
Transcriptomic Analysis to Understand the Nitrogen Stress Response Mechanism in BNI-Enabled Wheat

 ,
,  ,
,
Abstract
1. Introduction
2. Results
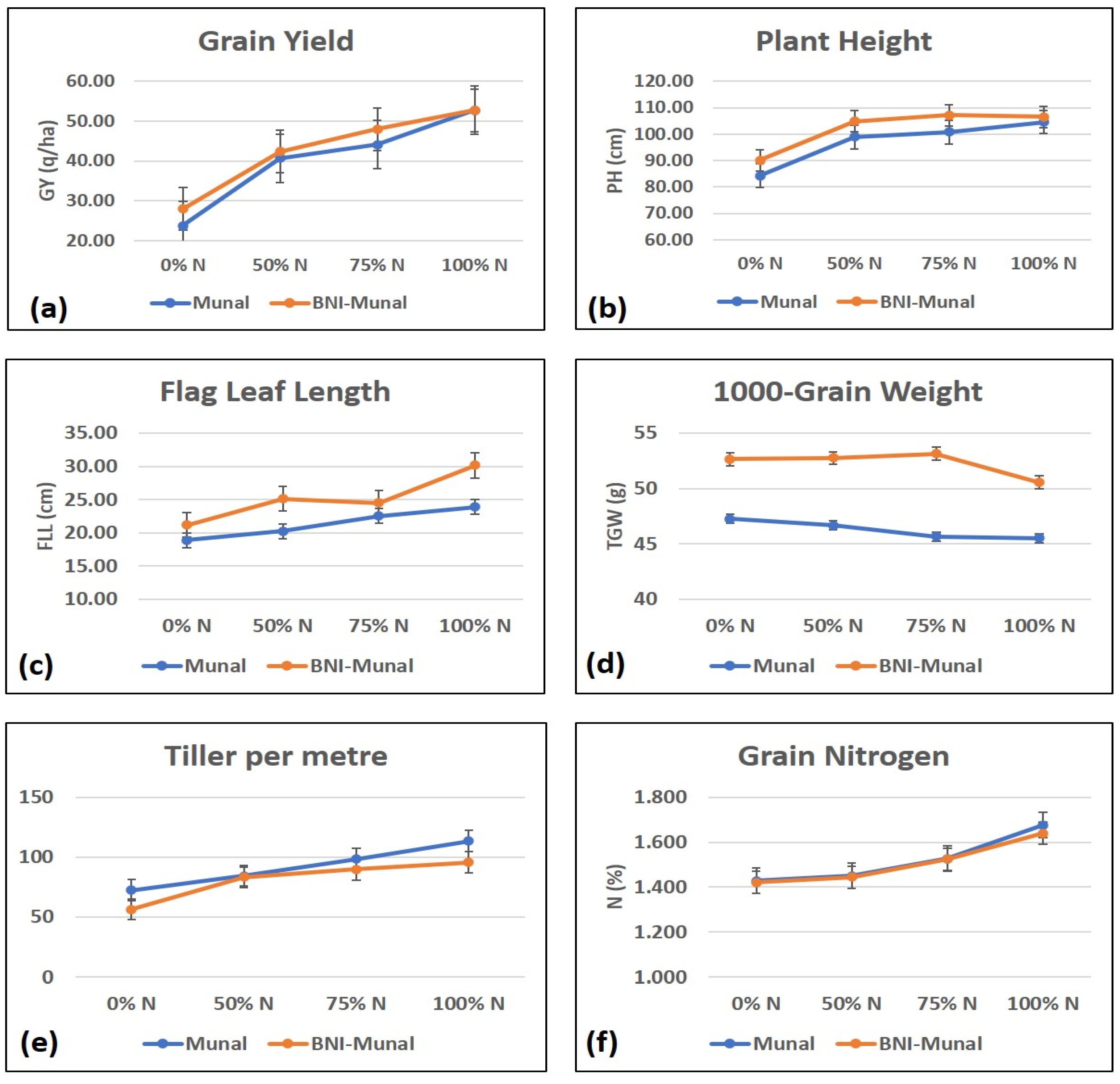
2.1. Performance of the Genotypes Based on Agro-Morphological Traits
2.2. RNA Extraction and Quantification
2.3. Library Construction
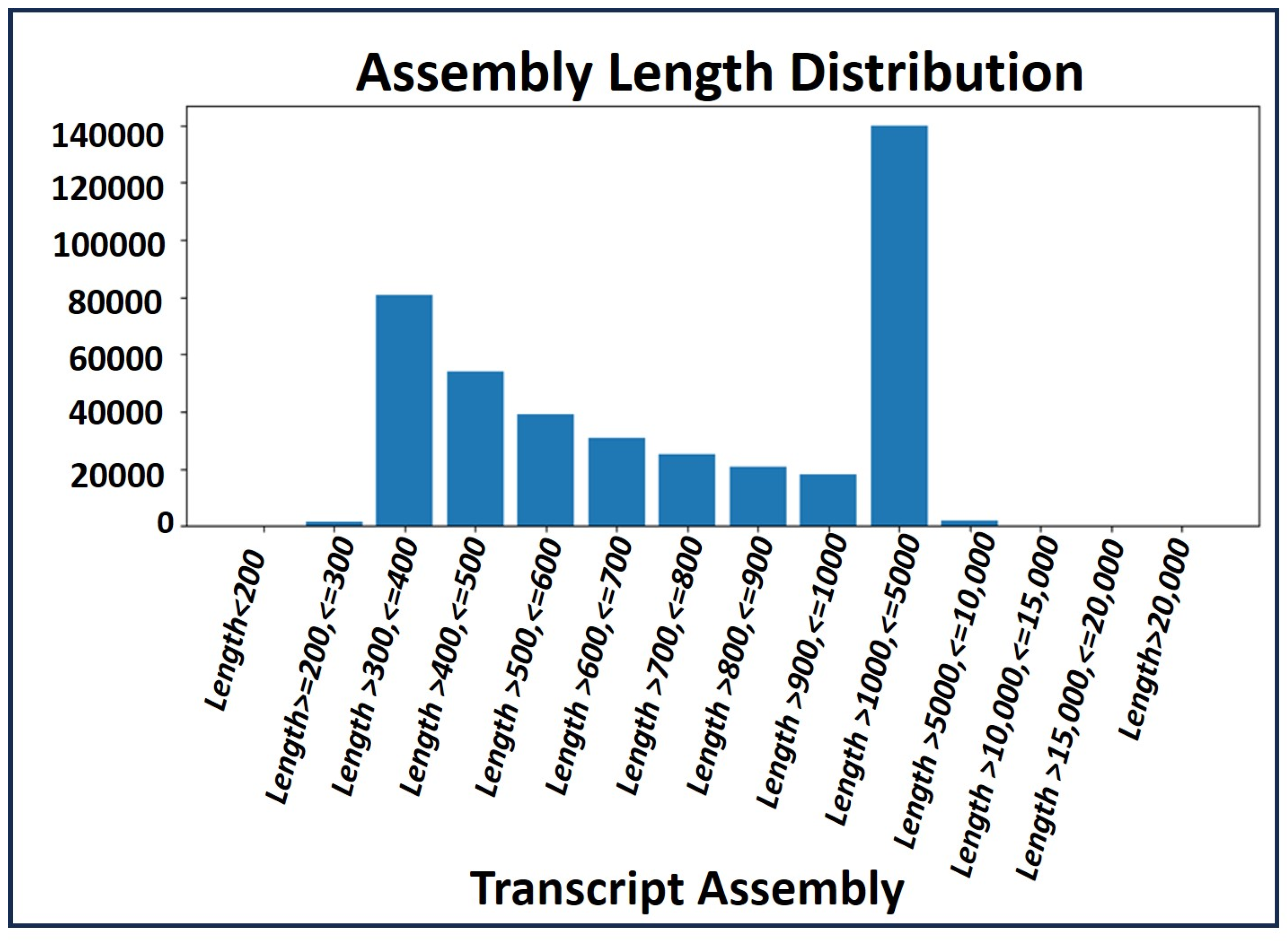
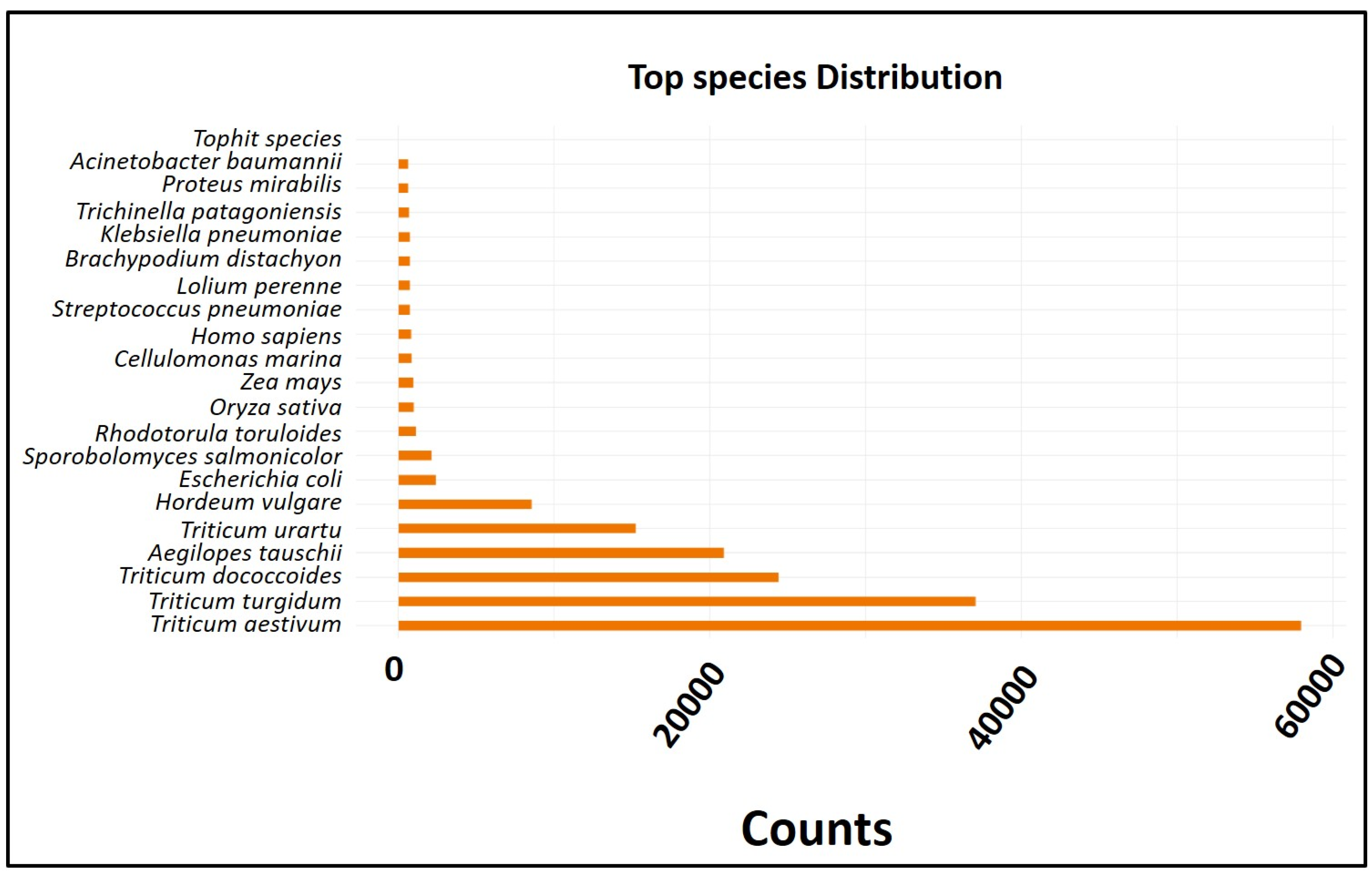
2.4. Transcript Profile of the RNA-Seq Data
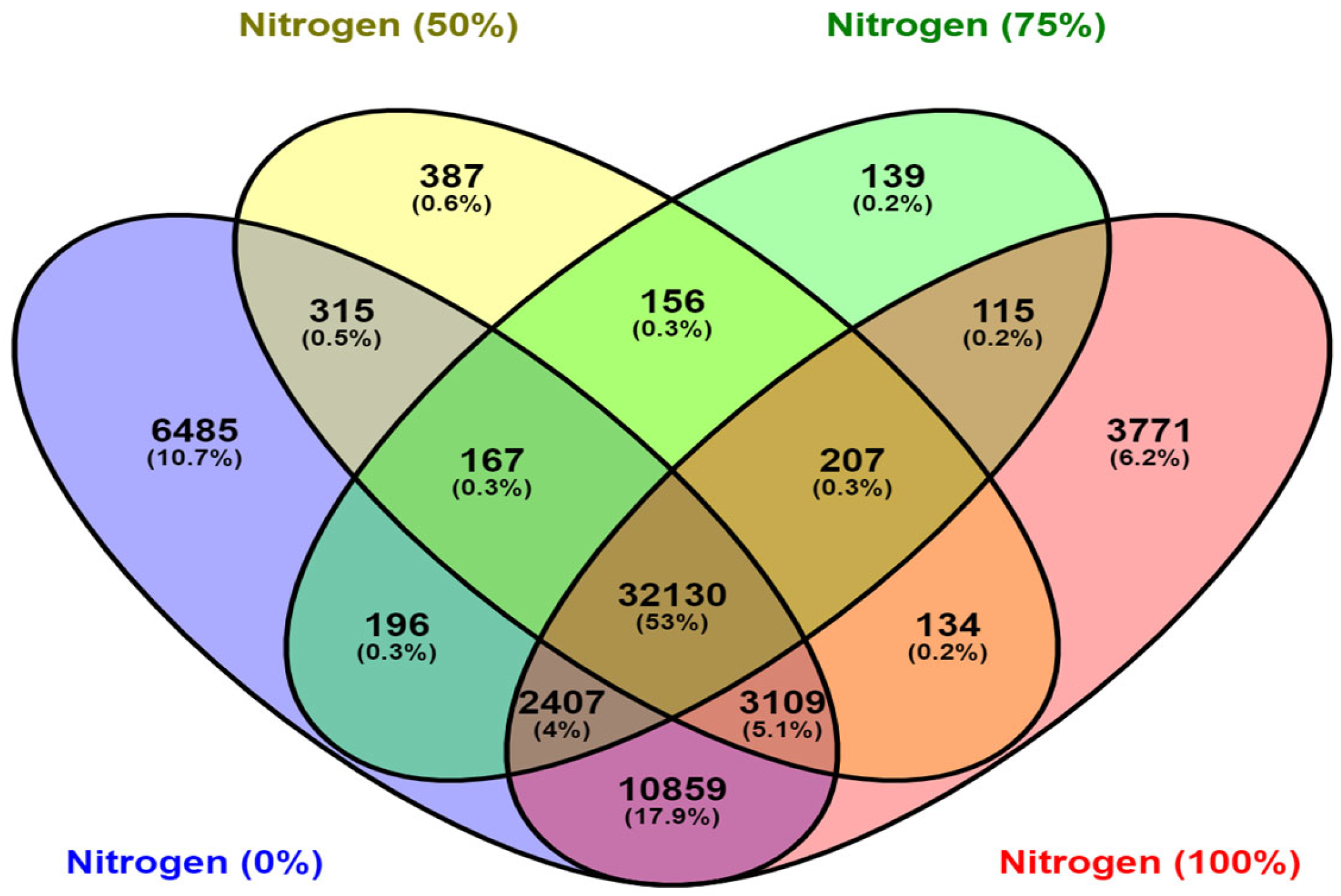
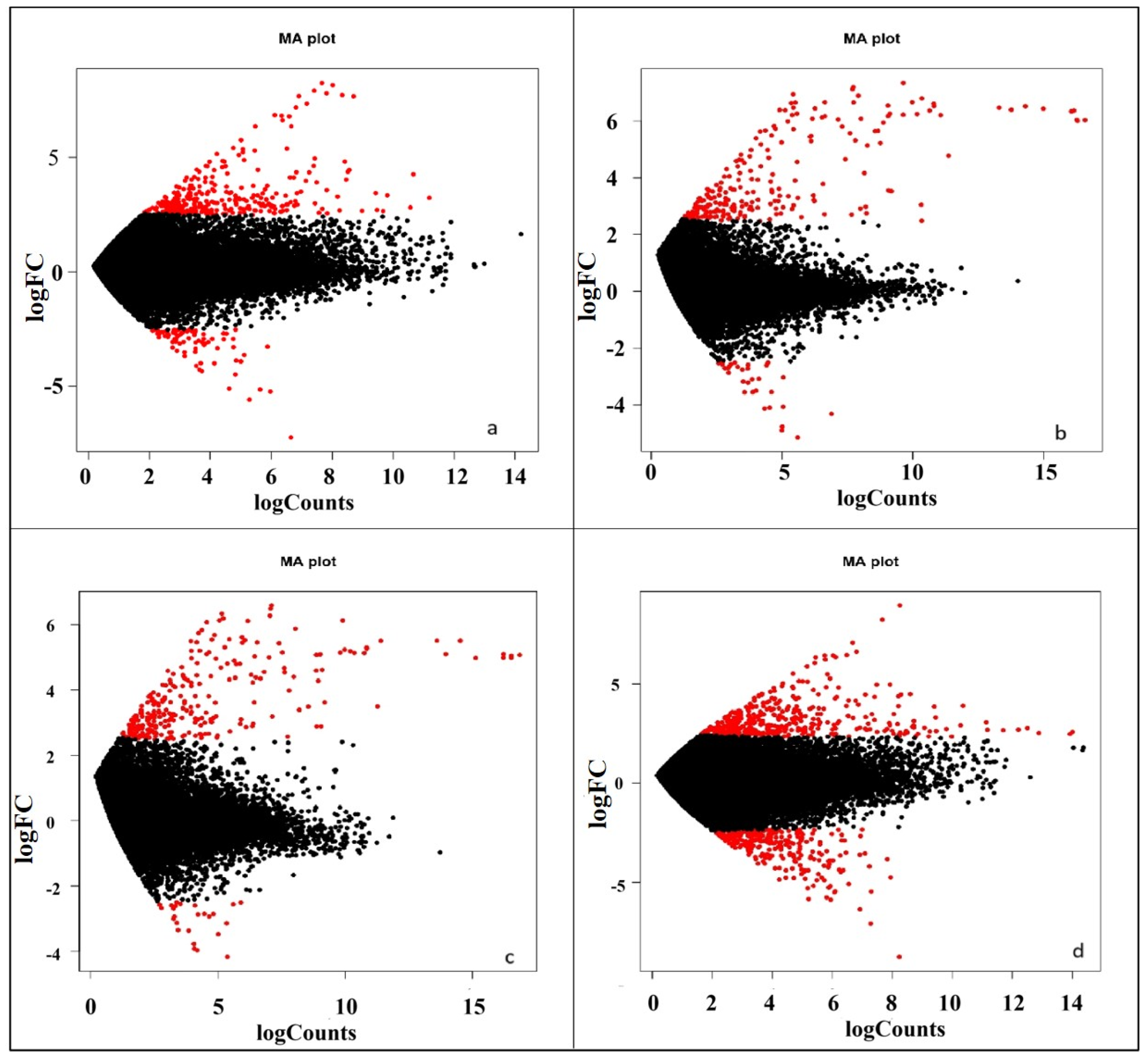
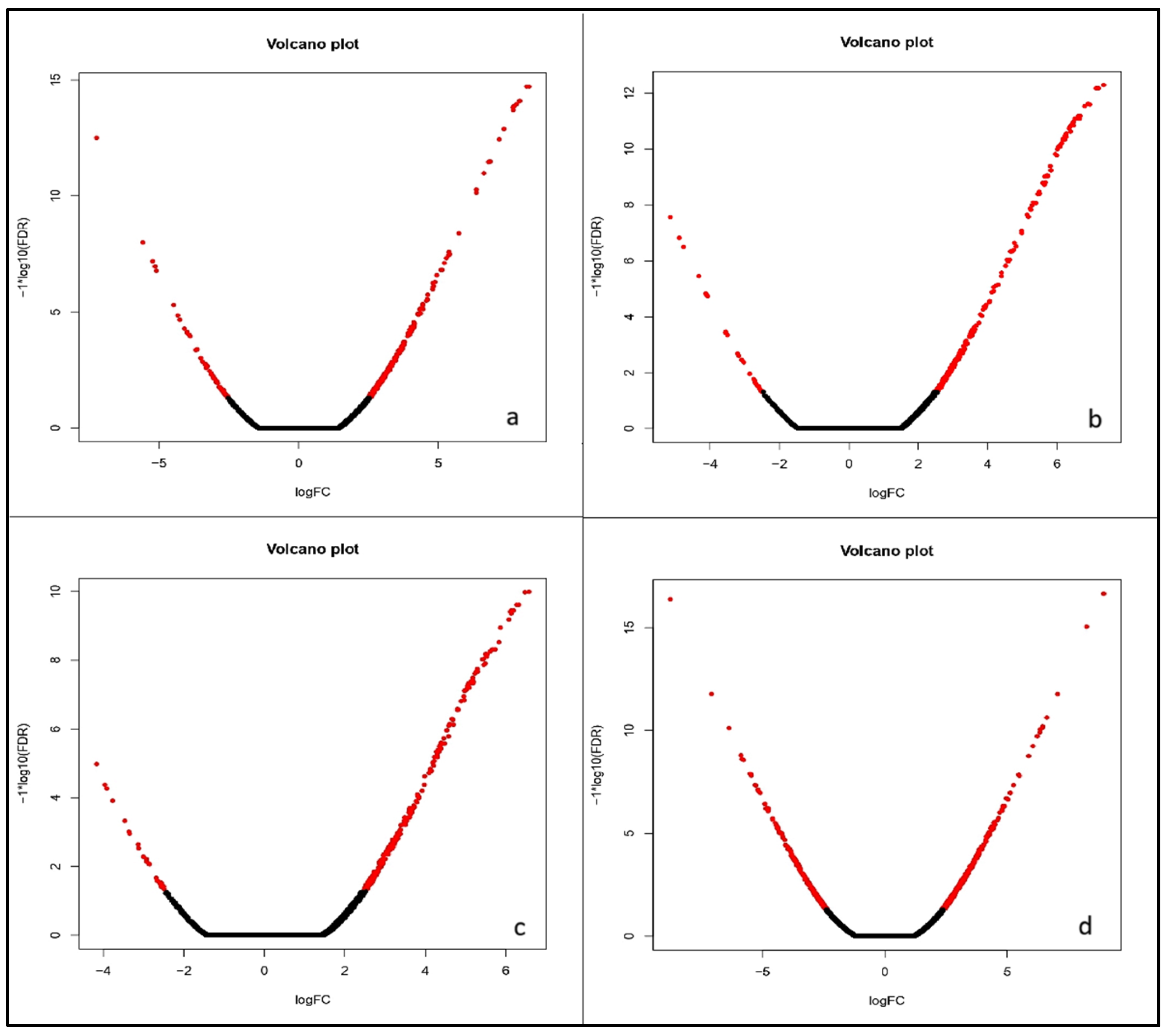
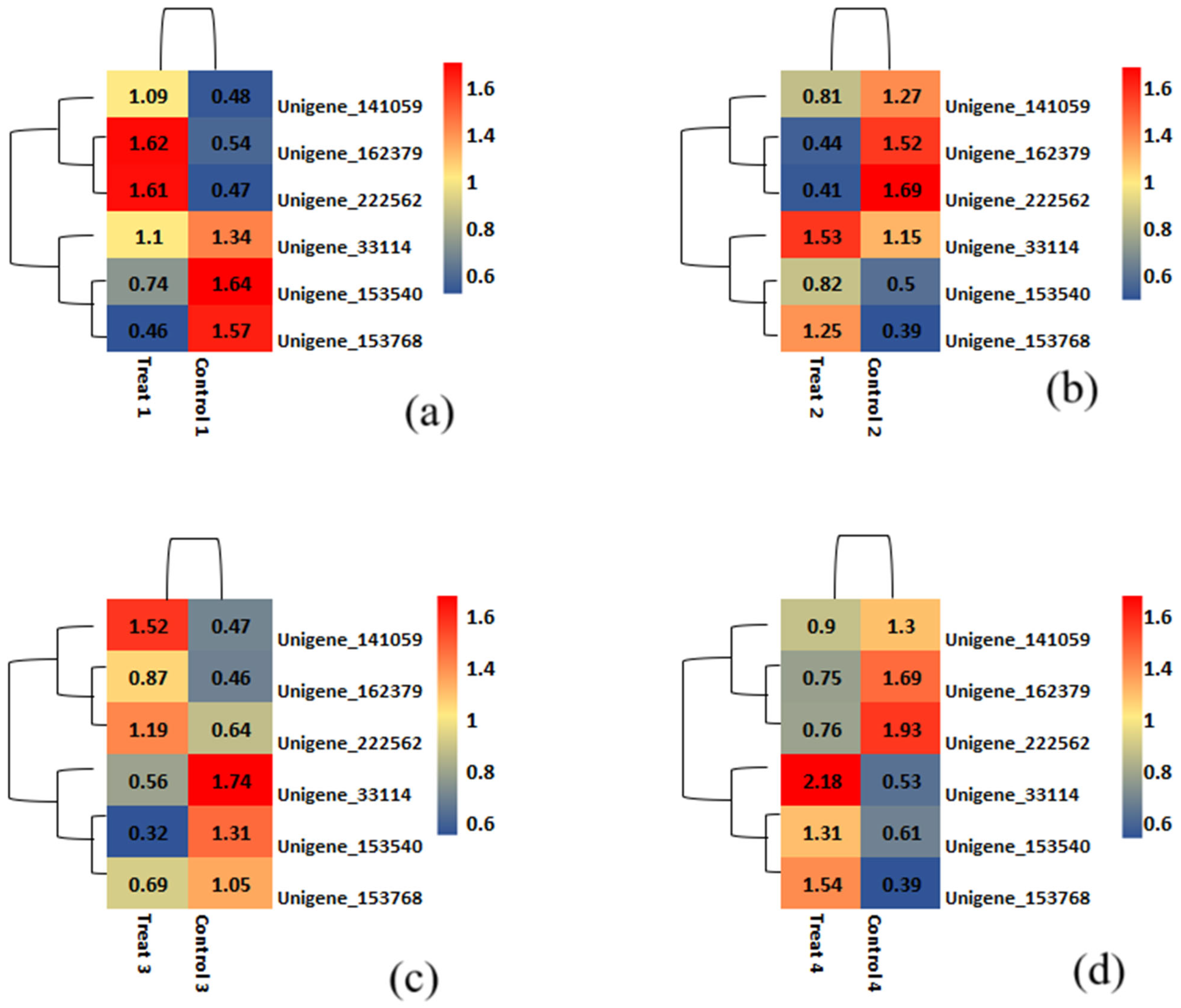
2.5. Gene Prediction and Differential Gene Expression Study
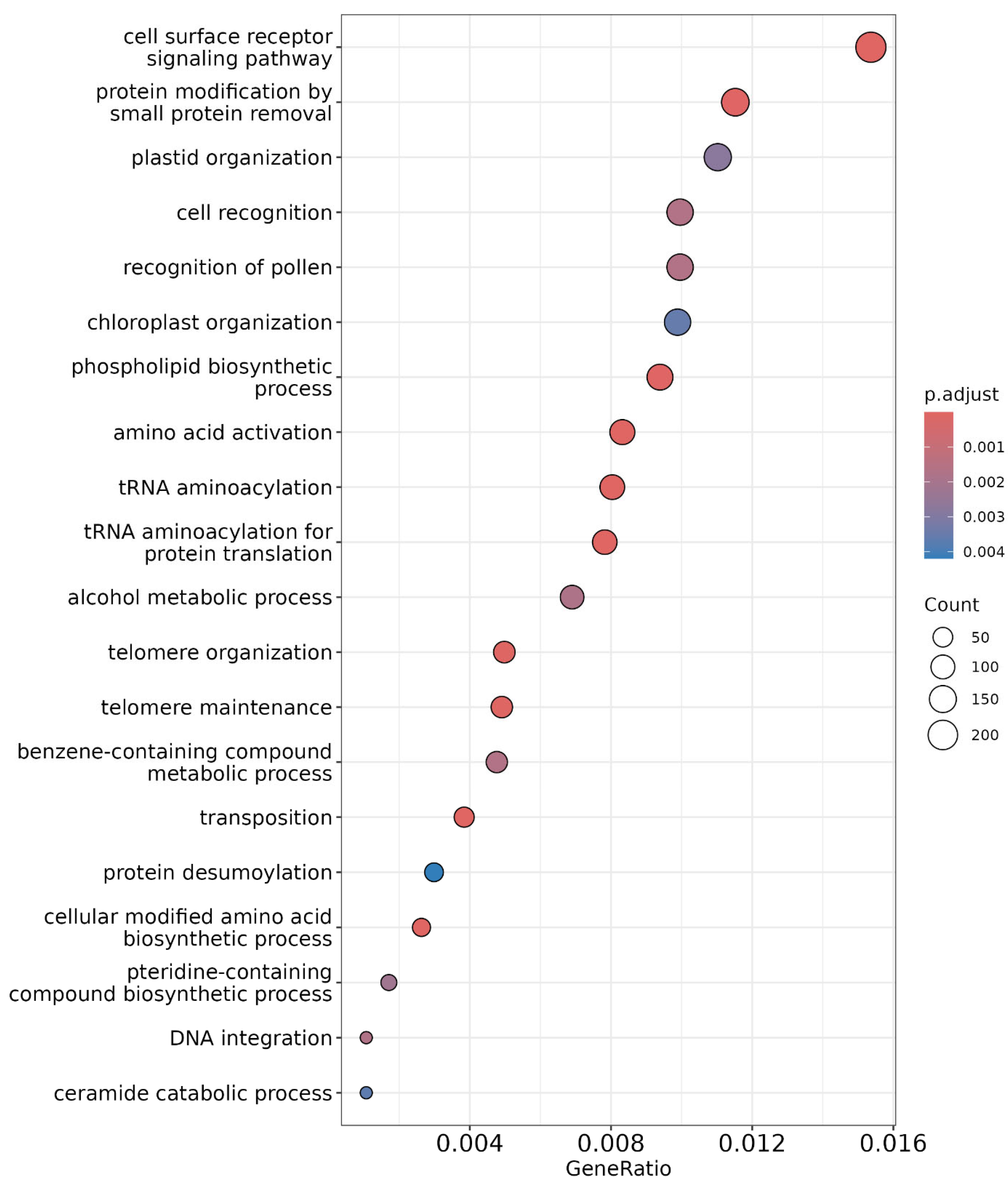
2.6. Gene Ontology
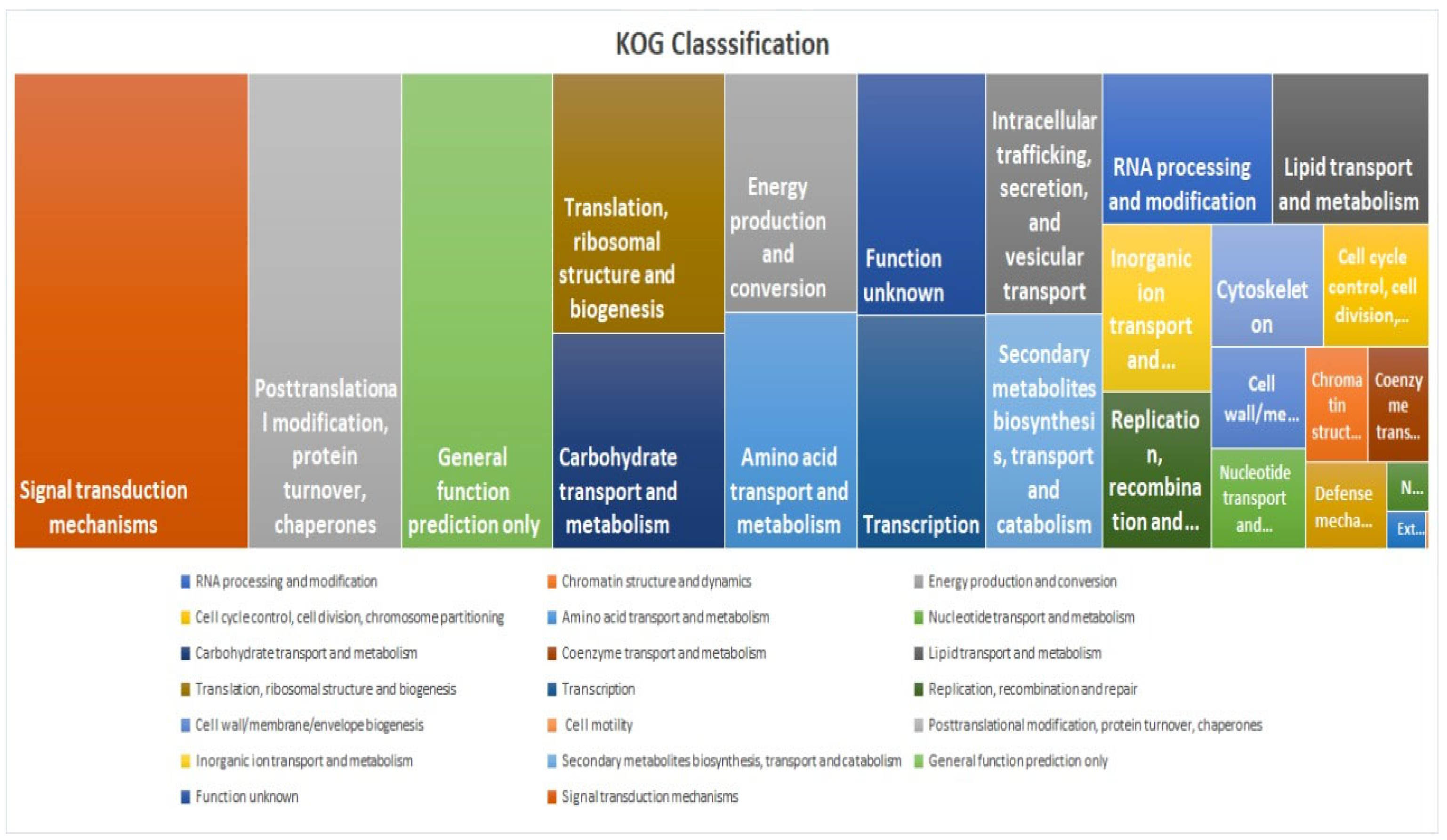
2.7. KOG Classification and Comparative Gene Annotation
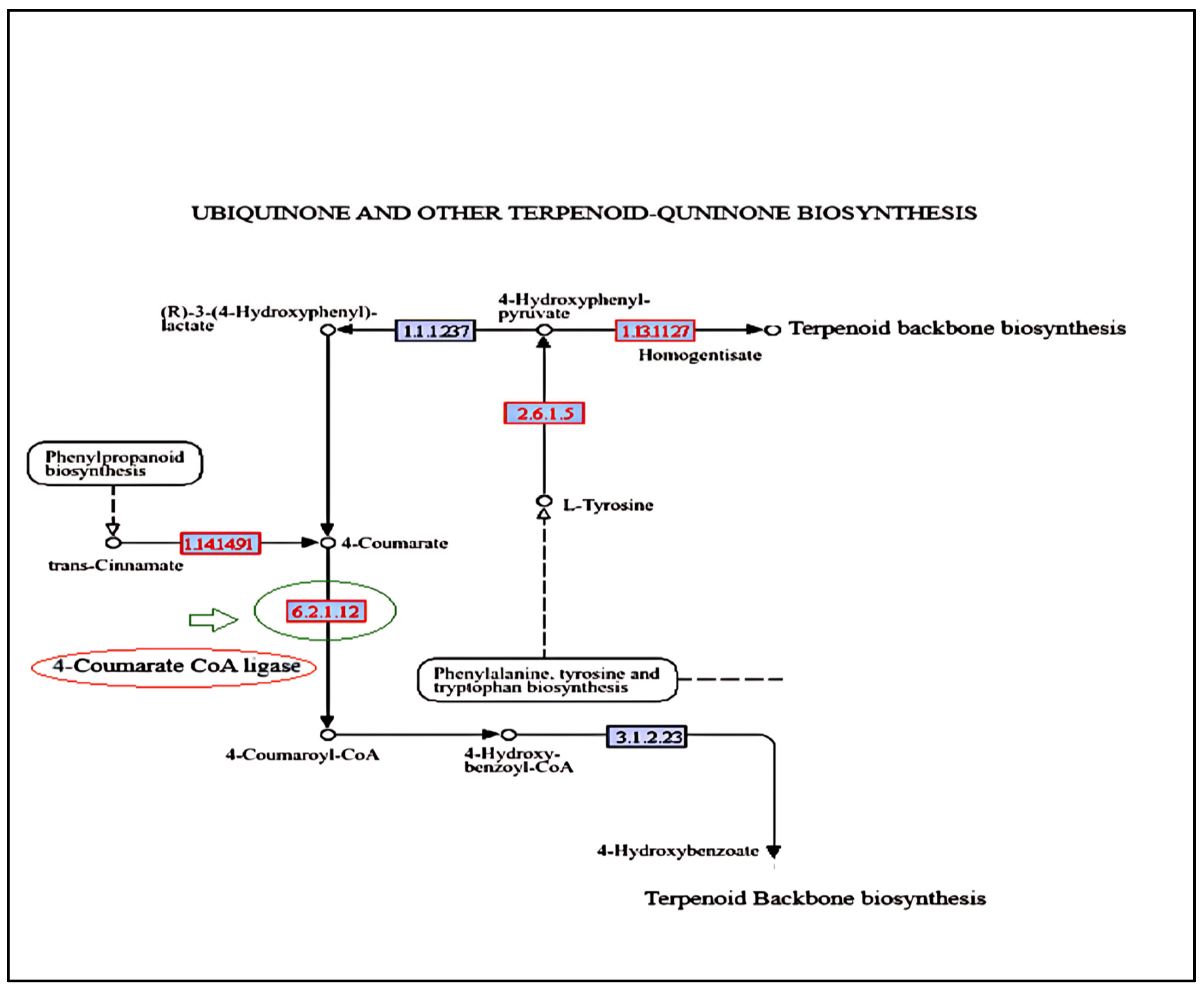
2.8. KEGG Pathway Analysis
2.9. Transcription Factors Identified
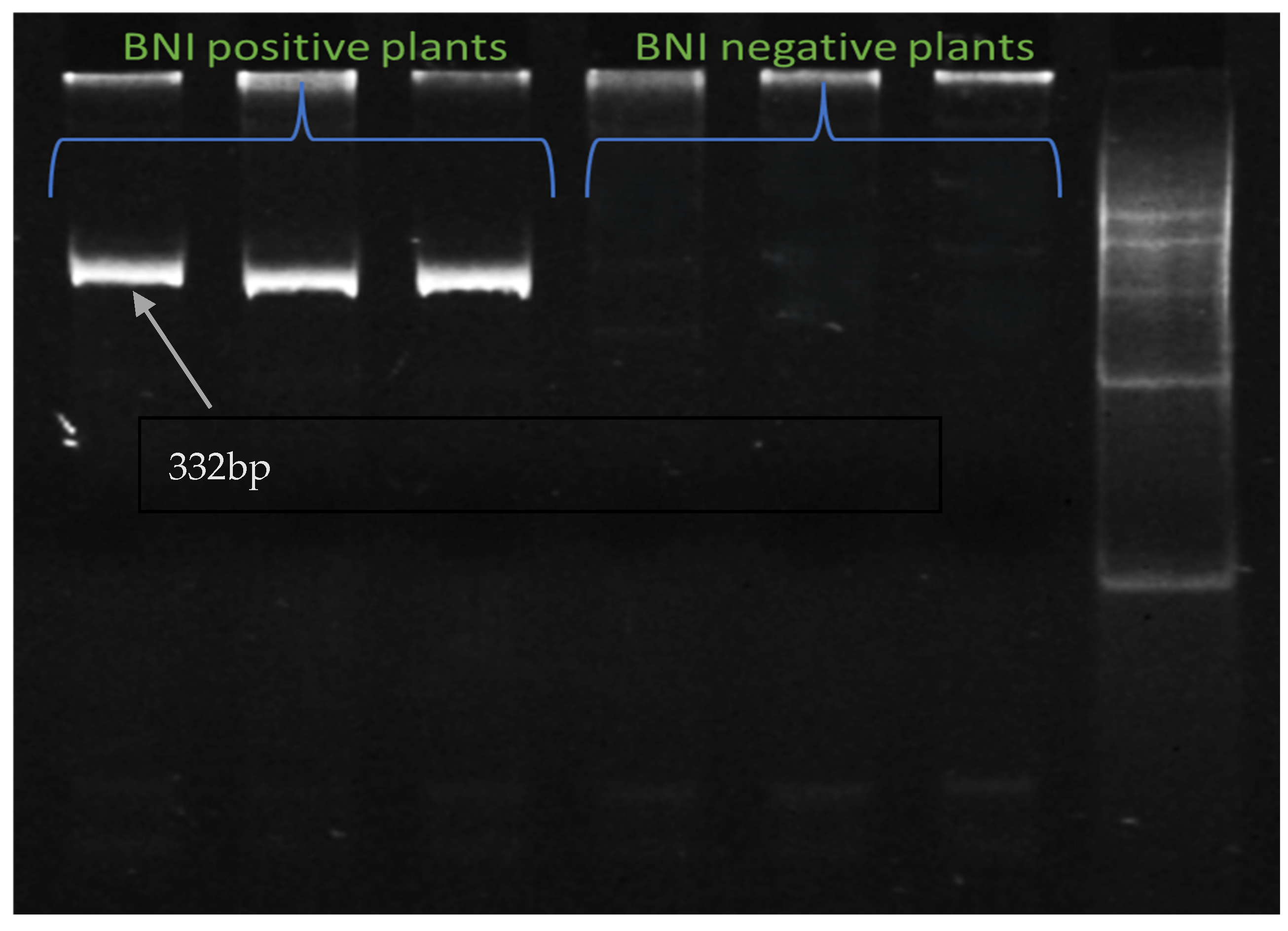
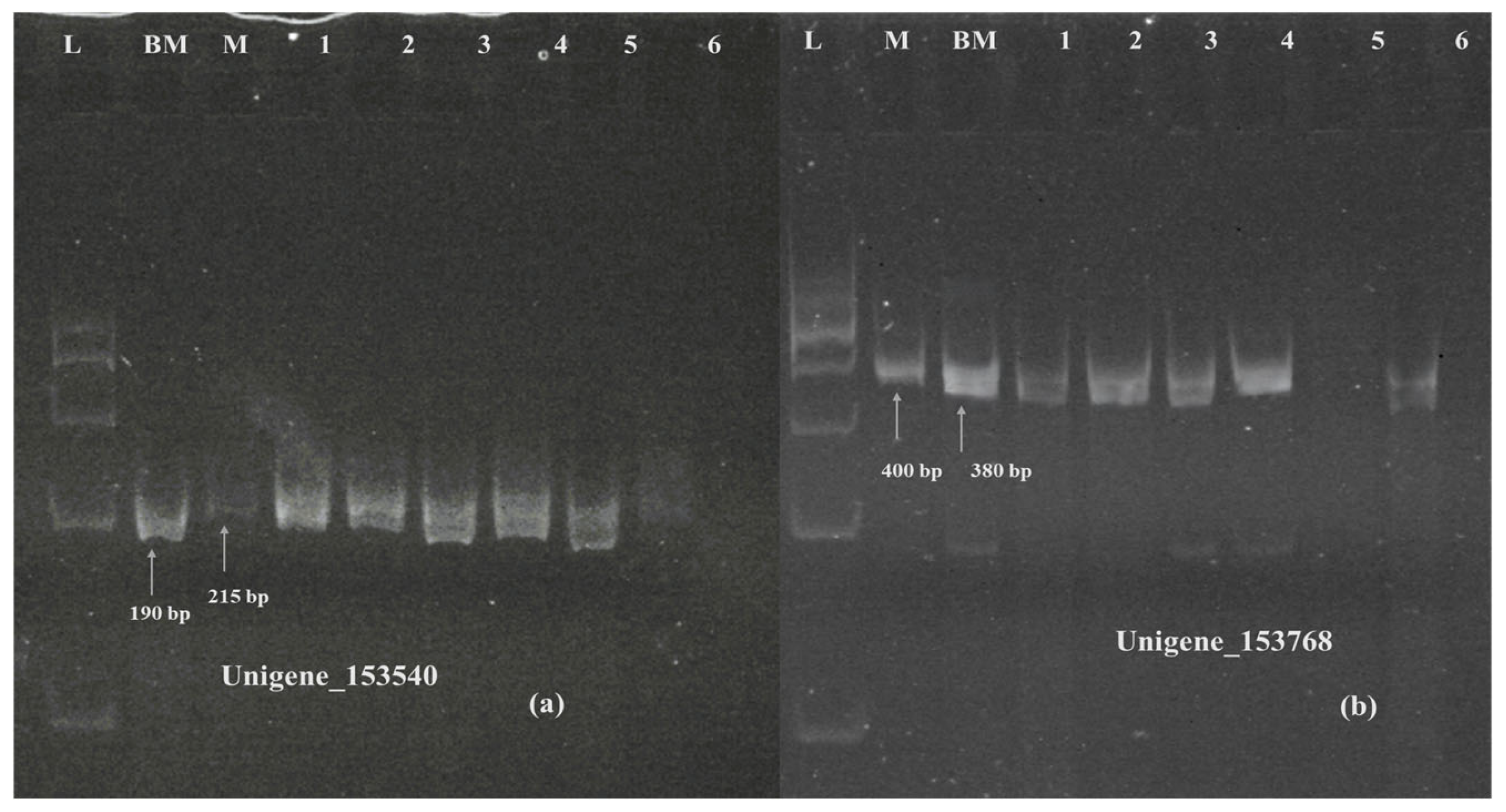
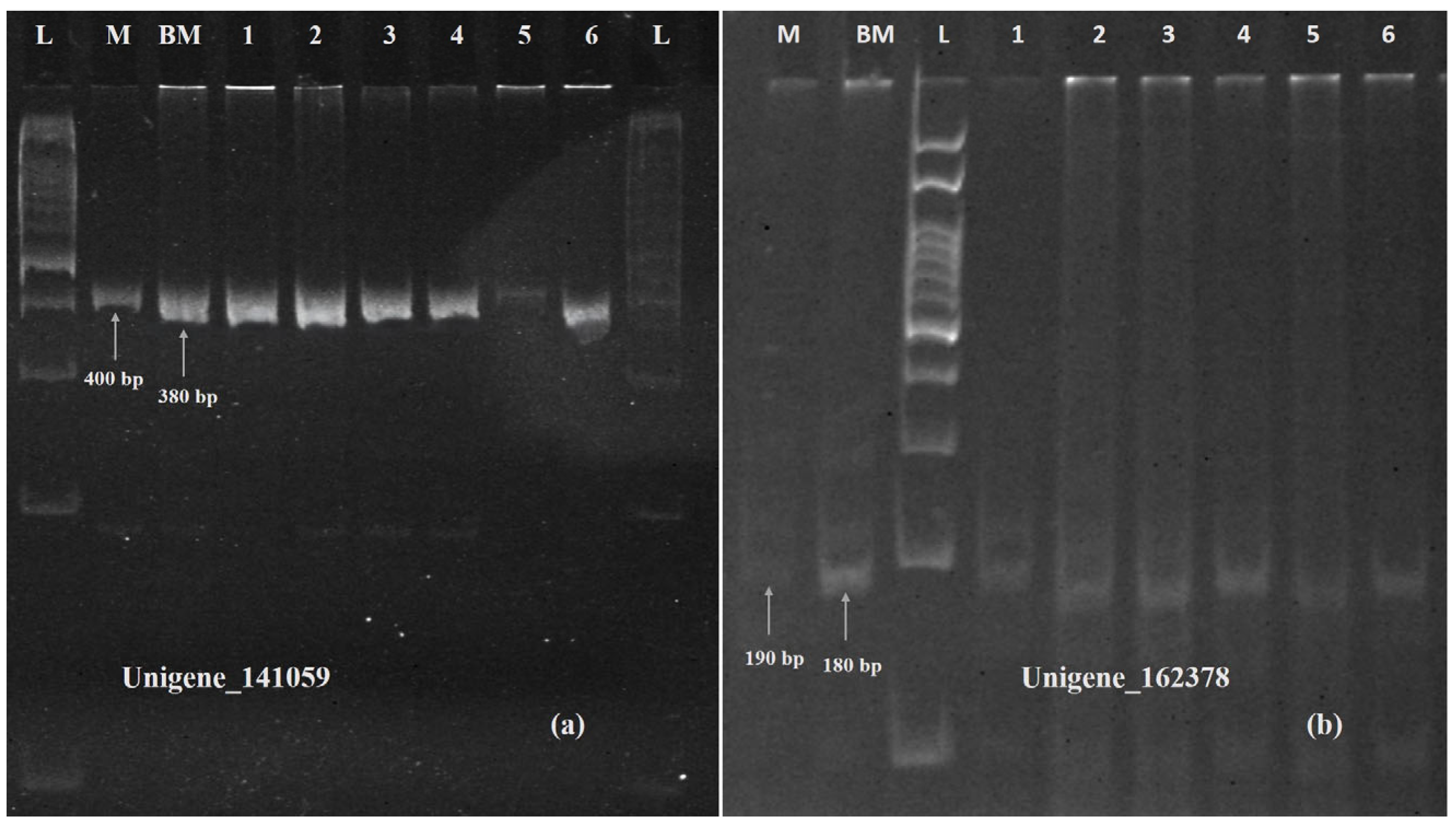
2.10. Validation of Plants with Genome-Specific DArT-Based Marker and Other Identified SSR Markers
3. Discussion
4. Materials and Methods
4.1. Plant Material, Nitrogen Dose Administration, and Data Collection of Agro-Morphological Traits
4.2. Sample Collection for Transcriptomic Analysis
4.3. RNA Isolation and Nitrogen Management Level
4.4. Library Construction and Sequencing
4.5. De Novo RNA-Seq Assembly and High-Quality Read Generation
4.6. Differential Gene Expression Analysis
4.7. Functional Gene Annotation and Gene Ontology (GO) Term Analysis
4.8. Enrichment Analysis Based on Gene Ontology Terms
4.9. KEGG Pathway Analysis
4.10. Validation and Polymerase Chain Reaction Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BNI | Biological nitrogen inhibition |
| NUE | Nitrogen use efficiency |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GO | Gene ontology |
References
- Cameron, K.C.; Di, H.J.; Moir, J.L. Nitrogen losses from the soil/plant system: A review. Ann. Appl. Biol. 2013, 162, 145–173. [Google Scholar] [CrossRef]
- Hina, N.S.; Athat, H.R.; Drewes, J.E.; Mishra, B.K. Global meta-analysis of nitrate leaching vulnerability in synthetic and organic fertilizers over the past four decades. Water 2024, 16, 457. [Google Scholar] [CrossRef]
- Syakila, A.; Kroeze, C. The global nitrous oxide budget revisited. Greenh. Gas. Meas. Manag. 2011, 1, 17–26. [Google Scholar] [CrossRef]
- Conley, D.J.; Paerl, H.W.; Howarth, R.W.; Boesch, D.F.; Seitzinger, S.P.; Havens, K.E.; Lancelot, C.; Likens, G.E. Controlling eutrophication: Nitrogen and phosphorus. Science 2009, 323, 1014–1015. [Google Scholar] [CrossRef]
- Raun, W.R.; Johnson, G.V. Improving nitrogen use efficiency for cereal production. Agron. J. 1999, 91, 357–363. [Google Scholar] [CrossRef]
- Cassman, K.G. Ecological intensification of cereal production systems: Yield potential, soil quality, and precision agriculture. Proc. Natl. Acad. Sci. USA 1999, 96, 5952–5959. [Google Scholar] [CrossRef]
- Ju, X.T.; Xing, G.X.; Chen, X.P.; Zhang, S.L.; Zhang, L.J.; Liu, X.J.; Cui, Z.L.; Yin, B.; Christie, P.; Zhu, Z.L.; et al. Reducing environmental risk by improving N management in intensive Chinese agricultural systems. Proc. Natl. Acad. Sci. USA 2009, 106, 3041–3046. [Google Scholar] [CrossRef]
- Peng, B.; Li, H.; Peng, X.; Zhang, Y.; Xu, N.; Liu, Y. Nitrogen assimilation and remobilization in plants: Progresses and perspectives. Plant Commun. 2019, 1, 100005. [Google Scholar] [CrossRef]
- Cox, W.J.; Reisenauer, H.M. Growth and ion uptake by wheat supplied nitrogen as nitrate, or ammonium, or both. Plant Soil 1973, 38, 363–380. [Google Scholar] [CrossRef]
- Torralbo, F.; González-Moro, M.B.; Baroja-Fernández, E.; Aranjuelo, I.; González-Murua, C. Differential regulation of stomatal conductance as a strategy to cope with ammonium fertilizer under ambient versus elevated CO2. Front. Plant Sci. 2019, 10, 597. [Google Scholar]
- Subbarao, G.V.; Sahrawat, K.L.; Nakahara, K.; Ishikawa, T.; Kishii, M.; Rao, I.M.; Hash, C.T.; George, T.S.; Srinivasa Rao, P.; Nardi, P.; et al. A paradigm shift towards low-nitrifying production systems: The role of biological nitrification inhibition (BNI). Ann. Bot. 2013, 112, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Subbarao, G.V.; Kishii, M.; Bozal-Leorri, A.; Ortiz-Monasterio, I.; Gao, X.; Ibba, M.I.; Karwat, H.; Gonzalez-Moro, M.B.; Gonzalez-Murua, C.; Yoshihashi, T.; et al. Enlisting wild grass genes to combat nitrification in wheat farming: A nature-based solution. Proc. Natl. Acad. Sci. USA 2021, 118, e2106595118. [Google Scholar] [CrossRef] [PubMed]
- Bozal-Leorri, A.; Subbarao, G.V.; Kishii, M.; Urmeneta, L.; Kommerell, V.; Karwat, H.; Braun, H.J.; Aparicio-Tejo, P.M.; Ortiz-Monasterio, I.; González-Murua, C.; et al. Biological nitrification inhibitor-trait enhances nitrogen uptake by suppressing nitrifier activity and improves ammonium assimilation in two elite wheat varieties. Front. Plant Sci. 2022, 13, 1034219. [Google Scholar] [CrossRef] [PubMed]
- Mishra, C.N.; Sharma, S.; Pawar, S.K.; Sabhyata; Mukesh; Kumar, A.; Kumar, S.; Mamrutha, H.M.; Tyagi, B.S.; Singh, G.P.; et al. Phenotyping and breeding strategies for incorporating BNI trait in wheat varieties. Plant Physiol. Rep. 2024, 29, 808–822. [Google Scholar] [CrossRef]
- Noureldin, N.A.; Saudy, H.S.; Ashwamy, F.; Saed, H.M. Grain yield response index of bread wheat cultivars as influenced by nitrogen levels. Ann. Agric. Sci. 2013, 58, 147–152. [Google Scholar] [CrossRef]
- Dong, Z.; Yang, X.; Liu, X.; Wang, B.; Ma, J.; Ren, J.; Qin, H.; Zhang, L.; Zhao, J.; Zhong, Z.; et al. Genome-wide impacts of alien chromatin introgression on wheat gene transcriptions. Sci. Rep. 2020, 10, 4801. [Google Scholar] [CrossRef]
- Chen, L.; Zhao, J.; Song, J.; Jameson, P.E.; Song, X. Genome-wide analysis and evolutionary perspective of the cytokinin dehydrogenase gene family in wheat (Triticum aestivum L.). Front. Genet. 2022, 13, 931659. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Zhang, Q. Genetic regulation of wheat plant architecture and nitrogen-use efficiency: Prospects for sustainable yield improvement. Adv. Sci. 2024, 11, 16433. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Zhang, Q. PPR proteins in plants: Roles, mechanisms, and prospects for rice research. Front. Plant Sci. 2024, 15, 1416742. [Google Scholar] [CrossRef]
- Dongwei, D.; Mingkun, M.; Xiaoyang, Z.; Yufang, L.; Kronzucker, H.J.; Weiming, S. Potential Secretory Transporters and Bisynthetic precursors of biological nitrification inhibitor 1,9-Decanediol in rice as revealed by transcriptome and metabolome amalyses. Rice Sci. 2024, 31, 87–102. [Google Scholar] [CrossRef]
- Subbarao, G.V.; Nakahara, K.; Hurtado, M.P.; Ono, H.; Moreta, D.E.; Salcedo, A.F.; Yoshihashi, T.; Ishikawa, T.; Ishitani, M.; Ohnishi-Kameyama, M.; et al. Evidence for biological nitrification inhibition in Brachiaria pastures. Proc. Natl. Acad. Sci. USA 2009, 106, 17302–17307. [Google Scholar] [CrossRef] [PubMed]
- Xuan, W.; Beeckman, T.; Xu, G. Plant nitrogen nutrition: Sensing and signaling. Curr. Opin. Plant. Biol. 2017, 39, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, A.; Chaturvedi, P.; Waldherr, S.; Subbarao, G.V.; Weckwerth, W. PANOMICS and the interface of root-soil microbiome and BNI. Trends Plant Sci. 2023, 28, 106–122. [Google Scholar] [CrossRef]
- Nunes-Nesi, A.; Fernie, A.R.; Stitt, M. Metabolic and signaling aspects underpinning the regulation of plant carbon nitrogen interactions. Mol. Plant 2010, 3, 973–996. [Google Scholar] [CrossRef]
- O’Brien, J.A.; Benková, E. Cytokinin cross-talking during biotic and abiotic stress responses. Front. Plant Sci. 2013, 4, 451. [Google Scholar] [CrossRef]
- Zhao, M.; Ding, H.; Zhu, J.K.; Zhang, F. Expanding plant transcriptomics to elucidate stress response mechanisms. Plant Physiol. 2017, 174, 1164–1174. [Google Scholar]
- Marschner, P.; Rengel, Z. (Eds.) Nutient Cycling in Terrestrial Ecosystems; Volume 10; Springer Science & Bussiness Media: Dordrencht, The Netherlands, 2007. [Google Scholar]
- Prasse, D.; Schmitz, R.A. Small RNAs involved in regulation of nitrogen metabolism. Microbiol Spectr. 2018, 6. [Google Scholar] [CrossRef]
- Denancé, N.; Szurek, B.; Noel, L.D. Emerging functions of nodulin-like proteins in non-nodulating plant species. Plant. Cell. Physiol. 2014, 55, 469–474. [Google Scholar] [CrossRef]
- Kumar, V.; Naresh, R.K.; Kumar, S.; Kumar, S.; Kumar, A.; Gupta, R.K.; Rathore, R.S.; Singh, S.P.; Dwivedi, A.; Tyagi, S.; et al. Efficient Nutrient Management Practices for Sustaining Soil Health and Improving Rice–Wheat Productivity: A Review. J. Pharmacogn. Phytochem. 2018, 7, 585–597. [Google Scholar]
- Ashikari, M.; Sakakibara, H.; Lin, S.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.R.; Qian, Q.; Kitano, H.; Matsuoka, M. Cytokinin oxidase regulates rice grain production. Science 2005, 309, 741–745. [Google Scholar] [CrossRef]
- Buchner, P.; Hawkesford, M.J. Complex Phylogeny and Gene Expression Patterns of Members of the NITRATE TRANSPORTER 1/PEPTIDE TRANSPORTER Family (NPF) in Wheat. J. Exp. Bot. 2014, 65, 5697–5710. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Naz, M.; Fan, X.; Xuan, W.; Miller, A.J.; Xu, X. Plant nitrate transporters: From gene function to application. J. Exp. Bot. 2017, 68, 2463–2475. [Google Scholar] [CrossRef] [PubMed]
- Tegeder, M.; Masclaux-Daubresse, C. Source and sink mechanism of nitrogen transport and use. New Phytol. 2018, 217, 35–53. [Google Scholar] [CrossRef]
- Fan, S.C.; Lin, C.S.; Hsu, P.K.; Lin, S.H.; Tsay, Y.F. The Arabidopsis nitrate transporter NRT1.7, expressed in phloem, is responsible for reallocation of nitrate to young leaves. Plant Cell 2009, 21, 2750–2761. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Hsu, P.-K.; Tsay, Y.-F. Uptake, allocation and signaling of nitrate. Trends Plant Sci. 2012, 17, 458–467. [Google Scholar] [CrossRef]
- Hsu, P.K.; Tsay, Y.F. Two phloem nitrate transporters, NRT1.11 and NRT1.12, are important for redistributing xylem-borne nitrate to enhance plant growth. Plant Physiol. 2013, 163, 844–856. [Google Scholar] [CrossRef]
- Yang, D.; Zhao, J.; Bi, C.; Li, L.; Wang, Z. Transcriptome and proteomics analysis of wheat seedling roots reveals that increasing NH4+/NO3–ratio induced root lignification and reduced nitrogen utilization. Front. Plant Sci. 2022, 12, 797260. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Z.; Sun, X.; Mu, X.; Chen, H.; Chen, F.; Yuan, L.; Mi, G. Interaction effect of nitrogen form and planting density on plant growth and nutrient uptake in maize seedlings. J. Integr. Agric. 2019, 20, 1120–1129. [Google Scholar] [CrossRef]
- Lei, L.; Li, Y.; Wang, Q.; Xu, Y.; Li, H.; Yang, H.; Zhao, Y.; Li, Y.; Wang, Y.; Li, Y.; et al. Nitrogen Use Efficiency Is Regulated by Interacting Proteins Relevant to Development in Wheat. Plant Biotechnol. J. 2018, 16, 1214–1226. [Google Scholar] [CrossRef]
- Hawkesford, M.J. Genetic Variation in Traits for Nitrogen Use Efficiency in Wheat. J. Exp. Bot. 2017, 68, 2627–2632. [Google Scholar] [CrossRef]
- Wani, S.H.; Kumar, V.; Shriram, V.; Sah, S.K. Phytohormones and their metabolic engineering for abiotic stress tolerance in crop plants. Crop J. 2016, 4, 162–176. [Google Scholar] [CrossRef]
- Zhang, M.; Zeng, H.; Afzal, M.R.; Gao, X.; Li, Y.; Subbarao, G.V.; Zhu, Y. BNI-release mechanisms in plant root systems: Current status of understanding. Biol. Fertil. Soils 2020, 58, 225–233. [Google Scholar] [CrossRef]
- Hammani, K.; Gobert, A.; Hleibieh, K.; Choulier, L.; Small, I.; Giegé, P. An Arabidopsis dual-localized pentatricopeptide repeat protein interacts with nuclear proteins involved in gene expression regulation. Plant Cell 2011, 23, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Aluko, O.O.; Kant, S.; Adedire, O.M.; Li, C.; Yuan, G.; Liu, H.; Wang, Q. Unlocking the potentials of nitrate transporters at improving plant nitrogen use efficiency. Front. Plant Sci. 2023, 14, 1074839. [Google Scholar] [CrossRef]
- Galuszka, P.; Frébort, I.; Šebela, M.; Sauer, P.; Jacobsen, S.; Peč, P. Cytokinin oxidase or dehydrogenase? Mechanism of cytokinin degradation in cereals. Eur. J. Biochem. 2001, 268, 450–461. [Google Scholar] [CrossRef]
- Fu, Q.; Li, S.; Yu, D. Identification of an Arabidopsis Nodulin-related protein in heat stress. Mol. Cells 2010, 29, 77–84. [Google Scholar] [CrossRef]
- Subbarao, G.V.; Nakahara, K.; Ishikawa, T.; Ono, H.; Yoshihashi, T.; Yoshihashi, T.; Zhu, Y.; Zakir, H.; Deshpande, S.P.; Hash, C.T.; et al. Genetic mitigation strategies to tackle agricultural ammonia emissions. Plant Sci. 2017, 256, 42–51. [Google Scholar] [CrossRef]
- ICAR-IIWBR. Progress Report of AICRP on Wheat and Barley 2020-21, Crop Improvement; Singh, G., Tyagi, B.S., Gupta, A., Singh, S.K., Kumar, S., Khan, H., Gupta, V., Gopalareddy, K., Sharma, A.K., Mishra, C.N., et al., Eds.; ICAR-Indian Institute of Wheat and Barley Research: Karnal, India, 2021; p. 227. [Google Scholar]
- de Mendiburu, F. Agricolae: Statistical Procedures for Agricultural Research. R Package Version 1.3-7. 2023. Available online: https://CRAN.R-project.org/package=agricolae (accessed on 28 February 2025).
- Chaudhary, S.; Chaudhary, P.S.; Vaishnani, T.A. Small RNA Extraction using Fractionation Approach and Library Preparation for NGS Platform. J. Adv. Res. Biotech. 2016, 1, 7. [Google Scholar] [CrossRef]
- Zadoks, J.C.; Chang, T.T.; Konzak, C.F. A decimal code for the growth stages of cereals. Weed Res. 1974, 14, 415–421. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New Perspectives on Genomes, Pathways, Diseases and Drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source of Variation | PH | FLL | TPM | 1000 GW | N | GY |
|---|---|---|---|---|---|---|
| Genotype | 0.099 | 0.005 * | 0.070 | 0.003 * | 0.841 | 0.447 |
| Treatment | 0.001 * | 0.005 * | 0.000 * | 0.878 | 0.081 | 0.000 * |
| Interaction | 0.943 | 0.562 | 0.734 | 0.966 | 0.997 | 0.960 |
| DGE Combination | Total DEGs | Down- Regulated | Up- Regulated | Significant Downregulated | Significant Upregulated | Significant DGE |
|---|---|---|---|---|---|---|
| C1-N (0%) | 55,668 | 27,550 | 28,118 | 73 | 298 | 371 |
| C2-N (50%) | 36,605 | 18,277 | 18,328 | 31 | 230 | 261 |
| C3-N (75%) | 35,517 | 17,662 | 17,855 | 26 | 277 | 303 |
| C4-N (100%) | 52,732 | 26,339 | 26,393 | 290 | 446 | 736 |
| Gene | Sequence Description (Accession ID) | logFC | p-Value | E-Value |
|---|---|---|---|---|
| Unigene_141059 | PPR protein (AtPPR), pentatricopeptide repeat-containing protein (XP_044339860.1) | 3.1 | 1.13 × 10−5 | 0 |
| Unigene_162379 | cytokinin dehydrogenase (XP_044355953.1) | 5 | 1.46 × 10−10 | 0 |
| Unigene_222562 | CRB-INRA-CFD-3358 nodulin-related protein (NrpA) (XP_044353808.1) | 6.4 | 8.42 × 10−15 | 1.6 |
| Unigene_153540 | NRT1/PTR FAMILY 2.13-like (XP_044353808.1) | −2.9 | 3.90 × 10−5 | 1.3 |
| Unigene_153768 | VRN-A1 (AAW73220.1) | −3.7 | 4.22 × 10−7 | 2.00 × 10−177 |
| Unigene_33114 | Triticum aestivum Glutathione hydrolase 1-like (XM_044476980.1) | −0.9 | 0.15 | 0 |
| Transcription Factors ID | TF Name | Basic Functions | Number of Unigenes |
|---|---|---|---|
| bHLH | Basic Helix-Loop-Helix | developmental process, cell differentiation, and stress response | 4872 |
| WRKY | stress response and hormonal signalling | 4071 | |
| FAR1 | Far-Red Impaired Response 1 | regulation of seed development in far-red light | 4003 |
| MYB_related | Myeloblastosis-related | cell cycle regulation, stress response and secondary metabolism | 3853 |
| NAC | NAM (No Apical Meristem from Petunia), ATAF-1/2 (Arabidopsis Transcription Activation Factor genes), and CUC2 (Cup-shaped Cotyledon from Arabidopsis thaliana) | plant development and defining organ boundary function | 3645 |
| ERF | Ethylene Response Factor | regulating gene expression in response to ethylene levels | 3255 |
| B3 | B3 Domain Transcription Factor | seed development and embryogenesis | 2725 |
| C2H2 | Cys2-His2 Zinc Finger | act as transcriptional activators and repressors | 2483 |
| bZIP | basic Leucine Zipper | stress response and hormonal signalling, and other developmental process | 1946 |
| GRAS | Gibberellic Acid Insensitive, Repressor of GA1 and Scarecrow | plant growth, particularly in gibberellin signalling and root development | 1901 |
| S.No. | Nitrogen Management Levels | DGE Comparisons | BNI Munal (Treated) | Munal (Control) |
|---|---|---|---|---|
| 1 | 0% N (T1 and T2) | C1 | T2 | T1 |
| 2 | 50% N (T3 and T4) | C2 | T4 | T3 |
| 3 | 75% N (T5 and T6) | C3 | T6 | T5 |
| 4 | 100% N (T7 and T8) | C4 | T8 | T7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, C.N.; Pawar, S.K.; Sharma, S.; Thakur, A.; Sabhyata, S.; Mishra, S.; Kumar, S.; Gupta, O.P.; Joshi, A.K.; Tiwari, R. Transcriptomic Analysis to Understand the Nitrogen Stress Response Mechanism in BNI-Enabled Wheat. Int. J. Mol. Sci. 2025, 26, 4610. https://doi.org/10.3390/ijms26104610
Mishra CN, Pawar SK, Sharma S, Thakur A, Sabhyata S, Mishra S, Kumar S, Gupta OP, Joshi AK, Tiwari R. Transcriptomic Analysis to Understand the Nitrogen Stress Response Mechanism in BNI-Enabled Wheat. International Journal of Molecular Sciences. 2025; 26(10):4610. https://doi.org/10.3390/ijms26104610
Chicago/Turabian StyleMishra, Chandra Nath, Sushma Kumari Pawar, Swati Sharma, Arun Thakur, Sabhyata Sabhyata, Shubham Mishra, Satish Kumar, Om Prakash Gupta, Arun Kumar Joshi, and Ratan Tiwari. 2025. "Transcriptomic Analysis to Understand the Nitrogen Stress Response Mechanism in BNI-Enabled Wheat" International Journal of Molecular Sciences 26, no. 10: 4610. https://doi.org/10.3390/ijms26104610
APA StyleMishra, C. N., Pawar, S. K., Sharma, S., Thakur, A., Sabhyata, S., Mishra, S., Kumar, S., Gupta, O. P., Joshi, A. K., & Tiwari, R. (2025). Transcriptomic Analysis to Understand the Nitrogen Stress Response Mechanism in BNI-Enabled Wheat. International Journal of Molecular Sciences, 26(10), 4610. https://doi.org/10.3390/ijms26104610