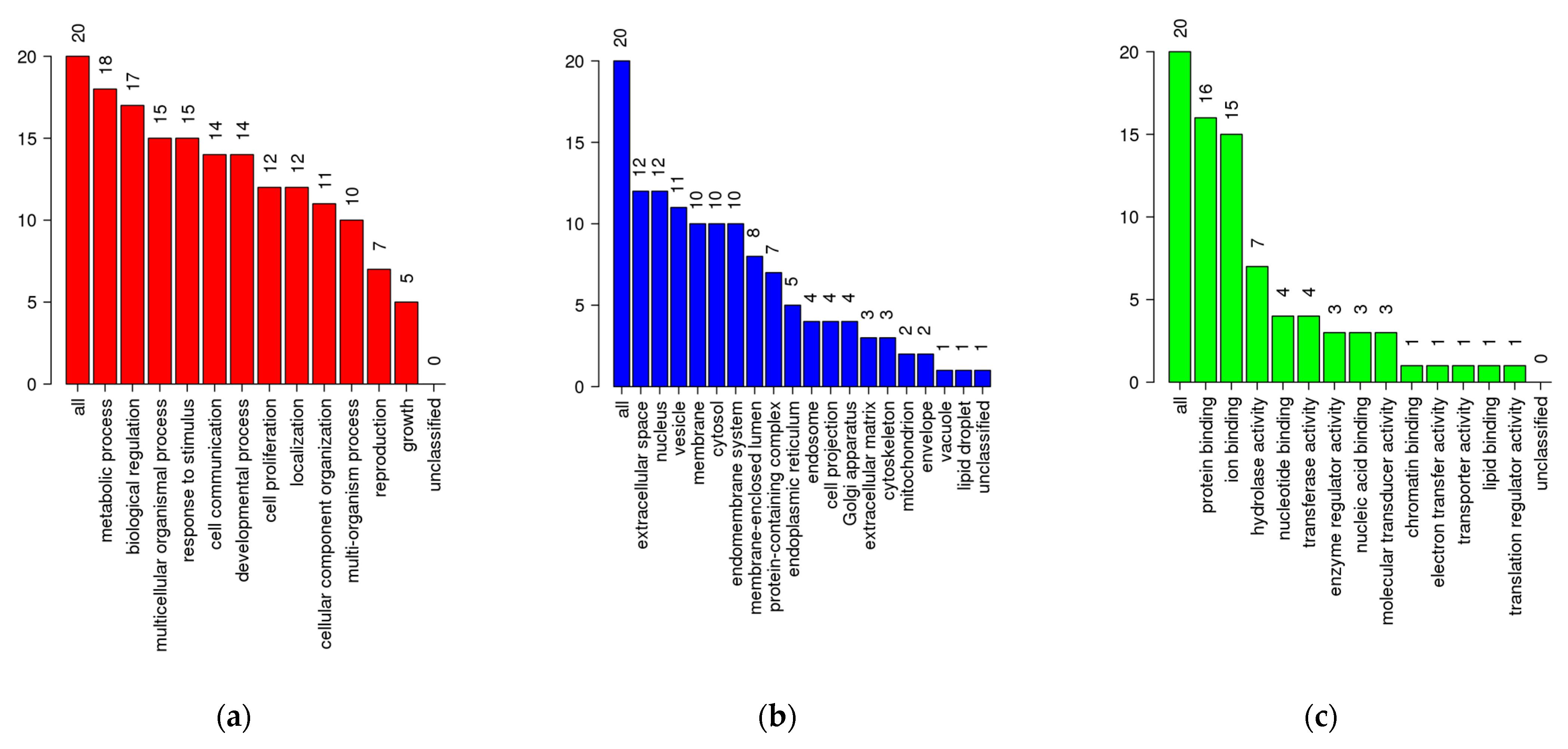
2.3. Network Pharmacology
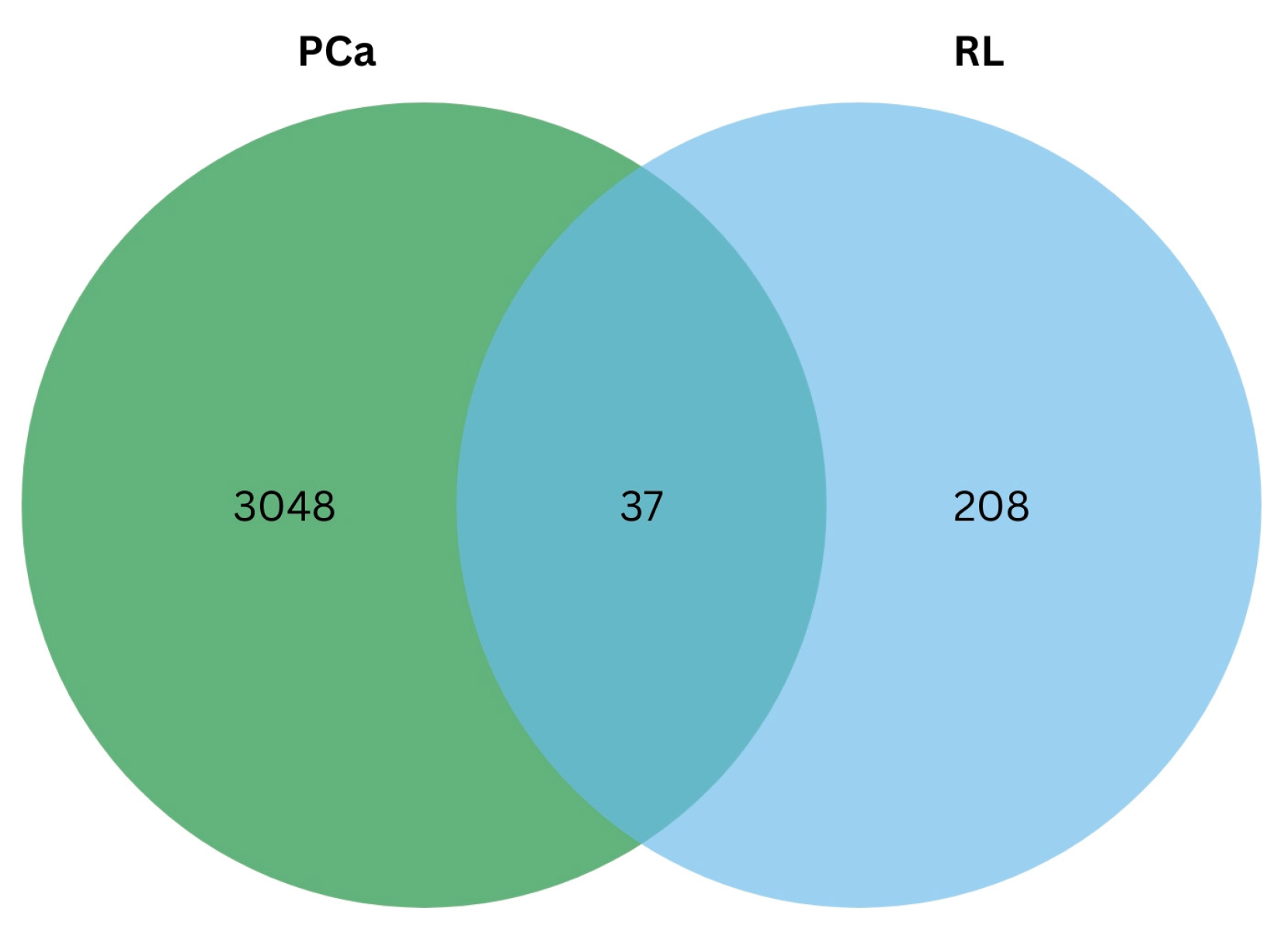
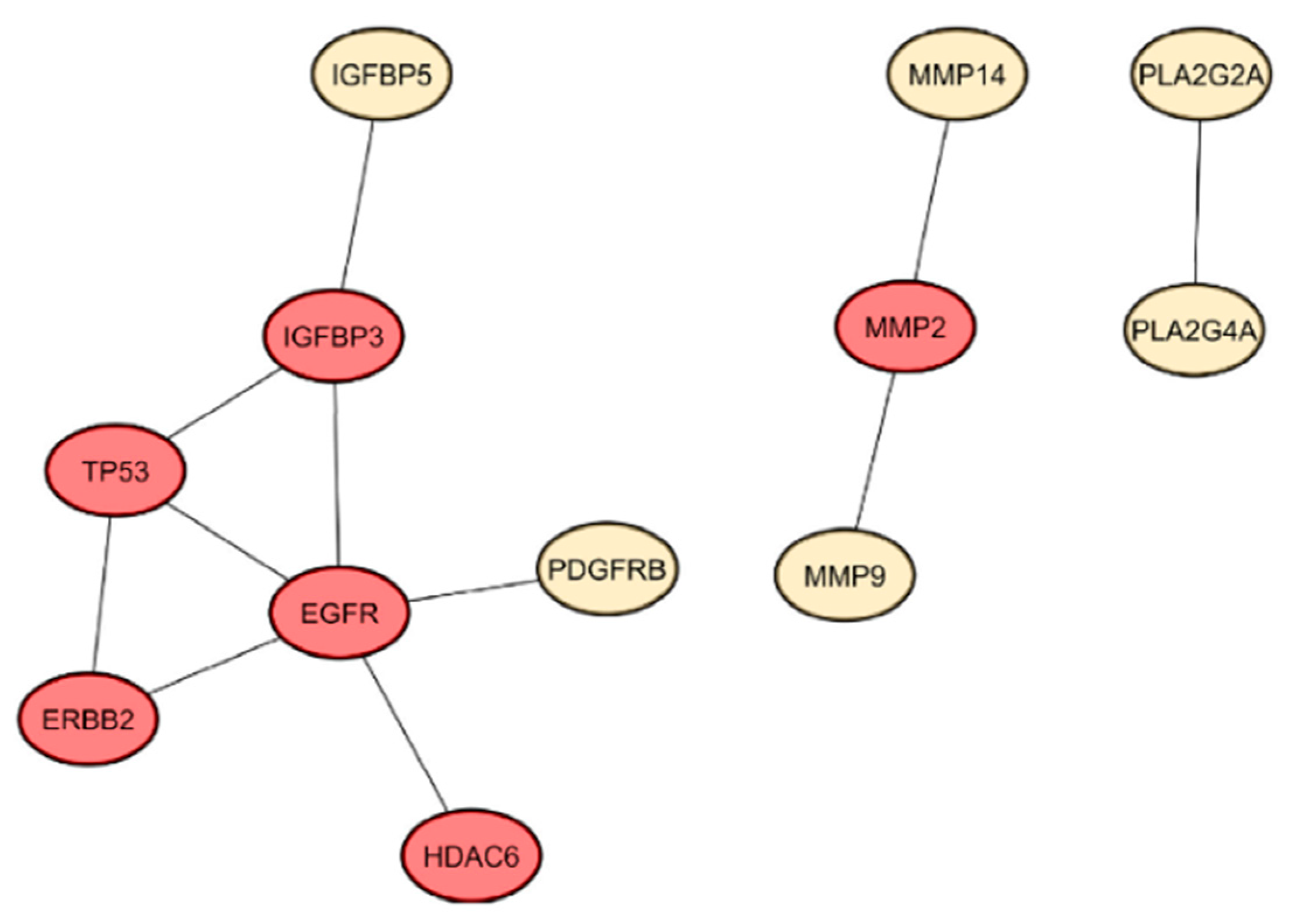
2.3.1. Protein–Protein Interaction Network of Identified Gene Targets
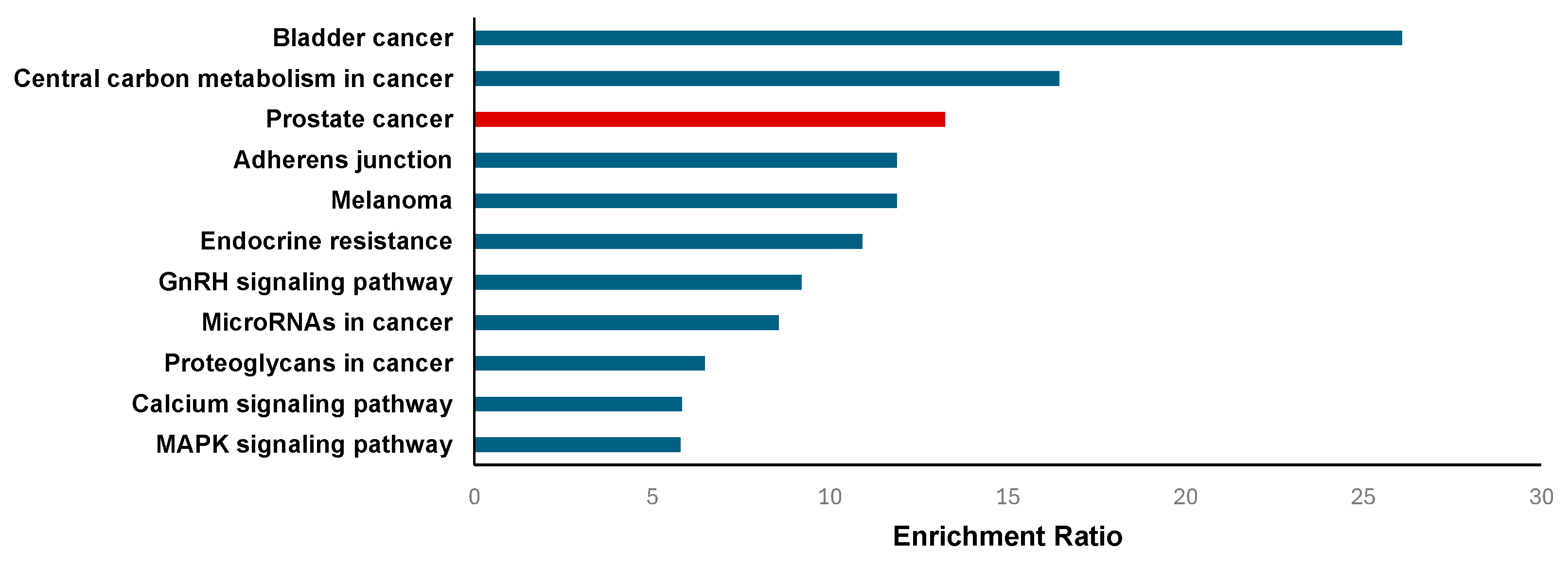
2.3.2. Enrichment Analysis
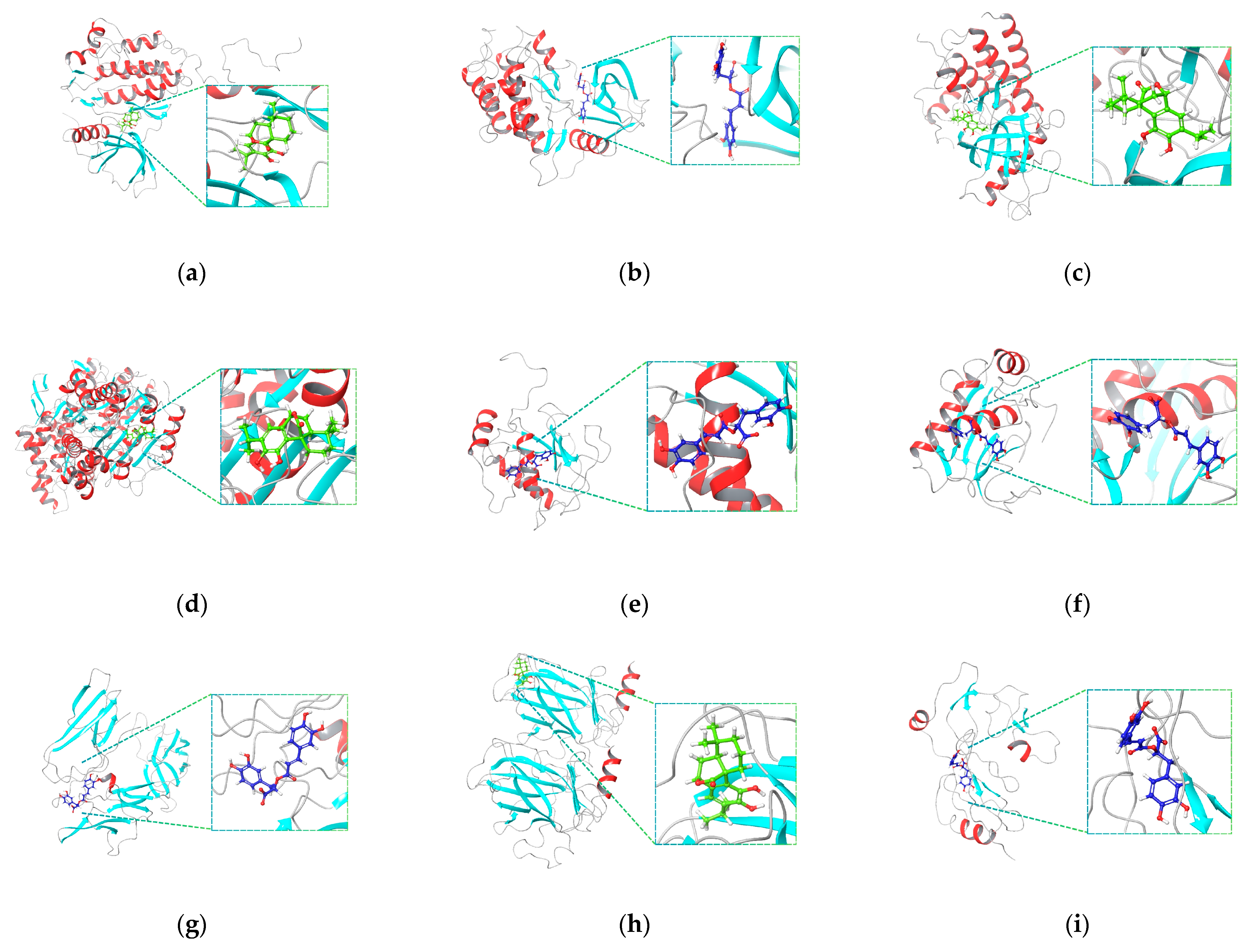
2.4. Molecular Docking Simulation and MM-GBSA Calculation
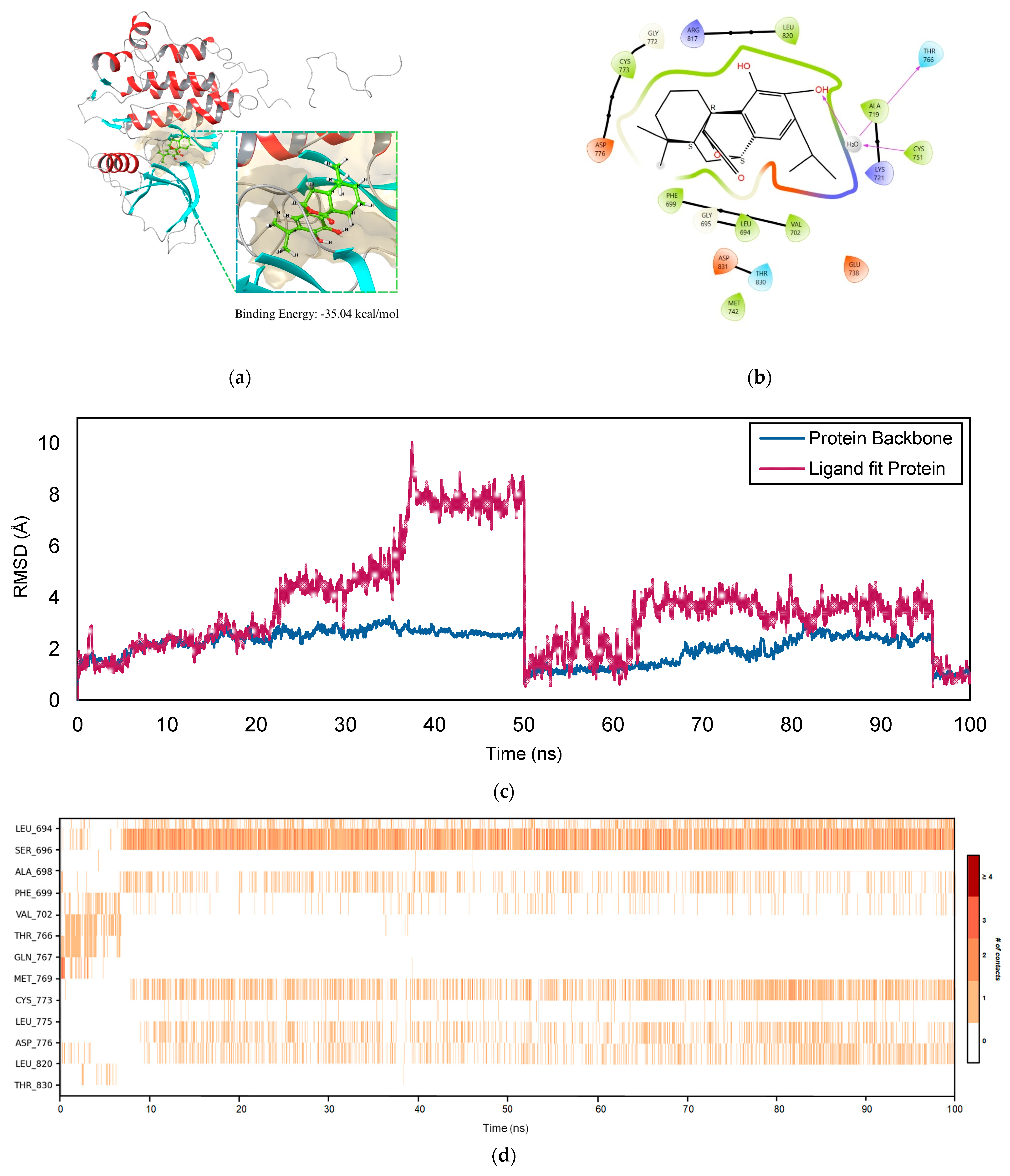
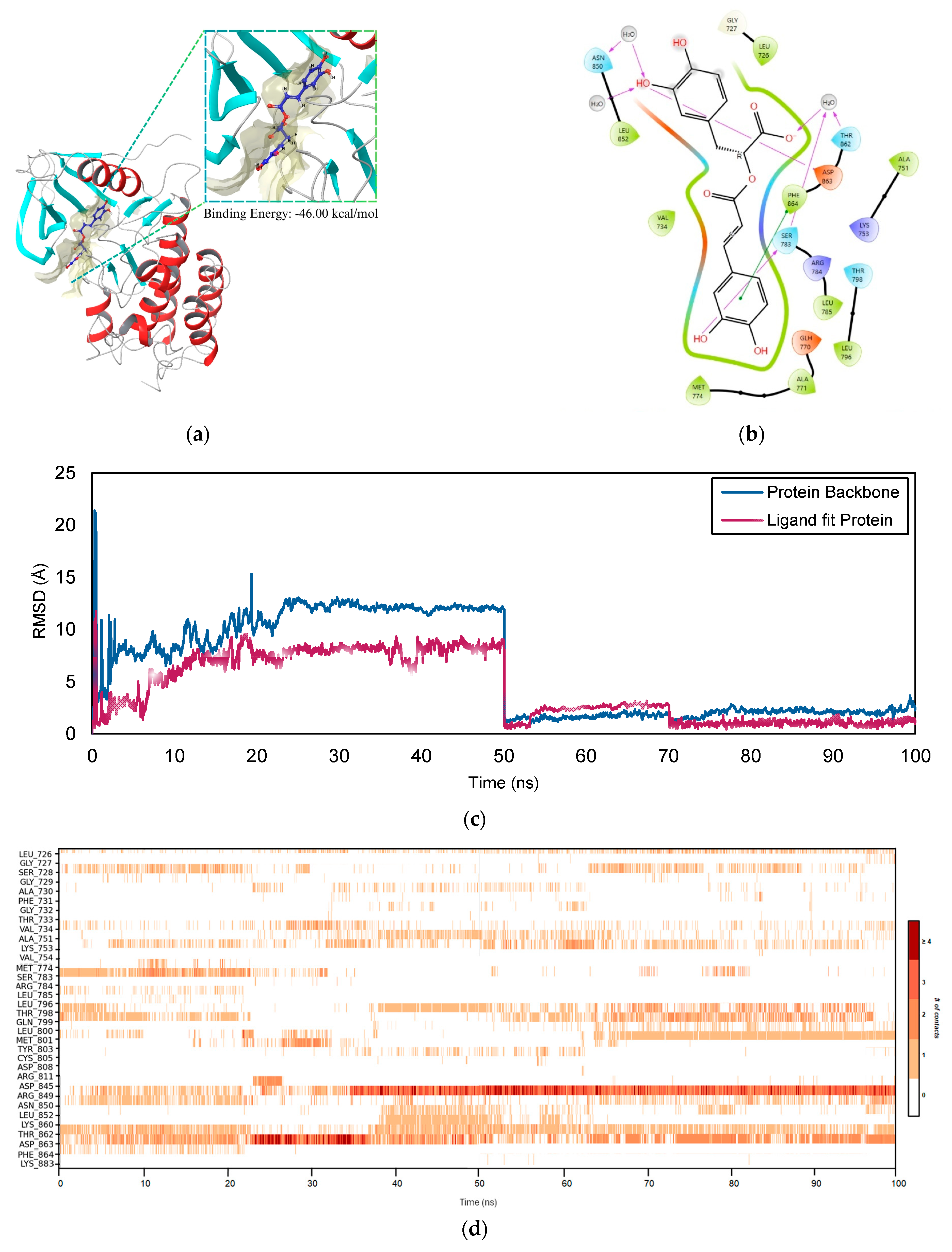
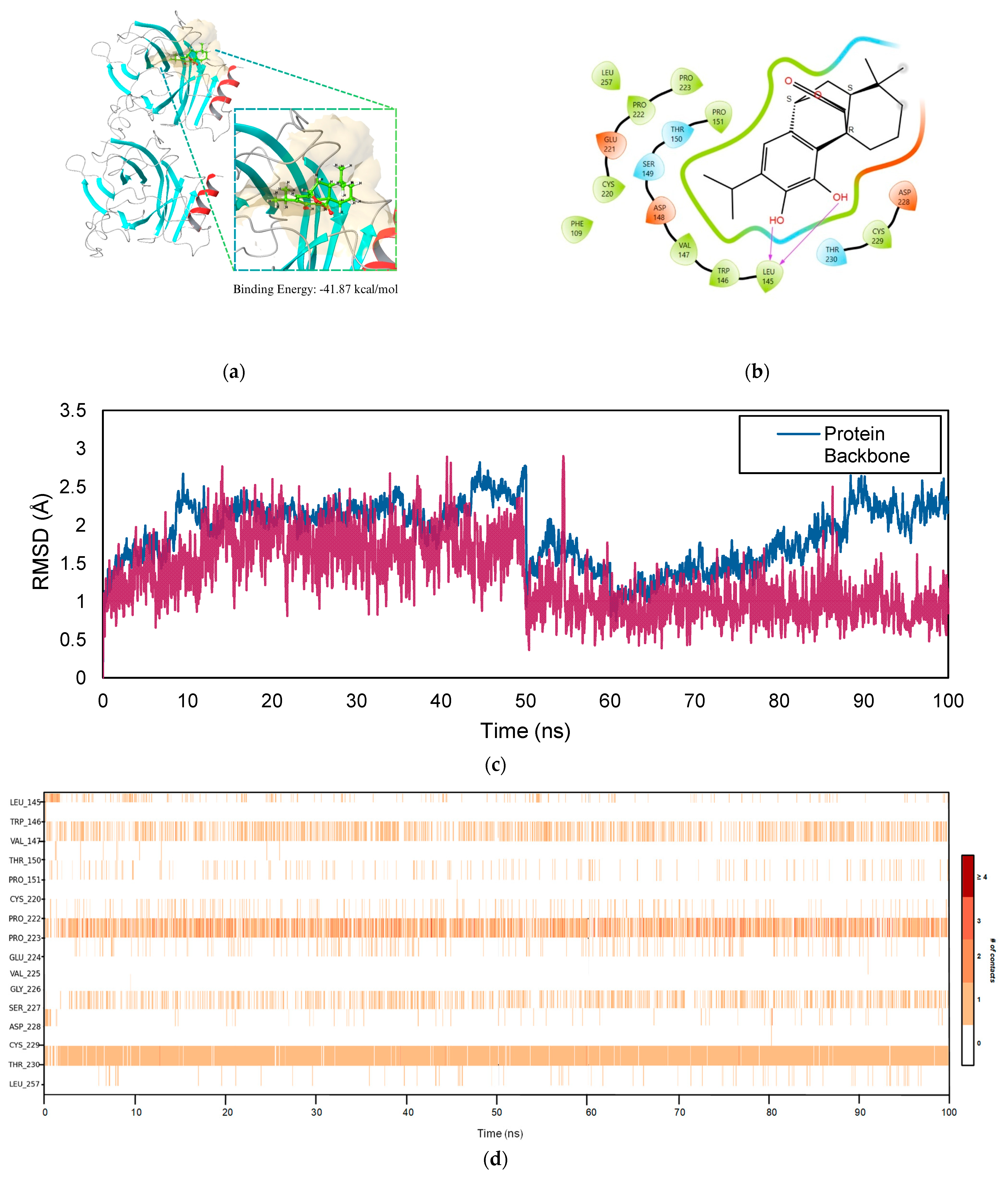
2.5. Molecular Dynamics Simulation
2.5.1. EGFR–COH Complex
2.5.2. ERBB2–RA Complex
2.5.3. TP53–COH Complex
2.6. ADMET Analysis
3. Discussion
4. Materials and Methods
4.1. Water and Ethanol Extractions of Rosemary Leaves
4.2. Liquid Chromatography-Mass Spectrometry (LC-MS) Analysis
4.3. Anti-Prostate Cancer Activity of Rosemary Leaf Extracts
4.3.1. Cell Culture
4.3.2. Cell Treatment
4.3.3. MTT Cell Viability Assay
4.4. Data Treatment and Statistical Analysis
4.5. Network Pharmacology
4.5.1. Screening and Identification of Gene Targets
4.5.2. Protein–Protein Interaction Network Construction
4.5.3. Gene Function and Pathway Enrichment Analysis
4.6. Molecular Docking
4.6.1. Docking Validation
4.6.2. Receptor–Ligand Docking
4.6.3. Molecular Mechanics Generalized Born Surface Area (MM-GBSA) Calculations
4.7. Molecular Dynamics Simulation
4.8. Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- White, A.K.; Thomson, C.S.; Forman, D.; Meryn, S. Men’s Health and the Excess Burden of Cancer in Men. Eur. Urol. Suppl. 2010, 9, 467–470. [Google Scholar] [CrossRef]
- Peate, I. Men and Cancer: The Gender Dimension. Br. J. Nurs. 2011, 20, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Faustino, A.; Oliveira, P.A. (Eds.) Prostate Cancer; MDPI: Basel, Switzerland, 2023; 150p. [Google Scholar] [CrossRef]
- Bissada, N.K. Prostate Cancer: An Overview. J. S. C. Med. Assoc. 2014, 96, 52. [Google Scholar] [CrossRef]
- Rivera-Izquierdo, M.; de Rojas, J.P.; Martínez-Ruiz, V.; Pérez-Gómez, B.; Sánchez, M.J.; Khan, K.S.; Jiménez-Moleón, J.J. Obesity as a Risk Factor for Prostate Cancer Mortality: A Systematic Review and Dose-Response Meta-Analysis of 280,199 Patients. Cancers 2021, 13, 4169. [Google Scholar] [CrossRef]
- Sirengo, J.L.; Alilah, D.A.; Mbete, D.A.; Keli, R. Estimation of Risk Factors Affecting Screening Outcomes of Prostate Cancer Using the Bayesian Ordinal Logistic Model. J. Probab. Stat. 2023, 2023, 4987764. [Google Scholar] [CrossRef]
- Raghallaigh, H.N.; Bott, S.R.J. The Role of Family History and Germline Genetics in Prostate Cancer Disease Profile and Screening. In Urologic Cancers [Internet]; Exon Publications: Brisbane, Australia, 2022; pp. 199–213. [Google Scholar] [CrossRef]
- Hamilton, W.; Sharp, D. Symptomatic Diagnosis of Prostate Cancer in Primary Care: A Structured Review. Br. J. Gen. Pract. 2004, 54, 617–621. [Google Scholar]
- Ferreira, V.V.; Angelo, I.; Thomas, B.; Ghosh, A.K. Cardiovascular Complications of Treatment for Prostate Cancer. Br. J. Hosp. Med. 2022, 83, 1–12. [Google Scholar] [CrossRef]
- Cheng, S.; Wang, S.; Liu, J.; Huang, J.; Liu, J. Research Progress in Focal Treatment of Prostate Cancer. Ann. Urol. Oncol. 2023, 6, 72–79. [Google Scholar] [CrossRef]
- Rice, M.A.; Malhotra, S.V.; Stoyanova, T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol. 2019, 9, 801. [Google Scholar] [CrossRef]
- AlZahrani, M.; Clemons, M.; Vandermeer, L.; Sienkiewicz, M.; Awan, A.A.; Hutton, B.; Pond, G.R.; Ng, T.L. Real-World Practice Patterns and Attitudes towards de-Escalation of Bone-Modifying Agents in Patients with Bone Metastases from Breast and Prostate Cancer: A Physician Survey. J. Bone Oncol. 2021, 26, 100339. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.K.; Kosoff, D.; Emamekhoo, H.; Lang, J.M.; Kyriakopoulos, C.E. PARP Inhibitors in Metastatic Prostate Cancer. Front. Oncol. 2023, 13, 1159557. [Google Scholar] [CrossRef] [PubMed]
- Younger, N.S.; Borno, H.T. Understanding the Current Therapeutic Landscape for Advanced Prostate Cancer. Int. J. Cancer Care Deliv. 2022, 2. [Google Scholar] [CrossRef]
- Salehi, B.; Fokou, P.V.T.; Yamthe, L.R.T.; Tali, B.T.; Adetunji, C.O.; Rahavian, A.; Mudau, F.N.; Martorell, M.; Setzer, W.N.; Rodrigues, C.F.; et al. Phytochemicals in Prostate Cancer: From Bioactive Molecules to Upcoming Therapeutic Agents. Nutrients 2019, 11, 1483. [Google Scholar] [CrossRef]
- Dincheva, I.; Badjakov, I.; Galunska, B. New Insights into the Research of Bioactive Compounds from Plant Origins with Nutraceutical and Pharmaceutical Potential. Plants 2023, 12, 258. [Google Scholar] [CrossRef]
- Ghasemzadeh, A.; Ghasemzadeh, N. Flavonoids and Phenolic Acids: Role and Biochemical Activity in Plants and Human. J. Med. Plants Res. 2011, 5, 6697–6703. [Google Scholar] [CrossRef]
- Bobach, C.; Schurwanz, J.; Franke, K.; Denkert, A.; Van Sung, T.; Kuster, R.; Mutiso, P.C.; Seliger, B.; Wessjohann, L.A. Multiple Readout Assay for Hormonal (Androgenic and Antiandrogenic) and Cytotoxic Activity of Plant and Fungal Extracts Based on Differential Prostate Cancer Cell Line Behavior. J. Ethnopharmacol. 2014, 155, 721–730. [Google Scholar] [CrossRef]
- Thomas-Charles, C.; Fennell, H. Anti-Prostate Cancer Activity of Plant-Derived Bioactive Compounds: A Review. Curr. Mol. Biol. Rep. 2019, 5, 140–151. [Google Scholar] [CrossRef]
- Choi, Y.J.; Choi, Y.K.; Lee, K.M.; Cho, S.G.; Kang, S.Y.; Ko, S.G. SH003 Induces Apoptosis of DU145 Prostate Cancer Cells by Inhibiting ERK-Involved Pathway. BMC Complement. Altern. Med. 2016, 16, 507. [Google Scholar] [CrossRef]
- Sitarek, P.; Merecz-Sadowska, A.; Śliwiński, T.; Zajdel, R.; Kowalczyk, T. An In Vitro Evaluation of the Molecular Mechanisms of Action of Medical Plants from the Lamiaceae Family as Effective Sources of Active Compounds against Human Cancer Cell Lines. Cancers 2020, 12, 2957. [Google Scholar] [CrossRef]
- Iqbal, J.; Abbasi, B.A.; Mahmood, T.; Kanwal, S.; Ali, B.; Shah, S.A.; Khalil, A.T. Plant-Derived Anticancer Agents: A Green Anticancer Approach. Asian Pac. J. Trop. Biomed. 2017, 7, 1129–1150. [Google Scholar] [CrossRef]
- Hammer, M.; Junghanns, W. Rosmarinus officinalis L.: Rosemary. In Medicinal, Aromatic and Stimulant Plants; Springer: Cham, Switzerland, 2020; pp. 501–521. [Google Scholar] [CrossRef]
- Berdahl, D.R.; McKeague, J. Rosemary and Sage Extracts as Antioxidants for Food Preservation. In Handbook of Antioxidants for Food Preservation; Woodhead Publishing: Cambridge, UK, 2015; pp. 177–217. [Google Scholar] [CrossRef]
- Pérez-Sánchez, A.; Barrajón-Catalán, E.; Ruiz-Torres, V.; Agulló-Chazarra, L.; Herranz-López, M.; Valdés, A.; Cifuentes, A.; Micol, V. Rosemary (Rosmarinus officinalis) Extract Causes ROS-Induced Necrotic Cell Death and Inhibits Tumor Growth in Vivo. Sci. Rep. 2019, 9, 808. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Tonacci, A.; Pioggia, G.; Musolino, C.; Gangemi, S. Anticancer Activity of Rosmarinus officinalis L.: Mechanisms of Action and Therapeutic Potentials. Nutrients 2020, 12, 1739. [Google Scholar] [CrossRef] [PubMed]
- Pappachan, F.; Suku, A.; Mohanan, S. Rosmarinus officinalis. In Herbs, Spices Their Roles Nutraceuticals Functional Foods; Academic Press: Cambridge, MA, USA, 2023; pp. 149–170. [Google Scholar] [CrossRef]
- Jaglanian, A.; Termini, D.; Tsiani, E. Rosemary (Rosmarinus officinalis L.) Extract Inhibits Prostate Cancer Cell Proliferation and Survival by Targeting Akt and MTOR. Biomed. Pharmacother. 2020, 131, 110717. [Google Scholar] [CrossRef]
- Jaglanian, A.; Tsiani, E. Inhibition of Prostate Cancer Cell Proliferation and Survival by Rosemary Extract. FASEB J. 2019, 33, 652.10. [Google Scholar] [CrossRef]
- Dagar, N.; Kale, A.; Jadhav, H.R.; Gaikwad, A.B. Nutraceuticals and Network Pharmacology Approach for Acute Kidney Injury: A Review from the Drug Discovery Aspect. Fitoterapia 2023, 168, 105563. [Google Scholar] [CrossRef]
- Xiang, C.; Liao, Y.; Chen, Z.; Xiao, B.; Zhao, Z.; Li, A.; Xia, Y.; Wang, P.; Li, H.; Xiao, T. Network Pharmacology and Molecular Docking to Elucidate the Potential Mechanism of Ligusticum Chuanxiong Against Osteoarthritis. Front. Pharmacol. 2022, 13, 854215. [Google Scholar] [CrossRef]
- Ma, S.; Hou, J.; Liu, S.; Zhu, F.; Wei, P.; Feng, C.; Chen, N. Lead Drug Discover Strategies from Natural Medicines Based on Network Pharmacology. Med. Res. Arch. 2023, 11. [Google Scholar] [CrossRef]
- Tang, J. Special Issue on Network Pharmacology Modeling for Drug Discovery. Processes 2023, 11, 1988. [Google Scholar] [CrossRef]
- Swari, D.A.M.A.; Santika, I.W.M.; Aman, I.G.M. ANTIFUNGAL ACTIVITIES OF ETHANOL EXTRACT OF ROSEMARY LEAF (Rosemarinus officinalis L.) AGAINST Candida Albicans. J. Pharm. Sci. Appl. 2020, 2, 28–35. [Google Scholar] [CrossRef]
- Salama, W.H.; Abdel-Aty, A.M.; Fahmy, A.S. Rosemary Leaves Extract: Anti-Snake Action against Egyptian Cerastes Cerastes Venom. J. Tradit. Complement. Med. 2018, 8, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Fazeli-Nasab, B.; Valizadeh, M.; Hassanzadeh, M.A.; Beigomi, M. Evaluation of the Antimicrobial Activity of Olive and Rosemary Leave Extracts Prepared with Different Solvents Against Antibiotic-Resistant Escherichia coli. Int. J. Infect. 2021, 8, e114498. [Google Scholar] [CrossRef]
- Ali Hasan, S.; Al-Rikaby, A.A. Study the Ability of Rosemary Leaf Ethanol Extract to Protect Male Rabbits’ Liver and Kidney against Poisoning Cypermethrin. Basrah J. Vet. Res. 2022, 21, 58–70. [Google Scholar] [CrossRef]
- Nguyen, H.C.; Nguyen, H.N.T.; Huang, M.Y.; Lin, K.H.; Pham, D.C.; Tran, Y.B.; Su, C.H. Optimization of Aqueous Enzyme-Assisted Extraction of Rosmarinic Acid from Rosemary (Rosmarinus officinalis L.) Leaves and the Antioxidant Activity of the Extract. J. Food Process. Preserv. 2021, 45, e15221. [Google Scholar] [CrossRef]
- Nieto, G.; Ros, G.; Castillo, J. Antioxidant and Antimicrobial Properties of Rosemary (Rosmarinus officinalis, L.): A Review. Medicines 2018, 5, 98. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene Set Analysis Toolkit with Revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Pudova, E.A.; Krasnov, G.S.; Kobelyatskaya, A.A.; Savvateeva, M.V.; Fedorova, M.S.; Pavlov, V.S.; Nyushko, K.M.; Kaprin, A.D.; Alekseev, B.Y.; Trofimov, D.Y.; et al. Gene Expression Changes and Associated Pathways Involved in the Progression of Prostate Cancer Advanced Stages. Front. Genet. 2021, 11, 613162. [Google Scholar] [CrossRef]
- Liu, K.; Chen, Y.; Feng, P.; Wang, Y.; Sun, M.; Song, T.; Tan, J.; Li, C.; Liu, S.; Kong, Q.; et al. Identification of Pathologic and Prognostic Genes in Prostate Cancer Based on Database Mining. Front. Genet. 2022, 13, 854531. [Google Scholar] [CrossRef]
- Song, Z.; Huang, Y.; Zhao, Y.; Ruan, H.; Yang, H.; Cao, Q.; Liu, D.; Zhang, X.; Chen, K. The Identification of Potential Biomarkers and Biological Pathways in Prostate Cancer. J. Cancer 2019, 10, 1398–1408. [Google Scholar] [CrossRef]
- Bhati, A.P.; Coveney, P.V. Large Scale Study of Ligand-Protein Relative Binding Free Energy Calculations: Actionable Predictions from Statistically Robust Protocols. J. Chem. Theory Comput. 2022, 18, 2687–2702. [Google Scholar] [CrossRef]
- Kallifatidis, G.; Hoy, J.J.; Lokeshwar, B.L. Bioactive Natural Products for Chemoprevention and Treatment of Castration-Resistant Prostate Cancer. Semin. Cancer Biol. 2016, 40–41, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Yesil-Celiktas, O.; Sevimli, C.; Bedir, E.; Vardar-Sukan, F. Inhibitory Effects of Rosemary Extracts, Carnosic Acid and Rosmarinic Acid on the Growth of Various Human Cancer Cell Lines. Plant Foods Hum. Nutr. 2010, 65, 158–163. [Google Scholar] [CrossRef] [PubMed]
- González-Vallinas, M.; Reglero, G.; Ramírez De Molina, A. Rosemary (Rosmarinus officinalis L.) Extract as a Potential Complementary Agent in Anticancer Therapy. Nutr. Cancer 2015, 67, 1223–1231. [Google Scholar] [CrossRef]
- Johnson, J.J. Carnosol: A Promising Anti-Cancer and Anti-Inflammatory Agent. Cancer Lett. 2011, 305, 1–7. [Google Scholar] [CrossRef]
- Leal, P.F.; Braga, M.E.M.; Sato, D.N.; Carvalho, J.E.; Marques, M.O.M.; Meireles, M.A.A. Functional Properties of Spice Extracts Obtained via Supercritical Fluid Extraction. J. Agric. Food Chem. 2003, 51, 2520–2525. [Google Scholar] [CrossRef]
- Vallverdú-Queralt, A.; Regueiro, J.; Martínez-Huélamo, M.; Rinaldi Alvarenga, J.F.; Leal, L.N.; Lamuela-Raventos, R.M. A Comprehensive Study on the Phenolic Profile of Widely Used Culinary Herbs and Spices: Rosemary, Thyme, Oregano, Cinnamon, Cumin and Bay. Food Chem. 2014, 154, 299–307. [Google Scholar] [CrossRef]
- Gioti, K.; Tenta, R. Bioactive Natural Products against Prostate Cancer: Mechanism of Action and Autophagic/Apoptotic Molecular Pathways. Planta Med. 2015, 81, 543–562. [Google Scholar] [CrossRef]
- Grande, F.; Giordano, F.; Occhiuzzi, M.A.; Rocca, C.; Ioele, G.; De Luca, M.; Ragno, G.; Panno, M.L.; Rizzuti, B.; Garofalo, A. Toward Multitasking Pharmacological COX-Targeting Agents: Non-Steroidal Anti-Inflammatory Prodrugs with Antiproliferative Effects. Molecules 2021, 26, 3940. [Google Scholar] [CrossRef]
- O’neill, E.J.; Den Hartogh, D.J.; Azizi, K.; Tsiani, E. Anticancer Properties of Carnosol: A Summary of In Vitro and In Vivo Evidence. Antioxidants 2020, 9, 961. [Google Scholar] [CrossRef]
- Petiwala, S.M.; Puthenveetil, A.G.; Johnson, J.J.; Heinrich, M.; Yang, H. Polyphenols from the Mediterranean Herb Rosemary (Rosmarinus officinalis) for Prostate Cancer. Front. Pharmacol. 2013, 4, 42634. [Google Scholar] [CrossRef]
- Petiwala, S.M.; Li, G.; Bosland, M.C.; Lantvit, D.D.; Petukhov, P.A.; Johnson, J.J. Carnosic Acid Promotes Degradation of the Androgen Receptor and Is Regulated by the Unfolded Protein Response Pathway in Vitro and in Vivo. Carcinogenesis 2016, 37, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Nadile, M.; Sze, N.S.K.; Fajardo, V.A.; Tsiani, E. Inhibition of Prostate Cancer Cell Survival and Proliferation by Carnosic Acid Is Associated with Inhibition of Akt and Activation of AMPK Signaling. Nutrients 2024, 16, 1257. [Google Scholar] [CrossRef] [PubMed]
- Alimirah, F.; Chen, J.; Basrawala, Z.; Xin, H.; Choubey, D. DU-145 and PC-3 Human Prostate Cancer Cell Lines Express Androgen Receptor: Implications for the Androgen Receptor Functions and Regulation. FEBS Lett. 2006, 580, 2294–2300. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.; Yousef, M.; Tsiani, E. Anticancer Effects of Rosemary (Rosmarinus officinalis L.) Extract and Rosemary Extract Polyphenols. Nutrients 2016, 8, 731. [Google Scholar] [CrossRef]
- Guo, W.H.; Zhang, K.; Yang, L.H. Potential Mechanisms of Pyrrosiae Folium in Treating Prostate Cancer Based on Network Pharmacology and Molecular Docking. Drug Dev. Ind. Pharm. 2022, 48, 189–197. [Google Scholar] [CrossRef]
- Nayana, P.; Manjunatha, H.; Andrade, P.K.; Vijayalaksmi, V. Investigating Drug-Target Interactions of Piperine in Prostate Cancer Using Network Pharmacology and Docking Studies. Res. J. Biotechnol. 2022, 17, 32–42. [Google Scholar] [CrossRef]
- Lu, D.; Shang, J.; Guo, X.; Zhang, Y. Assessing the Mechanism of Action of “Fructus Ligustri Lucidi-Cuscutae Semen” in Prostate Cancer Treatment Using Network Pharmacology and Molecular Docking. Comput. Math. Methods Med. 2022, 2022, 754361. [Google Scholar] [CrossRef]
- Mo, Y.; Chen, M.; Qin, H.; Liu, H.; Ye, Y. Pueraria Lobata Potentially Treating Prostate Cancer on Single-Cell Level by Network Pharmacology and AutoDock: Clinical Findings and Drug Targets. Comput. Math. Methods Med. 2022, 2022, 3758219. [Google Scholar] [CrossRef]
- Li, H.; Hung, A.; Yang, A.W.H. Herb-Target Virtual Screening and Network Pharmacology for Prediction of Molecular Mechanism of Danggui Beimu Kushen Wan for Prostate Cancer. Sci. Rep. 2021, 11, 6656. [Google Scholar] [CrossRef]
- Zhao, R.; Wang, Y.; Zhang, M.; Gu, X.; Wang, W.; Tan, J.; Wei, X.; Jin, N. Screening of Potential Therapy Targets for Prostate Cancer Using Integrated Analysis of Two Gene Expression Profiles. Oncol. Lett. 2017, 14, 5361–5369. [Google Scholar] [CrossRef]
- Chen, L.W.; Tuac, Y.; Li, S.; Leeman, J.E.; King, M.T.; Orio, P.F.; Nguyen, P.L.; D’Amico, A.V.; Aktan, C.; Sayan, M. Clinical Outcomes and Genomic Alterations in Gleason Score 10 Prostate Cancer. Cancers 2025, 17, 1055. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, G.; Nandakumar, S.; Hirani, R.; Nguyen, B.; Stopsack, K.H.; Kreitzer, C.; Rajanala, S.H.; Ghale, R.; Mazzu, Y.Z.; Pillarsetty, N.V.K.; et al. The Impact of PIK3R1 Mutations and Insulin–PI3K–Glycolytic Pathway Regulation in Prostate Cancer. Clin. Cancer Res. 2022, 28, 3603–3617. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Gorodetska, I.; Klusa, D.; Shi, Q.; Alves, T.C.; Pantel, K.; Dubrovska, A. Metabolic Regulation of Prostate Cancer Heterogeneity and Plasticity. Semin. Cancer Biol. 2022, 82, 94–119. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lu, Z.; Pan, D.; Zhang, Z.; He, H.; Wu, J.; Peng, N. Gene Expression Analysis Reveals Prognostic Biomarkers of the Tyrosine Metabolism Reprogramming Pathway for Prostate Cancer. J. Oncol. 2022, 2022, 5504173. [Google Scholar] [CrossRef]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular Characterization of Neuroendocrine Prostate Cancer and Identification of New Drug Targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489.e6. [Google Scholar] [CrossRef]
- Li, S.; Hou, J.; Xu, W. Screening and Identification of Key Biomarkers in Prostate Cancer Using Bioinformatics. Mol. Med. Rep. 2020, 21, 311–319. [Google Scholar] [CrossRef]
- Schitcu, V.H.; Raduly, L.; Zanoaga, O.; Jurj, A.; Munteanu, V.C.; Budisan, L.; Petrut, B.; Braicu, C.; Coman, I.; Berindan-Neagoe, I. TP53 Gene Implications in Prostate Cancer Evolution: Potential Role in Tumor Classification. Med. Pharm. Rep. 2023, 96, 384–391. [Google Scholar] [CrossRef]
- Singh, A.N.; Sharma, N. In-Silico Identification of Frequently Mutated Genes and Their Co-Enriched Metabolic Pathways Associated with Prostate Cancer Progression. Andrologia 2021, 53, e14236. [Google Scholar] [CrossRef]
- Pignon, J.C.; Koopmansch, B.; Nolens, G.; Delacroix, L.; Waltregny, D.; Winkler, R. Androgen Receptor Controls EGFR and ERBB2 Gene Expression at Different Levels in Prostate Cancer Cell Lines. Cancer Res. 2009, 69, 2941–2949. [Google Scholar] [CrossRef]
- Chen, L.; Mooso, B.A.; Jathal, M.K.; Madhav, A.; Johnson, S.D.; Van Spyk, E.; Mikhailova, M.; Zierenberg-Ripoll, A.; Xue, L.; Vinall, R.L.; et al. Dual EGFR/HER2 Inhibition Sensitizes Prostate Cancer Cells to Androgen Withdrawal by Suppressing ErbB3. Clin. Cancer Res. 2011, 17, 6218–6228. [Google Scholar] [CrossRef] [PubMed]
- Day, K.C.; Hiles, G.L.; Kozminsky, M.; Dawsey, S.J.; Paul, A.; Broses, L.J.; Shah, R.; Kunja, L.P.; Hall, C.; Palanisamy, N.; et al. HER2 and EGFR Overexpression Support Metastatic Progression of Prostate Cancer to Bone. Cancer Res. 2017, 77, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Lovaas, J.D.; Zhu, L.; Chiao, C.Y.; Byles, V.; Faller, D.V.; Dai, Y. SIRT1 Enhances Matrix Metalloproteinase-2 Expression and Tumor Cell Invasion in Prostate Cancer Cells. Prostate 2013, 73, 522–530. [Google Scholar] [CrossRef]
- Watson, G.W.; Wickramasekara, S.; Fang, Y.; Maier, C.S.; Williams, D.E.; Dashwood, R.H.; Perez, V.I.; Ho, E. HDAC6 Activity Is Not Required for Basal Autophagic Flux in Metastatic Prostate Cancer Cells. Exp. Biol. Med. 2016, 241, 1177–1185. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, L.; Hou, J.; Zou, Q.; Gao, Q.; Yao, W.; Yao, Q.; Zhang, J. Hierarchical Virtual Screening of the Dual MMP-2/HDAC-6 Inhibitors from Natural Products Based on Pharmacophore Models and Molecular Docking. J. Biomol. Struct. Dyn. 2019, 37, 649–670. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, Y.; Ressler, S.J.; Ittmann, M.M.; Ayala, G.E.; Dang, T.D.; Wang, F.; Rowley, D.R. FGFR1 Is Essential for Prostate Cancer Progression and Metastasis. Cancer Res. 2013, 73, 3716–3724. [Google Scholar] [CrossRef]
- Gong, Y.; Chippada-Venkata, U.D.; Oh, W.K. Roles of Matrix Metalloproteinases and Their Natural Inhibitors in Prostate Cancer Progression. Cancers 2014, 6, 1298. [Google Scholar] [CrossRef]
- Manzat Saplacan, R.M.; Balacescu, L.; Gherman, C.; Chira, R.I.; Craiu, A.; Mircea, P.A.; Lisencu, C.; Balacescu, O. The Role of PDGFs and PDGFRs in Colorectal Cancer. Mediat. Inflamm. 2017, 2017, 4708076. [Google Scholar] [CrossRef]
- Raica, M.; Cimpean, A.M. Platelet-Derived Growth Factor (PDGF)/PDGF Receptors (PDGFR) Axis as Target for Antitumor and Antiangiogenic Therapy. Pharmaceuticals 2010, 3, 572. [Google Scholar] [CrossRef]
- Mehta, H.H.; Gao, Q.; Galet, C.; Paharkova, V.; Wan, J.; Said, J.; Sohn, J.J.; Lawson, G.; Cohen, P.; Cobb, L.J.; et al. IGFBP-3 Is a Metastasis Suppression Gene in Prostate Cancer. Cancer Res. 2011, 71, 5154–5163. [Google Scholar] [CrossRef]
- Leslie, A.R.; Gao, A.C.; Lombard, A.P.; Ning, S. Abstract B076: IGFBP3 Promotes Resistance to Olaparib via Modulating EGFR Signaling in Advanced Prostate Cancer. Cancer Res. 2023, 83, B076. [Google Scholar] [CrossRef]
- Leslie, A.R.; Ning, S.; D’Abronzo, L.S.; Armstrong, C.; Sharifi, M.; Schaaf, Z.A.; Lou, W.; Evans, C.P.; Chen, H.-W.; Lombard, A.; et al. Abstract 3862: IGFBP3 Promotes Resistance to Olaparib via Modulating EGFR Signaling in Advanced Prostate Cancer. Cancer Res. 2023, 83, 3862. [Google Scholar] [CrossRef]
- de Silva, H.C.; Lin, M.Z.; Phillips, L.; Martin, J.L.; Baxter, R.C. IGFBP-3 Interacts with NONO and SFPQ in PARP-Dependent DNA Damage Repair in Triple-Negative Breast Cancer. Cell. Mol. Life Sci. 2019, 76, 2015–2030. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Yin, J.; Zhang, J.; Jiang, Y. Insulin-like Growth Factor Receptor Signaling in Tumorigenesis and Drug Resistance: A Challenge for Cancer Therapy. J. Hematol. Oncol. 2020, 13, 64. [Google Scholar] [CrossRef]
- Dong, L.; Qu, X.; Zhao, Y.; Wang, B. Prediction of Binding Free Energy of Protein-Ligand Complexes with a Hybrid Molecular Mechanics/Generalized Born Surface Area and Machine Learning Method. ACS Omega 2021, 6, 32938–32947. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Wargasetia, T.L.; Ratnawati, H.; Widodo, N.; Widyananda, M.H. Bioinformatics Study of Sea Cucumber Peptides as Antibreast Cancer Through Inhibiting the Activity of Overexpressed Protein (EGFR, PI3K, AKT1, and CDK4). Cancer Inform. 2021, 20, 11769351211031864. [Google Scholar] [CrossRef]
- Guterres, H.; Im, W. Improving Protein-Ligand Docking Results with High-Throughput Molecular Dynamics Simulations. J. Chem. Inf. Model. 2020, 60, 2189. [Google Scholar] [CrossRef]
- Aier, I.; Varadwaj, P.K.; Raj, U. Structural Insights into Conformational Stability of Both Wild-Type and Mutant EZH2 Receptor. Sci. Rep. 2016, 6, 34984. [Google Scholar] [CrossRef]
- Umar, K.; Zothantluanga, J.H.; Luckanagul, J.A.; Limpikirati, P.; Sriwidodo, S. Structure-Based Computational Screening of 470 Natural Quercetin Derivatives for Identification of SARS-CoV-2 Mpro Inhibitor. PeerJ 2023, 11, e14915. [Google Scholar] [CrossRef]
- Fusani, L.; Palmer, D.S.; Somers, D.O.; Wall, I.D. Exploring Ligand Stability in Protein Crystal Structures Using Binding Pose Metadynamics. J. Chem. Inf. Model. 2020, 60, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Zhang, R.; Peng, X.; Fang, L.; Chen, K.; Jestilä, J.S. Elucidation of Protein–Ligand Interactions by Multiple Trajectory Analysis Methods. Phys. Chem. Chem. Phys. 2024, 26, 6903–6915. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.J.B.; Huang, S.K.H.; De Castro-Cruz, K.A.; Leron, R.B.; Tsai, P.W. An In Vitro Evaluation and Network Pharmacology Analysis of Prospective Anti-Prostate Cancer Activity from Perilla Frutescens. Plants 2023, 12, 3006. [Google Scholar] [CrossRef] [PubMed]
- Merck KGaA. MTT Assay Protocol for Cell Viability and Proliferation. Available online: https://www.eurofins.de/medizinprodukte-pruefungen/validierte-methoden/mtt-test/ (accessed on 28 January 2024).
- Nurul, N.A.; Abas, F.; Othman, I.; Naidu, R. Diarylpentanoid (1,5-Bis(4-Hydroxy-3-Methoxyphenyl)-1,4-Pentadiene-3-One) (MS13) Exhibits Anti-Proliferative, Apoptosis Induction and Anti-Migration Properties on Androgen-Independent Human Prostate Cancer by Targeting Cell Cycle-Apoptosis and PI3K Signalling Pathways. Front. Pharmacol. 2021, 12, 707335. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An Enhanced Web Server for Large-Scale Expression Profiling and Interactive Analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated Data and New Features for Efficient Prediction of Protein Targets of Small Molecules. Nucleic Acids Res. 2019, 47, W357–W3664. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING Database in 2023: Protein–Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. CytoHubba: Identifying Hub Objects and Sub-Networks from Complex Interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef]
- Tang, Y.; Li, M.; Wang, J.; Pan, Y.; Wu, F.X. CytoNCA: A Cytoscape Plugin for Centrality Analysis and Evaluation of Protein Interaction Networks. Biosystems 2015, 127, 67–72. [Google Scholar] [CrossRef]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The Reactome Pathway Knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating Viruses and Cellular Organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor Package for Pathway-Based Data Integration and Visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Jung, D.; Yao, R. ShinyGO: A Graphical Gene-Set Enrichment Tool for Animals and Plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Belal, A.; Elanany, M.A.; Santali, E.Y.; Al-Karmalawy, A.A.; Aboelez, M.O.; Amin, A.H.; Abdellattif, M.H.; Mehany, A.B.M.; Elkady, H. Screening a Panel of Topical Ophthalmic Medications against MMP-2 and MMP-9 to Investigate Their Potential in Keratoconus Management. Molecules 2022, 27, 3584. [Google Scholar] [CrossRef]
- Shi, Z.; Yu, T.; Sun, R.; Wang, S.; Chen, X.Q.; Cheng, L.J.; Liu, R. Discovery of Novel Human Epidermal Growth Factor Receptor-2 Inhibitors by Structure-Based Virtual Screening. Pharmacogn. Mag. 2016, 12, 139. [Google Scholar] [CrossRef]
- Gabr, M.T.; El-Gohary, N.S.; El-Bendary, E.R.; El-Kerdawy, M.M. EGFR Tyrosine Kinase Targeted Compounds: In Vitro Antitumor Activity and Molecular Modeling Studies of New Benzothiazole and Pyrimido [2,1-b]Benzothiazole Derivatives. EXCLI J. 2014, 13, 573. [Google Scholar]
- Alzhrani, Z.M.M.; Alam, M.M.; Neamatallah, T.; Nazreen, S. Design, Synthesis and in Vitro Antiproliferative Activity of New Thiazolidinedione-1,3,4-Oxadiazole Hybrids as Thymidylate Synthase Inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 1116. [Google Scholar] [CrossRef]
- Luksch, H.; Romanowski, M.J.; Chara, O.; Tüngler, V.; Caffarena, E.R.; Heymann, M.C.; Lohse, P.; Aksentijevich, I.; Remmers, E.F.; Flecks, S.; et al. Naturally Occurring Genetic Variants of Human Caspase-1 Differ Considerably in Structure and the Ability to Activate Interleukin-1β. Hum. Mutat. 2013, 34, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound Databases. Nucleic Acids Res. 2016, 44, D1202. [Google Scholar] [CrossRef]
- Banerjee, P.; Kemmler, E.; Dunkel, M.; Preissner, R. ProTox-3.0—Prediction of TOXicity of Chemicals. Available online: https://tox.charite.de/protox3/index.php?site=home (accessed on 26 January 2025).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | PDB ID | Co-Crystallized Ligand ID | RMSD (Å) |
|---|---|---|---|
| EGFR | 1M17 | AQ4 | 1.2451 |
| ERRB2 | 3PP0 | 03Q | 0.6354 |
| FGFR1 | 7WCL | 8ZF | 1.7018 |
| HDAC6 | 5EDU | TSN | 0.7883 |
| MMP-2 | 1HOV | I52 | 1.3172 |
| MMP-9 | 1GKC | NFH | 1.4014 |
| PDGFRB | 3MJG | NDG | 1.3752 |
| TP53 (Y220C Mutation) | 2VUK | P38 | 0.3593 |
| IGFBP3 1 | 7WRQ | - | - |
| EGFR | ERBB2 | FGFR1 | HDAC6 | MMP-2 | MMP-9 | PDGFRB | TP53 | IGFBP3 3 | |
|---|---|---|---|---|---|---|---|---|---|
| COH | −35.04 | −14.13 | −37.11 | −49.41 | −25.91 | −30.05 | −29.83 | −41.87 | −42.85 |
| CA | −24.93 | 4.69 | −8.52 | −21.02 | 13.28 | −25.77 | −8.44 | −29.8 | −16.08 |
| RA | −30.36 | −46.00 | −30.22 | −38.12 | −39.77 | −42.28 | −35.27 | −34.47 | −47.23 |
| 5-FU 1 | −20.43 | −21.09 | −23.73 | −28.67 | −22.97 | −31.15 | −16.65 | −16.48 | −22.28 |
| RD 2 | −55.75 | −86.05 | −86.12 | −44.46 | −67.65 | −78.27 | −36.18 | −49.77 | - |
| Carnosol | Carnosic Acid | Rosmarinic Acid | |
|---|---|---|---|
| Molecular Weight | 330.423 | 332.439 | 360.32 |
| Hydrogen Bond Donors | 2 | 3 | 5 |
| Hydrogen Bond Acceptors | 4.5 | 3.5 | 7 |
| Solvent Accessible Surface Area (SASA) | 551.66 | 573.224 | 656.683 |
| Octanol-Water Partition Coefficient (LogP) | 2.991 | 3.758 | 1.213 |
| Percent Human Oral Absorption (%) | 100 | 89.53 | 38.34 |
| Lipinski’s Rule of Five (RO5) | 0 | 0 | 0 |
| Rule of Three (RO3) | 0 | 0 | 1 |
| Toxicity Category | Specific Toxicity or Pathway | Prediction and Probability | ||
|---|---|---|---|---|
| Carnosol | Carnosic Acid | Rosmarinic Acid | ||
| Organ Toxicity | Hepatotoxicity | Inactive (0.76) | Inactive (0.63) | Inactive (0.62) |
| Cardiotoxicity | Inactive (0.54) | Active (0.56) | Inactive (0.69) | |
| Toxicity End Points | Carcinogenicity | Inactive (0.62) | Inactive (0.60) | Inactive (0.66) |
| Mutagenicity | Inactive (0.88) | Inactive (0.84) | Inactive (0.85) | |
| BBB Barrier | Active (0.67) | Active (0.65) | Active (0.62) | |
| Tox21-Nuclear Receptor Signaling Pathways | Androgen Receptor (AR) | Inactive (0.96) | Inactive (0.87) | Inactive (0.94) |
| Estrogen Receptor Alpha (ER) | Inactive (0.74) | Inactive (0.64) | Inactive (0.74) | |
| PPAR-Gamma | Inactive (0.94) | Inactive (0.94) | Inactive (0.80) | |
| Tox21-Stress Response Pathways | Nrf2/ARE | Inactive (0.81) | Inactive (0.80) | Inactive (0.88) |
| Phosphoprotein (Tumor Suppressor) p53 | Inactive (0.78) | Inactive (0.82) | Inactive (0.73) | |
| Metabolism | Cytochrome CYP3A4 | Inactive (0.65) | Active (0.73) | Inactive (0.94) |
| Cytochrome CYP2E1 | Inactive (1.0) | Inactive (1.0) | Inactive (1.0) | |
| Oral Toxicity | Predicted LD50 (mg/kg) | 1500 | 287 | 5000 |
| Predicted Toxicity Class | 4 | 3 | 5 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Austria, S.F.B.; Lee, M.-J.; De Castro-Cruz, K.A.; Hsu, P.-H.; Hsieh, C.-Y.; Huang, S.K.-H.; Tsai, P.-W. In Vitro and In Silico Evaluation of the Potential Anti-Prostate Cancer Activity of Rosmarinus officinalis L. Leaf Extracts. Int. J. Mol. Sci. 2025, 26, 4650. https://doi.org/10.3390/ijms26104650
Austria SFB, Lee M-J, De Castro-Cruz KA, Hsu P-H, Hsieh C-Y, Huang SK-H, Tsai P-W. In Vitro and In Silico Evaluation of the Potential Anti-Prostate Cancer Activity of Rosmarinus officinalis L. Leaf Extracts. International Journal of Molecular Sciences. 2025; 26(10):4650. https://doi.org/10.3390/ijms26104650
Chicago/Turabian StyleAustria, Samantha Franchette B., Mon-Juan Lee, Kathlia A. De Castro-Cruz, Pang-Hung Hsu, Cheng-Yang Hsieh, Steven Kuan-Hua Huang, and Po-Wei Tsai. 2025. "In Vitro and In Silico Evaluation of the Potential Anti-Prostate Cancer Activity of Rosmarinus officinalis L. Leaf Extracts" International Journal of Molecular Sciences 26, no. 10: 4650. https://doi.org/10.3390/ijms26104650
APA StyleAustria, S. F. B., Lee, M.-J., De Castro-Cruz, K. A., Hsu, P.-H., Hsieh, C.-Y., Huang, S. K.-H., & Tsai, P.-W. (2025). In Vitro and In Silico Evaluation of the Potential Anti-Prostate Cancer Activity of Rosmarinus officinalis L. Leaf Extracts. International Journal of Molecular Sciences, 26(10), 4650. https://doi.org/10.3390/ijms26104650