

Type 1 Diabetes Mellitus and Vitamin D

Abstract
1. Introduction
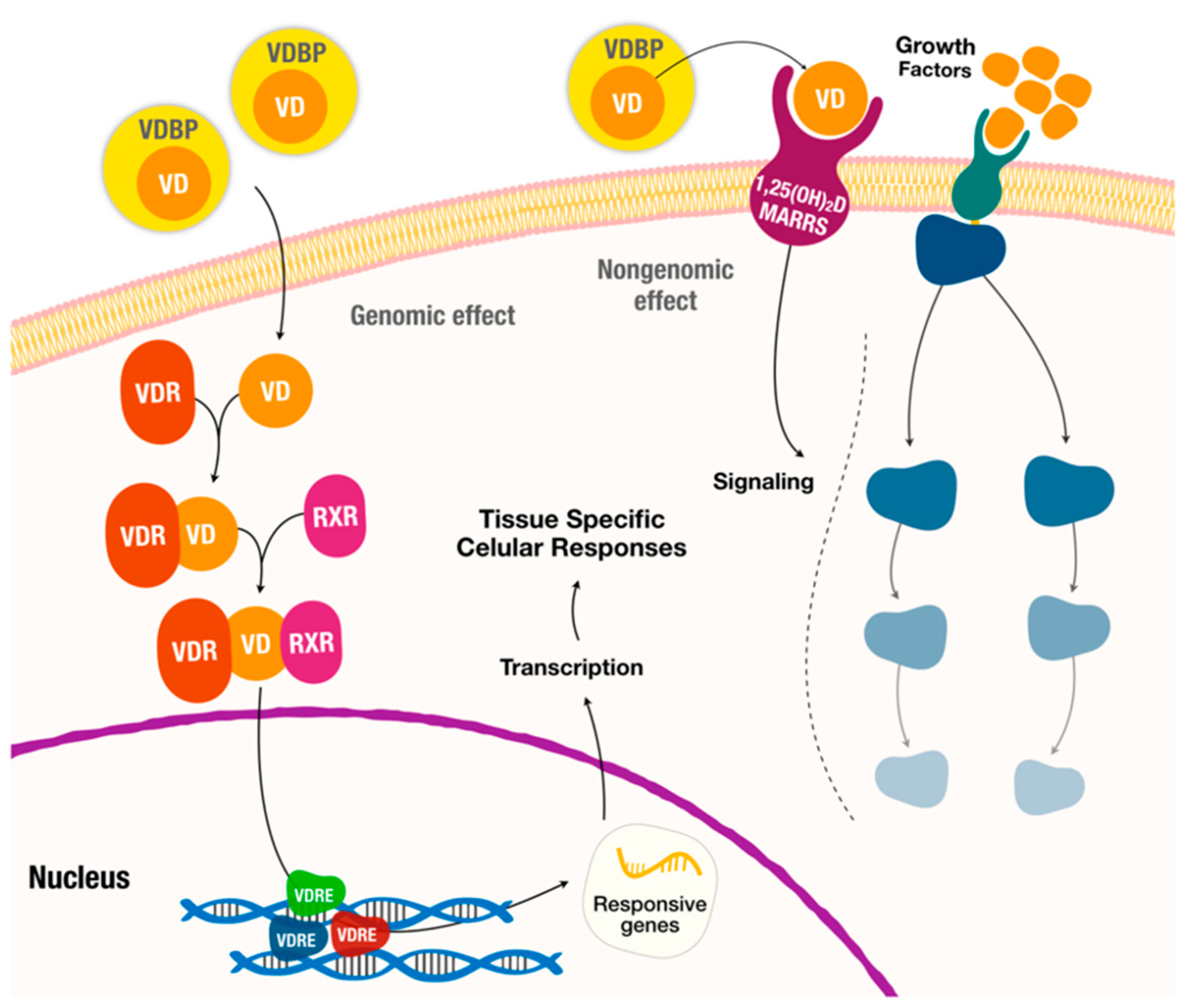
2. Vitamin D Synthesis and Metabolism
3. Pathogenesis and Natural History of Type 1 Diabetes Mellitus
4. Immunomodulatory Effects of Vitamin D in Autoimmune Diseases
5. Vitamin D Status and the Risk of Type 1 Diabetes Mellitus
6. Vitamin D Supplementation in Type 1 Diabetes Mellitus

{kind=link}
{kind=link}
| Author, Year, and Country | Study Design | Supplementation Dosage Duration | Significant Findings |
|---|---|---|---|
| Gabbay et al., 2012 (Brazil) [74] | Randomized, double blind, placebo-controlled, prospective trial | Cholecalciferol (2000 IU/d for 18 months) | Decrease in Hb1Ac levels Decrease in autoantibody titers Stimulated C-peptide enhancement Increase in Treg percentage |
| Ataie-Jafari et al., 2013 (Iran) [89] | Randomized, single blind, placebo-controlled trial | Alfacalcidol (0.25 μg/twice daily for 6 months) | Improved stimulated C-peptide |
| Federico et al., 2014 (Italy) [85] | Pilot interventional study | Calcidiol (10–30 μg/d for one year) | Decrease in insulin requirements Stability of fasting C-peptide levels Inhibition of GAD-65 antibodies |
| Treiber et al., 2015 (Austria) [75] | Randomized, double blind, placebo-controlled, prospective trial | Cholecalciferol (70 IU/kg/d for 12 months) | Decrease in Hb1Ac levels Stimulated C-peptide enhancement Reduction in daily insulin doses Increase in Treg percentage |
| Panjiyar et al., 2018 (India) [76] | Prospective, case-control, interventional study | Cholecalciferol (3000 IU/d for one year) | Decrease in Hb1Ac levels Reduction in daily insulin doses Stimulated C-peptide enhancement |
| Cadario et al., 2019 (Italy) [81] | Case study | Cholecalciferol (1000 IU/d) plus EPA + DHA (50–60 mg/kg/d) for 12 months | Decrease in insulin requirements |
| Reddy et al., 2022 (India) [82] | Pilot study | Cholecalciferol (2000 IU/d) plus lansoprazole (15–30 mg) for six months | Decrease in insulin requirements Slower fasting peptide-C decline |
| Pinheiro et al., 2023 (Brazil, Italy) [83] | Case-control study | Cholecalciferol (5000 IU/d) plus sitagliptin (50 mg/day) for 12 months | Longer duration of the remission phase |
| Nwosu et al., 2022 (Massachusetts, USA) [84] | Randomized, double blind, placebo-controlled, prospective trial | Ergocalciferol (50,000 IU/wk for 2 months, then fortnightly for 10 months) | Decrease in insulin requirements Reduction in TNF-α concentration |
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| 25(OH)D | 25-hydroxicholecalciferol |
| 1,25(OH)2D | 1,25-hydroxicholecalciferol |
| APCs | antigen-presenting cells |
| anti-IA2 | antibodies to insulinoma-associated antigen-2 |
| CD8+ T cells | cytotoxic T cells |
| DCs | dendritic cells |
| DHA | docosahexaenoic acid |
| EPA | eicosapentaenoic acid |
| GAD65 | antibodies to glutamic acid decarboxylase |
| Hb1Ac | hemoglobina glicosilada |
| HLA | human leukocyte antigen |
| IAAs | anti-insulin antibodies |
| ICAs | antibodies against insulin-producing islet cells |
| IFN-γ | interferon-γ |
| IL | interleukin |
| MARRS | membrane-associated rapid response steroid-binding protein |
| NK | natural killer cells |
| PPIs | proton pump inhibitor |
| PUFAs | polyunsaturated fatty acids |
| ROS | reactive oxygen species |
| RXR | retinoid X receptor |
| T1DM | Type 1 diabetes mellitus |
| TGF-β | transforming growth factor beta |
| Th1 | CD4+ type 1 T helper |
| Th2 | CD4+ type 2 T helper |
| Th17 | CD4+ type 17 T helper |
| Treg | CD4+ T regulatory cells |
| TNF-α | tumor necrosis factor α |
| VDR | vitamin D receptors |
| VDBP | vitamin D binding protein |
| VDRE | vitamin D response elements |
| ZnT8 | antibodies to zinc transporter 8 |
References
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.C.; Harjutsalo, V.; Rosenbauer, J.; Neu, A.; Cinek, O.; Skrivarhaug, T.; Rami-Merhar, B.; Soltesz, G.; Svensson, J.; Parslow, R.C. Trends and cyclical variation in the incidence of childhood type 1 diabetes in 26 European centres in the 25 year period 1989–2013: A multicentre prospective registration study. Diabetologia 2019, 62, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. The vitamin D deficiency pandemic: Approaches for diagnosis, treatment and prevention. Rev. Endocr. Metab. Disord. 2017, 18, 153–165. [Google Scholar] [CrossRef]
- Chen, Y.L.; Huang, Y.C.; Qiao, Y.C.; Ling, W.; Pan, Y.H.; Geng, L.J.; Xiao, J.L.; Zhang, X.X.; Zhao, H.L. Climates on incidence of childhood type 1 diabetes mellitus in 72 countries. Sci. Rep. 2017, 7, 12810. [Google Scholar] [CrossRef]
- Park, C.Y.; Shin, S.; Han, S.N. Multifaceted Roles of Vitamin D for Diabetes: From Immunomodulatory Functions to Metabolic Regulations. Nutrients 2024, 16, 3185. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Song, A.; Jin, Y.; Xia, Q.; Song, G.; Xing, X. A dose–response meta-analysis between serum concentration of 25-hydroxyvitamin D and risk of type 1 diabetes mellitus. Eur. J. Clin. Nutr. 2021, 75, 1010–1023. [Google Scholar] [CrossRef]
- Franchi, B.; Piazza, M.; Sandri, M.; Mazzei, F.; Maffeis, C.; Boner, A.L. Vitamin D at the onset of type 1 diabetes in Italian children. Eur. J. Pediatr. 2014, 173, 477–482. [Google Scholar] [CrossRef]
- Feng, R.; Li, Y.; Li, G.; Li, Z.; Zhang, Y.; Li, Q.; Sun, C. Lower serum 25 (OH) D concentrations in type 1 diabetes: A meta-analysis. Diabetes Res. Clin. Pract. 2015, 108, e71–e75. [Google Scholar] [CrossRef]
- Al-Zubeidi, H.; Leon-Chi, L.; Newfield, R.S. Low vitamin D level in pediatric patients with new onset type 1 diabetes is common, especially if in ketoacidosis. Pediatr. Diabetes 2016, 17, 1592–1598. [Google Scholar] [CrossRef]
- Rasoul, M.A.; Al-Mahdi, M.; Al-Kandari, H.; Dhaunsi, G.S.; Haider, M.Z. Low serum vitamin-D status is associated with high prevalence and early onset of type-1 diabetes mellitus in Kuwaiti children. BMC Pediatr. 2016, 6, 95. [Google Scholar] [CrossRef]
- Marino, R.; Misra, M. Extra-Skeletal Effects of Vitamin D. Nutrients 2019, 11, 1460. [Google Scholar] [CrossRef] [PubMed]
- Botelho, J.; Machado, V.; Proença, L.; Delgado, A.S.; Mendes, J.J. Vitamin D deficiency and oral health: A comprehensive review. Nutrients 2020, 12, 1471. [Google Scholar] [CrossRef] [PubMed]
- Daskalopoulou, M.; Pylli, M.; Giannakou, K. Vitamin D Deficiency as a Possible Cause of Type 1 Diabetes in Children and Adolescents up to 15 Years Old: A Systematic Review. Rev. Diabet. Stud. 2022, 18, 58–67. [Google Scholar] [CrossRef]
- Wu, J.; Atkins, A.; Downes, M.; Wei, Z. Vitamin D in Diabetes: Uncovering the Sunshine Hormone’s Role in Glucose Metabolism and Beyond. Nutrients 2023, 15, 1997. [Google Scholar] [CrossRef]
- Jacobs, A.; Warnants, M.; Vollmuth, V.; Winkler, C.; Weiss, A.; Ziegler, A.G.; Lundgren, M.; Elding Larsson, H.; Kordonouri, O.; von dem Berge, T.; et al. Vitamin D insufficiency in infants with increased risk of developing type 1 diabetes: A secondary analysis of the POInT Study. BMJ Paediatr. Open 2024, 8, e002212. [Google Scholar] [CrossRef]
- Yang, X.; Chai, M.; Lin, M. Proportion of vitamin D deficiency in children/adolescents with type 1 diabetes: A systematic review and meta-analysis. BMC Pediatr. 2024, 24, 192. [Google Scholar] [CrossRef]
- Liu, C.; Wang, J.; Wan., Y.; Xia., X.; Pan., J.; Gu., W.; Li., M. Serum vitamin D deficiency in children and adolescents is associated with type 1 diabetes mellitus. Endocr. Connect. 2018, 7, 1275–1279. [Google Scholar] [CrossRef] [PubMed]
- Norris, J.M.; Lee, H.S.; Frederiksen, B.; Erlund, I.; Uusitalo, U.; Yang, J.; Lernmark, Å.; Simell, O.; Toppari, J.; Rewers, M.; et al. Plasma 25-Hydroxyvitamin D Concentration and Risk of Islet Autoimmunity. Diabetes 2018, 67, 146–154. [Google Scholar] [CrossRef]
- Rasoul, M.A.; Haider, M.Z.; Al-Mahdi, M.; Al-Kandari, H.; Dhaunsi, G.S. Relationship of four vitamin D receptor gene polymorphisms with type 1 diabetes mellitus susceptibility in Kuwaiti children. BMC Pediatr. 2019, 19, 71. [Google Scholar] [CrossRef]
- Fletcher, J.; Bishop, E.M.; Harrison, S.R.; Swift, A.; Cooper, S.C.; Dimeloe, S.K.; Raza, K.; Hewison, M. Autoimmune disease and interconnections with vitamin D. Endocr. Connect. 2022, 11, e210554. [Google Scholar] [CrossRef]
- Holick, M.F.; Binkley, N.C.; Bischoff-Ferrari, H.A.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Murad, M.H.; Weaver, C.M. Endocrine Society. Evaluation, treatment, and prevention of vitamin D deficiency: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1911–1930. [Google Scholar] [CrossRef] [PubMed]
- Hii, C.S.; Ferrante, A. The Non-Genomic Actions of Vitamin D. Nutrients 2016, 8, 135. [Google Scholar] [CrossRef]
- Zmijewski, M.A. Nongenomic Activities of Vitamin D. Nutrients 2022, 14, 5104. [Google Scholar] [CrossRef]
- Paschou, S.A.; Papadopoulou-Marketou, N.; Chrousos, G.P.; Kanaka-Gantenbein, C. On type 1 diabetes mellitus pathogenesis. Endocr. Connect. 2018, 7, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Willcox, A.; Gillespie, K.M. Histology of Type 1 Diabetes Pancreas. Methods. Mol. Biol. 2016, 1433, 105–117. [Google Scholar]
- Noble, J.A.; Valdes, A.M. Genetics of the HLA region in the prediction of type 1 diabetes. Curr. Diab. Rep. 2011, 11, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Pociot, F.; Lernmark, A. Genetic risk factorsk for type 1 diabetes. Lancet 2016, 387, 2331–2339. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, L.M.; Zou, Y.; Zhang, S.; Xiong, F.; Wang, C.Y. Implication of epigenetic factors in the pathogenesis of type 1 diabetes. Chin. Med. J. 2021, 134, 1031–1042. [Google Scholar] [CrossRef]
- Zorena, K.; Michalska, M.; Kurpas, M.; Jaskulak, M.; Murawska, A.; Rostami, S. Environmental Factors and the Risk of Developing Type 1 Diabetes—Old Disease and New Data. Biology 2022, 11, 608. [Google Scholar] [CrossRef]
- Lamb, M.M.; Miller, M.; Seifert, J.A.; Frederiksen, B.; Kroehl, M.; Rewers, M.; Norris, J.M. The effect of childhood cow’s milk intake and HLA-DR genotype on risk of islet autoimmunity and type 1 diabetes: The diabetes autoimmunity study in the young. Pediatr. Diabetes 2015, 16, 31–38. [Google Scholar] [CrossRef]
- Neuman, V.; Plachy, L.; Pruhova, S.; Sumnik, Z. Dietary Components in the Pathogenesis and Prevention of Type 1 Diabetes in Children. Horm. Res. Paediatr. 2024, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Wong, F.S.; Wen, L. Type 1 diabetes and gut microbiota: Friend or foe? Pharmacol. Res. 2015, 98, 9–15. [Google Scholar] [CrossRef]
- Zhou, H.; Sun, L.; Zhang, S.; Zhao, X.; Gang, X.; Wang, G. Evaluating the Causal Role of Gut Microbiota in Type 1 Diabetes and Its Possible Pathogenic Mechanisms. Front. Endocrinol. 2020, 11, 125. [Google Scholar] [CrossRef] [PubMed]
- Del Chierico, F.; Rapini, N.; Deodati, A.; Matteoli, M.C.; Cianfarani, S.; Putignani, L. Pathophysiology of Type 1 Diabetes and Gut Microbiota Role. Int. J. Mol. Sci. 2022, 23, 14650. [Google Scholar] [CrossRef]
- Fliegerová, K.O.; Mahayri, T.M.; Sechovcová, H.; Mekadim, C.; Mrázek, J.; Jarošíková, R.; Dubský, M.; Fejfarová, V. Diabetes and gut microbiome. Front. Microbiol. 2025, 15, 1451054. [Google Scholar] [CrossRef]
- Wang, Z.; Lu, Q.; Wang, Z. Epigenetic alterations in cellular immunity: New insights into autoimmune diseases. Cell Physiol. Biochem. 2017, 41, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Toni, G.; Tascini, G.; Santi, E.; Berioli, M.G.; Principi, N. Environmental Factors Associated With Type 1 Diabetes. Front. Endocrinol. 2019, 10, 592. [Google Scholar] [CrossRef]
- LaMarca, V.; Gianchecchi, E.; Fierabracci, A. Type 1 Diabetes and Its Multi-Factorial Pathogenesis: The Putative Role of NK Cells. Int. J. Mol. Sci. 2018, 19, 794. [Google Scholar] [CrossRef]
- Sosenko, J.M.; Skyler, J.S.; Palmer, J.P.; Krischer, J.P.; Yu, L.; Mahon, J.; Eisenbarth, G. The prediction of type 1 diabetes by multiple autoantibody levels and their incorporation into an autoantibody risk score in relatives of type 1 diabetic patients. Diabetes Care 2013, 36, 2615–2620. [Google Scholar] [CrossRef]
- Infante, M.; Ricordi, C.; Sanchez, J.; Clare-Salzler, M.J.; Padilla, N.; Fuenmayor, V.; Chavez, C.; Alvarez, A.; Baidal, D.; Alejandro, R.; et al. Influence of Vitamin D on Islet Autoimmunity and Beta-Cell Function in Type 1 Diabetes. Nutrients 2019, 11, 2185. [Google Scholar] [CrossRef]
- He, L.P.; Song, Y.X.; Zhu, T.; Gu, W.; Liu, C.W. Progress in the Relationship between Vitamin D Deficiency and the Incidence of Type 1 Diabetes Mellitus in Children. J. Diabetes Res. 2022, 2022, 5953562. [Google Scholar] [CrossRef]
- Marino, K.R.; Lundberg, R.L.; Jasrotia, A.; Maranda, L.S.; Thompson, M.J.; Barton, B.A.; Alonso, L.C.; Nwosu, B.U. A predictive model for lack of partial clinical remission in new-onset pediatric type 1 diabetes. PLoS ONE 2017, 12, e0176860. [Google Scholar] [CrossRef] [PubMed]
- Fonolleda, M.; Murillo, M.; Vázquez, F.; Bel, J.; Vives-Pi, M. Remission Phase in Paediatric Type 1 Diabetes: New Understanding and Emerging Biomarkers. Horm. Res. Paediatr. 2017, 88, 307–315. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial oxidative stress— a causative factor and therapeutic target in many diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef] [PubMed]
- Gurgul-Convey, E.; Mehmeti, I.; Lortz, S.; Lenzen, S. Cytokine toxicity in insulin-producing cells is mediated by nitro-oxidative stress-induced hydroxyl radical formation in mitochondria. J. Mol. Med. 2011, 89, 785–798. [Google Scholar] [CrossRef]
- Chen, J.; Stimpson, S.E.; Fernandez-Bueno, G.A.; Mathews, C.E. Mitochondrial Reactive Oxygen Species and Type 1 Diabetes. Antioxid. Redox Signal. 2018, 29, 1361–1372. [Google Scholar] [CrossRef] [PubMed]
- Pinzaru, A.D.; Lupu, A.; Chisnoiu, T.; Baciu, G.; Baciu, A.P.; Baciu, C.; Lupu, V.V.; Balasa, A.L.; Chirila, S.; Panculescu, F.G.; et al. Comparative Study of Oxidative Stress Responses in Pediatric Type 1 Diabetes and Transient Hyperglycemia. Int. J. Mol. Sci. 2025, 26, 1701. [Google Scholar] [CrossRef]
- Prietl, B.; Treiber, G.; Pieber, T.R.; Amrein, K. Vitamin D and immune function. Nutrients 2013, 5, 2502–2521. [Google Scholar] [CrossRef]
- Dankers, W.; Colin, E.M.; van Hamburg, J.P.; Lubberts, E. Vitamin D in autoimmunity: Molecular mechanism and therapeutic potential. Front. Immunol. 2017, 7, 697. [Google Scholar] [CrossRef]
- Rak, K.; Bronkowska, M. Immunomodulatory Effect of Vitamin D and Its Potential Role in the Prevention and Treatment of Type 1 Diabetes Mellitus—A Narrative Review. Molecules 2018, 24, 53. [Google Scholar] [CrossRef] [PubMed]
- Cyprian, F.; Lefkou, E.; Varoudi, K.; Girardi, G. Immunomodulatory Effects of Vitamin D in Pregnancy and Beyond. Front. Immunol. 2019, 10, 2739. [Google Scholar] [CrossRef]
- Gallo, D.; Baci, D.; Kustrimovic, N.; Lanzo, N.; Patera, B.; Tanda, M.L.; Piantanida, E.; Mortara, L. How Does Vitamin D Affect Immune Cells Crosstalk in Autoimmune Diseases? Int. J. Mol. Sci. 2023, 24, 4689. [Google Scholar] [CrossRef]
- Ghaseminejad-Raeini, A.; Ghaderi, A.; Sharafi, A.; Nematollahi-Sani, B.; Moossavi, M.; Derakhshani, A.; Sarab, G.A. Immunomodulatory actions of vitamin D in various immune-related disorders: A comprehensive review. Front. Immunol. 2023, 14, 950465. [Google Scholar] [CrossRef]
- Galdo-Torres, D.; Andreu, S.; Caballero, O.; Hernández-Ruiz, I.; Ripa, I.; Bello-Morales, R.; López-Guerrero, J.A. Immune Modulatory Effects of Vitamin D on Herpesvirus Infections. Int. J. Mol. Sci. 2025, 26, 1767. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhang, Y.; Kwong, J.S.; Zhang, C.; Li, S.; Sun, F.; Niu, Y.; Du, L. The methodological quality assessment tools for preclinical and clinical studies, systematic review and meta-analysis, and clinical practice guideline: A systematic review. J. Evid. Based Med. 2015, 8, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Manousaki, D.; Harroud, A.; Mitchell, R.E.; Ross, S.; Forgetta, V.; Timpson, N.J.; Smith, G.D.; Polychronakos, C.; Richards, J.B. Vitamin D levels and risk of type 1 diabetes: A Mendelian randomization study. PLoS Med. 2021, 18, e1003536. [Google Scholar]
- Najjar, L.; Sutherland, J.; Zhou, A.; Hyppönen, E. Vitamin D and Type 1 Diabetes Risk: A. Systematic Review and Meta-Analysis of Genetic Evidence. Nutrients 2021, 13, 4260. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, Q.; Xu, N.; Xu, K.; Wang, J.; He, W.; Yang, T. Association between two polymorphisms (FokI and BsmI) of vitamin D receptor gene and type 1 diabetes mellitus in Asian population: A meta-analysis. PLoS ONE 2014, 9, e89325. [Google Scholar] [CrossRef]
- Habibian, N.; Amoli, M.M.; Abbasi, F.; Rabbani, A.; Alipour, A.; Sayarifard, F.; Rostami, P.; Dizaji, S.P.; Saadati, B.; Setoodeh, A. Role of vitamin D and vitamin D receptor gene polymorphisms on residual beta cell function in children with type 1 diabetes mellitus. Pharmacol. Rep. 2019, 71, 282–288. [Google Scholar] [CrossRef]
- Ran, Y.; Hu, S.; Yu, X.; Li, R. Association of vitamin D receptor gene polymorphism with type 1 diabetes mellitus risk in children: A protocol for systematic review and meta-analysis. Medicine 2021, 100, e26637. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, I.M.; Joner, G.; Jenum, P.A.; Eskild, A.; Torjesen, P.A.; Stene, L.C. Maternal serum levels of 25-hydroxy-vitamin D during pregnancy and risk of type 1 diabetes in the offspring. Diabetes 2012, 61, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.E.; Reinert, L.; Kinnunen, L.; Harjutsalo, V.; Koskela, P.; Surcel, H.M.; Lamberg-Allardt, C.; Tuomilehto, J. Serum 25-hydroxyvitamin D level during early pregnancy and type 1 diabetes risk in the offspring. Diabetologia 2012, 55, 1291–1294. [Google Scholar] [CrossRef]
- Granfors, M.; Augustin, H.; Ludvigsson, J.; Brekke, H.K. No association between use of multivitamin supplement containing vitamin D during pregnancy and risk of Type 1 Diabetes in the child. Pediatr. Diabetes 2016, 17, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Silvis, K.; Aronsson, C.A.; Liu, X.; Uusitalo, U.; Yang, J.; Tamura, R.; Lernmark, Å.; Rewers, M.; Hagopian, W.; She, J.X.; et al. Maternal dietary supplement use and development of islet autoimmunity in the offspring: TEDDY study. Pediatr. Diabetes 2019, 20, 86–92. [Google Scholar] [CrossRef]
- Dong, J.Y.; Zhang, W.G.; Chen, J.J.; Zhang, Z.L.; Han, S.F.; Qin, L.Q. Vitamin D intake and risk of type 1 diabetes: A meta-analysis of observational studies. Nutrients 2013, 5, 3551–3562. [Google Scholar] [CrossRef]
- Zipitis, C.S.; Akobeng, A.K. Vitamin D supplementation in early childhood and risk of type 1 diabetes: A systematic review and meta-analysis. Arch. Dis. Child. 2008, 93, 512–517. [Google Scholar] [CrossRef]
- Raab, J.; Giannopoulou, E.Z.; Schneider, S.; Warncke, K.; Krasmann, M.; Winkler, C.; Ziegler, A.G. Prevalence of vitamin D deficiency in pre-type 1 diabetes and its association with disease progression. Diabetologia 2014, 57, 902–908. [Google Scholar] [CrossRef]
- Simpson, M.; Brady, H.; Yin, X.; Seifert, J.; Barriga, K.; Hoffman, M.; Bugawan, T.; Barón, A.E.; Sokol, R.J.; Eisenbarth, G.; et al. No association of vitamin D intake or 25-hydroxyvitamin D levels in childhood with risk of islet autoimmunity and type 1 diabetes: The diabetes autoimmunity study in the young (DAISY). Diabetologia 2011, 54, 2779–2788. [Google Scholar] [CrossRef]
- Mäkinen, M.; Mykkänen, J.; Koskinen, M.; Simell, V.; Veijola, R.; Hyöty, H.; Ilonen, J.; Knip, M.; Simell, O.; Toppari, J. Serum 25-hydroxyvitamin D concentrations in children progressing to autoimmunity and clinical type 1 diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 723–729. [Google Scholar] [CrossRef]
- Giulietti, A.; Gysemans, C.; Stoffedvls, K.; van Etten, E.; Decallonne, B.; Overbergh, L.; Mathieu, C. Vitamin D deficiency in early life accelerates Type 1 diabetes in non-obese diabetic mice. Diabetologia 2004, 47, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Kachapati, K.; Adams, D.; Bednar, K.; Ridgway, W.M. The non-obese diabetic (NOD) mouse as a model of human type 1 diabetes. Methods Mol. Biol. 2012, 933, 3–16. [Google Scholar]
- Yu, J.; Sharma, P.; Girgis, C.M.; Gunton, J.E. Vitamin D and Beta Cells in Type 1 Diabetes: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 14434. [Google Scholar] [CrossRef]
- Gabbay, M.A.; Sato, M.N.; Finazzo, C.; Duarte, A.J.; Dib, S.A. Effect of cholecalciferol as adjunctive therapy with insulin on protective immunologic profile and decline of residual β-cell function in new-onset type 1 diabetes mellitus. Arch. Pediatr. Adolesc. Med. 2012, 166, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Treiber, G.; Prietl, B.; Fröhlich-Reiterer, E.; Lechner, E.; Ribitsch, A.; Fritsch, M. Cholecalciferol supplementation improves suppressive capacity of regulatory T-cells in young patients with new-onset type 1 diabetes mellitus—A randomized clinical trial. Clin. Immunol. 2015, 161, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Panjiyar, R.P.; Dayal, D.; Attri, S.V.; Sachdeva, N.; Sharma, R.; Bhalla, A.K. Sustained serum 25-hydroxyvitamin D concentrations for one year with cholecalciferol supplementation improves glycaemic control and slows the decline of residual β cell function in children with type 1 diabetes. Pediatr. Endocrinol. Diabetes Metab. 2018, 2018, 111–117. [Google Scholar] [CrossRef]
- Mishra, A.; Dayal, D.; Sachdeva, N.; Attri, S.V. Effect of 6-months’ vitamin D supplementation on residual beta cell function in children with type 1 diabetes: A case control interventional study. J. Pediatr. Endocrinol. Metab. 2016, 29, 395–400. [Google Scholar] [CrossRef]
- Sharma, S.; Biswal, N.; Bethou, A.; Rajappa, M.; Kumar, S.; Vinayagam, V. Does Vitamin D Supplementation Improve Glycaemic Control In Children With Type 1 Diabetes Mellitus?—A Randomized Controlled Trial. J. Clin. Diagn. Res. 2017, 11, SC15. [Google Scholar] [CrossRef]
- Perchard, R.; Magee, L.; Whatmore, A.; Ivison, F.; Murray, P.; Stevens, A. A pilot interventional study to evaluate the impact of cholecalciferol treatment on HbA1c in type 1 diabetes (T1D). Endocr. Connect. 2017, 6, 225–231. [Google Scholar] [CrossRef]
- Li, X.; Liao, L.; Yan, X.; Huang, G.; Lin, J.; Lei, M. Protective effects of 1-alpha-hydroxyvitamin D3 on residual beta-cell function in patients with adult-onset latent autoimmune diabetes (LADA). Diabetes Metab. Res. Rev. 2009, 25, 411–416. [Google Scholar] [CrossRef]
- Cadario, F.; Pozzi, E.; Rizzollo, S.; Stracuzzi, M.; Beux, S.; Giorgis, A.; Carrera, D.; Fullin, F.; Riso, S.; Rizzo, A.M.; et al. Vitamin D and ω-3 Supplementations in Mediterranean Diet During the 1st Year of Overt Type 1 Diabetes: A Cohort Study. Nutrients. 2019, 11, 2158. [Google Scholar] [CrossRef]
- Reddy, R.; Dayal, D.; Sachdeva, N.; Attri, S.V.; Gupta, V.K. Combination therapy with lansoprazole and cholecalciferol is associated with a slower decline in residual beta-cell function and lower insulin requirements in children with recent onset type 1 diabetes: Results of a pilot study. Einstein 2022, 20, eAO0149. [Google Scholar] [CrossRef]
- Pinheiro, M.M.; Pinheiro, F.M.M.; de Arruda, M.M.; Beato, G.M.; Verde, G.A.C.L.; Bianchini, G.; Casalenuovo, P.R.M.; Argolo, A.A.A.; de Souza, L.T.; Pessoa, F.G.; et al. Association between sitagliptinplus vitamin D3 (VIDPP-4i) use and clinical remission in patients with new-onset type 1 diabetes: A retrospectivecase-control study. Arch. Endocrinol. Metab. 2023, 67, e000652. [Google Scholar] [CrossRef]
- Nwosu, B.U.; Parajuli, S.; Jasmin, G.; Fleshman, J.; Sharma, R.B.; Alonso, L.C.; Lee, A.F.; Barton, B.A. Ergocalciferol in new-onset type 1 diabetes: A randomized controlled trial. J. Endocrine. Soc. 2022, 6, bvab179. [Google Scholar] [CrossRef] [PubMed]
- Federico, G.; Focosi, D.; Marchi, B.; Randazzo, E.; De Donno, M.; Vierucci, F. Administering 25-hydroxyvitamin D3 in vitamin D-deficient young type 1A diabetic patients reduces reactivity against islet autoantigens. Clin. Nutr. 2014, 33, 1153–1156. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, C.; Pitocco, D.; Napoli, N.; di Stasio, E.; Maggi, D.; Manfrini, S. No protective effect of calcitriol on beta-cell function in recent-onset type 1 diabetes: The IMDIAB XIII trial. Diabetes Care 2010, 33, 1962–1963. [Google Scholar] [CrossRef]
- Walter, M.; Kaupper, T.; Adler, K.; Foersch, J.; Bonifacio, E.; Ziegler, A.G. No effect of the 1alpha,25-dihydroxyvitamin D3 on beta-cell residual function and insulin requirement in adults with new-onset type 1 diabetes. Diabetes Care 2010, 33, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Rizka, A.; Setiati, S.; Harimurti, K.; Sadikin, M.; Mansur, I.G. Effect of Alfacalcidol on Inflammatory markers and T Cell Subsets in Elderly with Frailty Syndrome: A Double Blind Randomized Controlled Trial. Acta Med. Indones. 2018, 50, 215–221. [Google Scholar]
- Ataie-Jafari, A.; Loke, S.C.; Rahmat, A.B.; Larijani, B.; Abbasi, F.; Leow, M.K. A randomized placebo-controlled trial of alphacalcidol on the preservation of beta cell function in children with recent onset type 1 diabetes. Clin. Nutr. 2013, 32, 911–917. [Google Scholar] [CrossRef]
- Mastali, V.P.; Hoseini, R.; Azizi, M. The effect of short-term vitamin D on the antioxidant capacity following exhaustive aerobic exercise. Afri. Health. Sci. 2023, 23, 584–589. [Google Scholar] [CrossRef]
- Tagliferri, S.; Porri, D.; De Giuseppe, R.; Manuelli, M.; Alessio, F.; Cena, H. The controversial role of vitamin D as an antioxidant: Results from randomised controlled trials. Nutr. Res. Rev. 2019, 32, 99–105. [Google Scholar] [CrossRef] [PubMed]

| Immune Cell Type | Vitamin-D-Induced Effect |
|---|---|
| Macrophages | ▼ Pro-inflammatory IL-1, IL-6, IL-8, IL-12 ▲ Anti-inflammatory IL-10 |
| Natural killer cells | ▼ Pro-inflammatory IFN-γ ▲ Anti-inflammatory IL-4 |
| Dendritic cells | ▼ Pro-inflammatory IL-2, IL-6, IL-12 ▲ Anti-inflammatory IL-10 ▼ DCs’ differentiation (tolerogenic DCs) ▼ Antigen-presenting cells (T-cell anergy) |
| CD4+ T cells | ▼ Hyperactivation ▼ Th1, Th17 ▲ Th2, Treg ▲ Anti-inflammatory IL-4, IL-5, IL-10, TGF-β ▼ Pro-inflammatory IL-2, IFN-γ, TNF-α, IL-17, IL-21 |
| CD8+ T cells | ▼ Hyperactivation |
| B cells | ▼ B cell proliferation and differentiation into plasma cells ▼ Memory B cell formation ▼ Autoantibody production |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durá-Travé, T.; Gallinas-Victoriano, F. Type 1 Diabetes Mellitus and Vitamin D. Int. J. Mol. Sci. 2025, 26, 4593. https://doi.org/10.3390/ijms26104593
Durá-Travé T, Gallinas-Victoriano F. Type 1 Diabetes Mellitus and Vitamin D. International Journal of Molecular Sciences. 2025; 26(10):4593. https://doi.org/10.3390/ijms26104593
Chicago/Turabian StyleDurá-Travé, Teodoro, and Fidel Gallinas-Victoriano. 2025. "Type 1 Diabetes Mellitus and Vitamin D" International Journal of Molecular Sciences 26, no. 10: 4593. https://doi.org/10.3390/ijms26104593
APA StyleDurá-Travé, T., & Gallinas-Victoriano, F. (2025). Type 1 Diabetes Mellitus and Vitamin D. International Journal of Molecular Sciences, 26(10), 4593. https://doi.org/10.3390/ijms26104593