
Facing the Challenge to Mimic Breast Cancer Heterogeneity: Established and Emerging Experimental Preclinical Models Integrated with Omics Technologies


Abstract
1. Introduction
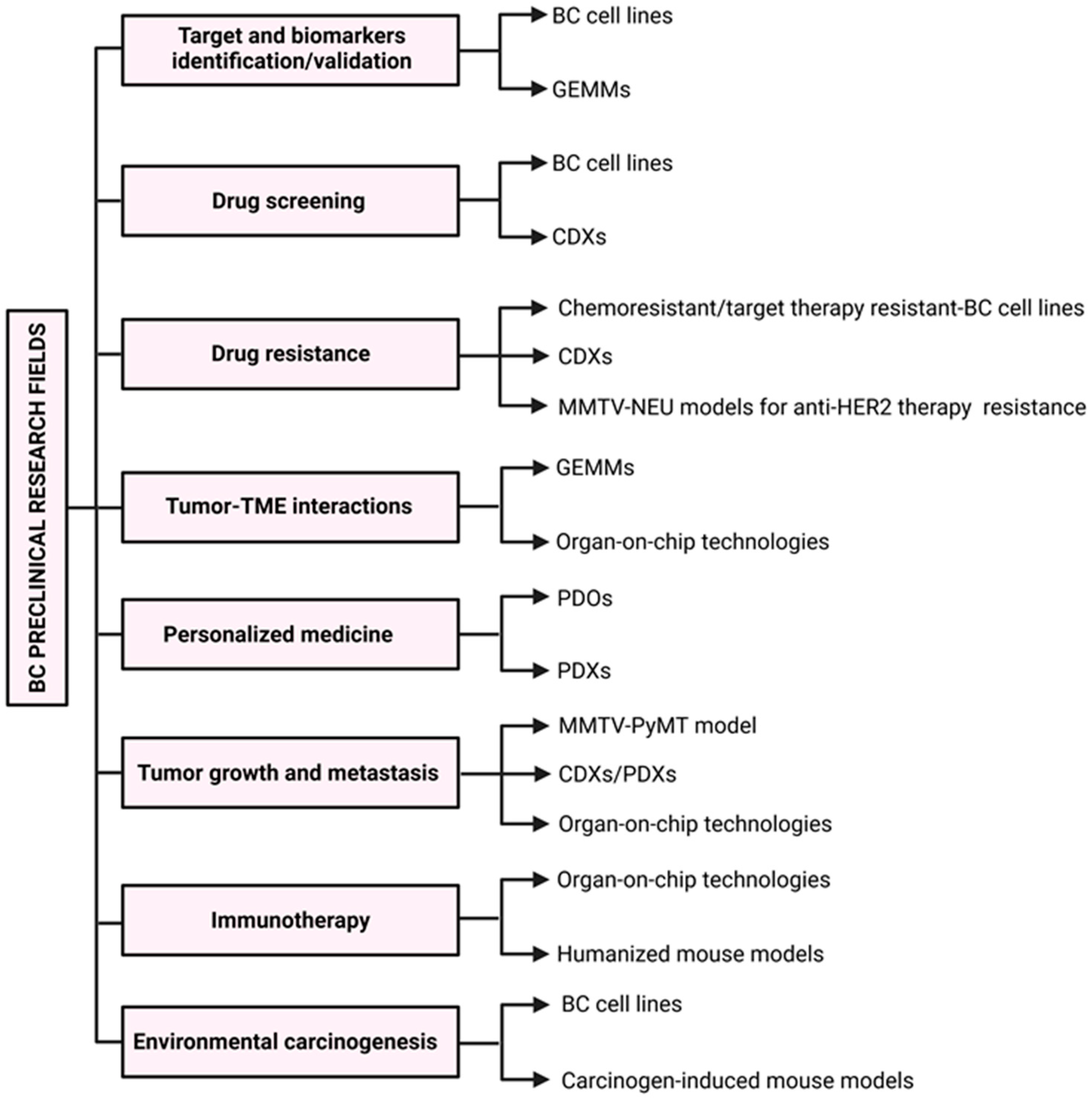
2. In Vitro Models
2.1. BC Cell Lines
2.1.1. Luminal BC Cell Lines
2.1.2. HER2-Positive BC Cell Lines
2.1.3. Basal BC Cell Lines
2.1.4. Characteristics, Strengths, and Weaknesses of Widely Used BC Cell Lines
2.2. Patient-Derived Organoids
2.3. Organ-on-Chip Technologies
3. Murine Models of BC
3.1. Carcinogen-Induced Mouse Models
3.2. GEMMs
3.2.1. MMTV-PyMT
3.2.2. MMTV-NEU
3.2.3. MMTV-WNT1
3.2.4. MMTV-TGFα
3.2.5. Knockout Models
3.2.6. Current Challenges and Future Perspectives
3.3. Xenograft Models
3.3.1. CDX Models
3.3.2. PDX Models
3.3.3. Humanized Mouse Models
4. Ethical and Financial Issues in Choosing Animal Models
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BC | Breast Cancer |
| ER | Estrogen Receptor |
| PR | Progesterone Receptor |
| HER2 | Human Epidermal Growth Factor 2 |
| ASCO-CAP | American Society of Clinical Oncology—College of American Pathologists |
| TNBC | Triple-Negative Breast Cancer |
| TME | Tumor Microenvironment |
| scRNA-seq | Single-cell RNA sequencing |
| PAM50 | Prediction Analysis for Microarrays |
| DepMap | Cancer Dependency Map |
| CCLE | Cancer Cell Line Encyclopedia |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| shRNA | short-hairpin RNA |
| PARP | Poly (ADP-ribose) polymerase |
| IC50 | Half-maximal Inhibitory Concentration |
| SNV | Single Nucleotide Variant |
| WT | Wild-Type |
| GDSC | Genomics of Drug Sensitivity in Cancer |
| CNVs | Copy Number Variations |
| ECM | Extracellular Matrix |
| PDOs | Patient-Derived Organoids |
| GPNMB | glycoprotein nonmetastatic melanoma protein B |
| (CAR)-T | chimeric antigen receptor T |
| CDXs | Cell-line-derived xenografts |
| PDXs | patient-derived xenografts |
| NOD | Non-Obese Diabetic |
| NSG | NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ |
| NOG | NOD.Cg-PrkdcscidIl2rgtm1Sug/Jic |
| PDX-MI | Patient-Derived Xenograft Minimal Information |
| GEMMs | Genetically Engineered Mouse Models |
| MMTV-LTR | Mammary Tumor Virus Long Terminal Repeat |
| WAP | Whey Acidic Protein |
| BLG | Bovine β-Lactoglobulin |
| PyMT | Polyomavirus Middle T Antigen |
| MAPK | Mitogen-Activated Protein Kinase |
| PI3K | Phosphatidylinositol-4,5-bisphosphate 3-kinase |
| Shc | Src homology and collagen |
| Grb2 | Growth factor receptor-bound protein 2 |
| PIP3 | Phosphatidylinositol (3,4,5)-trisphosphate |
| PDK1 | 3-phosphoinositide-dependent kinase 1 |
| mTOR | Mammalian Target of Rapamycin |
| TICs | tumor-initiating cells |
| TGFα | Transforming growth factor α |
| EGF | Epidermal Growth Factor |
| EGFR | Epidermal Growth Factor Receptor |
| KO | Knockout |
| loxP | Locus of X-over, P1 |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| Cas9 | CRISPR-associated protein 9 |
| DSBs | Double-Stranded DNA Breaks |
| CAFs | Cancer-Associated Fibroblasts |
| SEARCHBreast | Sharing Experimental Animal Resources: Coordinating Holdings—Breast |
| Hu-PBL | Humanized Peripheral Blood Lymphocytes |
| BLT | Bone marrow–Liver–Thymus |
| GvHD | graft-versus-host disease |
| MISHUM | Minimal information for standardization of humanized mouse models |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, P.; Disalvatore, D.; Rotmensz, N.; Curigliano, G.; Colleoni, M.; Dellapasqua, S.; Pruneri, G.; Mastropasqua, M.G.; Luini, A.; Bassi, F.; et al. Proposed new clinicopathological surrogate definitions of luminal A and luminal B (HER2-negative) intrinsic breast cancer subtypes. Breast Cancer Res. 2014, 16, R65. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Pineda, E.; Adamo, B.; Galván, P.; Fernández, A.; Gaba, L.; Díez, M.; Viladot, M.; Arance, A.; Muñoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24 (Suppl. 2), S26–S35. [Google Scholar] [CrossRef]
- Wolff, A.C.; Hammond, M.E.H.; Allison, K.H.; Harvey, B.E.; Mangu, P.B.; Bartlett, J.M.S.; Bilous, M.; Ellis, I.O.; Fitzgibbons, P.; Hanna, W.; et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline focused update. J. Clin. Oncol. 2018, 36, 2105–2122. [Google Scholar] [CrossRef]
- Ferrando-Díez, A.; Felip, E.; Pous, A.; Bergamino Sirven, M.; Margelí, M. Targeted therapeutic options and future perspectives for HER2-positive breast cancer. Cancers 2022, 14, 3305. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Carey, L.A.; Dees, E.C.; Sawyer, L.; Gatti, L.; Moore, D.T.; Collichio, F.; Ollila, D.W.; Sartor, C.I.; Graham, M.L.; Perou, C.M. The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef] [PubMed]
- Lv, K.; He, T. Cancer-associated fibroblasts: Heterogeneity, tumorigenicity and therapeutic targets. Mol. Biomed. 2024, 5, 70. [Google Scholar] [CrossRef]
- Sotiriou, C.; Wirapati, P.; Loi, S.; Harris, A.; Fox, S.; Smeds, J.; Nordgren, H.; Farmer, P.; Praz, V.; Haibe-Kains, B.; et al. Gene expression profiling in breast cancer: Understanding the molecular basis of histologic grade to improve prognosis. J. Natl. Cancer Inst. 2006, 98, 262–272. [Google Scholar] [CrossRef]
- Sørlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.-F.; Rueda, O.M.; Vollan, H.-K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.-J.; et al. The somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [PubMed]
- Lasfargues, E.Y.; Ozzello, L. Cultivation of human breast carcinomas. J. Natl. Cancer Inst. 1958, 21, 1131–1147. [Google Scholar]
- Soule, H.D.; Vazquez, J.; Long, A.; Albert, S.; Brennan, M. A human cell line from a pleural effusion derived from a breast carcinoma. J. Natl. Cancer Inst. 1973, 51, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F.; Kurvari, V.; Virmani, A.; Gollahon, L.; Sakaguchi, M.; Westerfield, M.; Kodagoda, D.; Stasny, V.; Cunningham, H.T.; Wistuba, I.I.; et al. Characterization of paired tumor and non-tumor cell lines established from patients with breast cancer. Int. J. Cancer 1998, 78, 766–774. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Zhang, S.; Yazdanparast, A.; Li, M.; Pawar, A.V.; Liu, Y.; Inavolu, S.M.; Cheng, L. Comprehensive comparison of molecular portraits between cell lines and tumors in breast cancer. BMC Genom. 2016, 17 (Suppl. 7), 525. [Google Scholar] [CrossRef]
- Subik, K.; Lee, J.F.; Baxter, L. The Expression Patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by Immunohistochemical Analysis in Breast Cancer Cell Lines. Breast Cancer Basic Clin. Res. 2010, 4, 117822341000400004, Erratum in Breast Cancer 2010, 12, 35–41. [Google Scholar] [CrossRef]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast cancer cell line classification and its relevance with breast tumor subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Asselin-Labat, M.-L.; Sutherland, K.D.; Barker, H.; Thomas, R.; Shackleton, M.; Forrest, N.C.; Hartley, L.; Robb, L.; Grosveld, F.G.; Van Der Wees, J.; et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat. Cell Biol. 2007, 9, 201–209. [Google Scholar] [CrossRef]
- Charafe-Jauffret, E.; Ginestier, C.; Monville, F.; Finetti, P.; Adélaïde, J.; Cervera, N.; Fekairi, S.; Xerri, L.; Jacquemier, J.; Birnbaum, D.; et al. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 2006, 25, 2273–2284. [Google Scholar] [CrossRef]
- Hollestelle, A.; Nagel, J.H.A.; Smid, M.; Lam, S.; Elstrodt, F.; Wasielewski, M.; Ng, S.S.; French, P.J.; Peeters, J.K.; Rozendaal, M.J.; et al. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res. Treat. 2010, 121, 53–64. [Google Scholar] [CrossRef]
- Ring, A.; Dowsett, M. Mechanisms of tamoxifen resistance. Endocr.-Related Cancer 2004, 11, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, E.E.; McDaniel, R.E.; Maximov, P.Y.; Fan, P.; Jordan, V.C. Models and mechanisms of acquired antihormone resistance in breast cancer: Significant clinical progress despite limitations. Horm. Mol. Biol. Clin. Investig. 2012, 9, 143–163. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, Y.; Zhang, C.; Chu, J.; Wu, Y.; Li, Y.; Liu, J.; Li, S.; Shi, Q.; Jin, L.; et al. Tamoxifen-resistant breast cancer cells are resistant to DNA-damaging chemotherapy because of upregulated BARD1 and BRCA1. Nat. Commun. 2018, 9, 1595. [Google Scholar] [CrossRef]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.-P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Miles, D.; Kim, S.-B.; Im, Y.-H.; Im, S.-A.; Semiglazov, V.; Ciruelos, E.; Schneeweiss, A.; Loi, S.; Monturus, E.; et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): End-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Álvarez, M.; Luque, M.; Morales-Gallego, M.; Cristóbal, I.; Ramírez-Merino, N.; Rangel, Y.; Izarzugaza, Y.; Eroles, P.; Albanell, J.; Madoz-Gúrpide, J.; et al. Generation and characterization of trastuzumab/pertuzumab-resistant HER2-positive breast cancer cell lines. Int. J. Mol. Sci. 2023, 25, 207. [Google Scholar] [CrossRef]
- Wissner, A.; Mansour, T.S. The development of HKI-272 and related compounds for the treatment of cancer. Arch. Pharm. 2008, 341, 465–477. [Google Scholar] [CrossRef]
- Canonici, A.; Gijsen, M.; Mullooly, M.; Bennett, R.; Bouguern, N.; Pedersen, K.; O’brien, N.A.; Roxanis, I.; Li, J.-L.; Bridge, E.; et al. Neratinib overcomes trastuzumab resistance in HER2 amplified breast cancer. Oncotarget 2013, 4, 1592–1605. [Google Scholar] [CrossRef]
- Arshad, M.; Azad, A.; Chan, P.Y.K.; Vigneswara, V.; Feldinger, K.; Nafi, S.N.M.; Laporte-Maguire, E.; De Santo, C.; Zuo, J.; Shaaban, A.M.; et al. Neratinib could be effective as monotherapy or in combination with trastuzumab in HER2-low breast cancer cells and organoid models. Br. J. Cancer 2024, 130, 1990–2002. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Easton, D.F.; Pharoah, P.D.P.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-panel sequencing and the prediction of breast-cancer risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- DepMap: The Cancer Dependency Map Project at Broad Institute. Available online: https://depmap.org/portal/ (accessed on 1 October 2024).
- Genomics of Drug Sensitivity in Cancer. Available online: https://www.cancerrxgene.org/ (accessed on 1 October 2024).
- Gambardella, G.; Viscido, G.; Tumaini, B.; Isacchi, A.; Bosotti, R.; di Bernardo, D. A single-cell analysis of breast cancer cell lines to study tumour heterogeneity and drug response. Nat. Commun. 2022, 13, 1714. [Google Scholar] [CrossRef]
- Heiser, L.M.; Sadanandam, A.; Kuo, W.-L.; Benz, S.C.; Goldstein, T.C.; Ng, S.; Gibb, W.J.; Wang, N.J.; Ziyad, S.; Tong, F.; et al. Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2724–2729. [Google Scholar] [CrossRef] [PubMed]
- Forozan, F.; Mahlamäki, E.H.; Monni, O.; Chen, Y.; Veldman, R.; Jiang, Y.; Gooden, G.C.; Ethier, S.P.; Kallioniemi, A.; Kallioniemi, O.P. Comparative genomic hybridization analysis of 38 breast cancer cell lines: A basis for interpreting complementary DNA microarray data. Cancer Res. 2000, 60, 4519–4525. [Google Scholar] [PubMed]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Dekkers, J.F.; van Vliet, E.J.; Sachs, N.; Rosenbluth, J.M.; Kopper, O.; Rebel, H.G.; Wehrens, E.J.; Piani, C.; Visvader, J.E.; Verissimo, C.S.; et al. Long-term culture, genetic manipulation and xenotransplantation of human normal and breast cancer organoids. Nat. Protoc. 2021, 16, 1936–1965. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e10. [Google Scholar] [CrossRef]
- Chen, P.; Zhang, X.; Ding, R.; Yang, L.; Lyu, X.; Zeng, J.; Lei, J.H.; Wang, L.; Bi, J.; Shao, N.; et al. Patient-Derived Organoids Can Guide Personalized-Therapies for Patients with Advanced Breast Cancer. Adv. Sci. 2021, 8, 2101176. [Google Scholar] [CrossRef]
- Saeki, S.; Kumegawa, K.; Takahashi, Y.; Yang, L.; Osako, T.; Yasen, M.; Otsuji, K.; Miyata, K.; Yamakawa, K.; Suzuka, J.; et al. Transcriptomic intratumor heterogeneity of breast cancer patient-derived organoids may reflect the unique biological features of the tumor of origin. Breast Cancer Res. 2023, 25, 21. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.-H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid modeling of the tumor immune microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [PubMed]
- Zumwalde, N.A.; Haag, J.D.; Sharma, D.; Mirrielees, J.A.; Wilke, L.G.; Gould, M.N.; Gumperz, J.E. Analysis of Immune Cells from Human Mammary Ductal Epithelial Organoids Reveals Vδ2+ T Cells That Efficiently Target Breast Carcinoma Cells in the Presence of Bisphosphonate. Cancer Prev. Res. 2016, 9, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Huh, D.; Kim, H.J.; Fraser, J.P.; Shea, D.E.; Khan, M.; Bahinski, A.; Hamilton, G.A.; Ingber, D.E. Microfabrication of human organs-on-chips. Nat. Protoc. 2013, 8, 2135–2157. [Google Scholar] [CrossRef]
- Ingber, D.E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 2022, 23, 467–491. [Google Scholar] [CrossRef]
- Han, S.J.; Kwon, S.; Kim, K.S. Challenges of applying multicellular tumor spheroids in preclinical phase. Cancer Cell Int. 2021, 21, 152. [Google Scholar] [CrossRef]
- Cauli, E.; Polidoro, M.A.; Marzorati, S.; Bernardi, C.; Rasponi, M.; Lleo, A. Cancer-on-chip: A 3D model for the study of the tumor microenvironment. J. Biol. Eng. 2023, 17, 53. [Google Scholar] [CrossRef]
- Truong, D.D.; Kratz, A.; Park, J.G.; Barrientos, E.S.; Saini, H.; Nguyen, T.; Pockaj, B.; Mouneimne, G.; LaBaer, J.; Nikkhah, M. A Human Organotypic Microfluidic Tumor Model Permits Investigation of the Interplay between Patient-Derived Fibroblasts and Breast Cancer Cells. Cancer Res. 2019, 79, 3139–3151. [Google Scholar] [CrossRef]
- Hao, S.; Ha, L.; Cheng, G.; Wan, Y.; Xia, Y.; Sosnoski, D.M.; Mastro, A.M.; Zheng, S. A Spontaneous 3D Bone-on-a-Chip for Bone Metastasis Study of Breast Cancer Cells. Small 2018, 14, e1702787. [Google Scholar] [CrossRef] [PubMed]
- Zarin, B.; Rafiee, L.; Abdollahi, S.; Vatani, M.; Hassani, M.; Sanati-Nezhad, A.; Javanmard, S.H. Studying breast cancer lung metastasis using a multi-compartment microfluidic device with a mimetic tumor-stroma interaction model. Transl. Oncol. 2025, 53, 102303. [Google Scholar] [CrossRef] [PubMed]
- Low, L.A.; Mummery, C.; Berridge, B.R.; Austin, C.P.; Tagle, D.A. Organs-on-chips: Into the next decade. Nat. Rev. Drug Discov. 2021, 20, 345–361. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, D.; Wang, Y.; Lin, S.; Jiang, Y. A novel 3D breast-cancer-on-chip platform for therapeutic evaluation of drug delivery systems. Anal. Chim. Acta 2018, 1036, 97–106. [Google Scholar] [CrossRef]
- Maulana, T.I.; Teufel, C.; Cipriano, M.; Roosz, J.; Lazarevski, L.; Hil, F.E.v.D.; Scheller, L.; Orlova, V.; Koch, A.; Hudecek, M.; et al. Breast cancer-on-chip for patient-specific efficacy and safety testing of CAR-T cells. Cell Stem Cell 2024, 31, 989–1002.e9. [Google Scholar] [CrossRef] [PubMed]
- Breschi, A.; Gingeras, T.R.; Guigó, R. Comparative transcriptomics in human and mouse. Nat. Rev. Genet. 2017, 18, 425–440. [Google Scholar] [CrossRef]
- National Human Genome Research Institute. Available online: https://www.genome.gov/genetics-glossary (accessed on 1 January 2025).
- Liu, Y.; Yin, T.; Feng, Y.; Cona, M.M.; Huang, G.; Liu, J.; Song, S.; Jiang, Y.; Xia, Q.; Swinnen, J.V.; et al. Mammalian models of chemically induced primary malignancies exploitable for imaging-based preclinical theragnostic research. Quant. Imaging Med. Surg. 2015, 5, 708–729. [Google Scholar] [CrossRef]
- IARC Monographs on the Identification of Carcinogenic Hazards to Humans. Available online: https://monographs.iarc.who.int (accessed on 1 January 2025).
- Rehm, S. Chemically induced mammary gland adenomyoepitheliomas and myoepithelial carcinomas of mice. Immunohistochemical and ultrastructural features. Am. J. Pathol. 1990, 136, 575–584. [Google Scholar]
- Budán, F.; Varjas, T.; Nowrasteh, G.; Prantner, I.; Varga, Z.; Ember, A.; Cseh, J.; Gombos, K.; Pázsit, E.; Gobel, G.; et al. Early modification of c-myc, Ha-ras and p53 expressions by chemical carcinogens (DMBA, MNU). In Vivo 2009, 23, 591–598. [Google Scholar]
- Abba, M.C.; Zhong, Y.; Lee, J.; Kil, H.; Lu, Y.; Takata, Y.; Simper, M.S.; Gaddis, S.; Shen, J.; Aldaz, C.M. DMBA induced mouse mammary tumors display high incidence of activating Pik3caH1047 and loss of function Pten mutations. Oncotarget 2016, 7, 64289–64299. [Google Scholar] [CrossRef]
- Plante, I. Dimethylbenz(a)anthracene-induced mammary tumorigenesis in mice. Methods Cell Biol. 2021, 163, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Wagner, E.F.; Palmiter, R.D. The origins of oncomice: A history of the first transgenic mice genetically engineered to develop cancer. Genes Dev. 2007, 21, 2258–2270. [Google Scholar] [CrossRef] [PubMed]
- Menezes, M.E.; Das, S.K.; Emdad, L.; Windle, J.J.; Wang, X.-Y.; Sarkar, D.; Fisher, P.B. Genetically engineered mice as experimental tools to dissect the critical events in breast cancer. Adv. Cancer Res. 2014, 121, 331–382. [Google Scholar] [CrossRef]
- Wagner, K.U.; Wall, R.J.; St-Onge, L.; Gruss, P.; Wynshaw-Boris, A.; Garrett, L.; Li, M.; Furth, P.A.; Hennighausen, L. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 1997, 25, 4323–4330. [Google Scholar] [CrossRef]
- Graff, S.; Moore, D.H. Isolation of mouse mammary carcinoma virus. Cancer 1949, 2, 755–762. [Google Scholar] [CrossRef]
- Callahan, R.; Smith, G.H. MMTV-induced mammary tumorigenesis: Gene discovery, progression to malignancy and cellular pathways. Oncogene 2000, 19, 992–1001. [Google Scholar] [CrossRef]
- Guy, C.T.; Cardiff, R.D.; Muller, W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: A transgenic mouse model for metastatic disease. Mol. Cell. Biol. 1992, 12, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Davie, S.A.; Maglione, J.E.; Manner, C.K.; Young, D.; Cardiff, R.D.; MacLeod, C.L.; Ellies, L.G. Effects of FVB/NJ and C57Bl/6J strain backgrounds on mammary tumor phenotype in inducible nitric oxide synthase deficient mice. Transgenic Res. 2007, 16, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.J.; Sinn, E.; Pattengale, P.K.; Wallace, R.; Leder, P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 1988, 54, 105–115. [Google Scholar] [CrossRef]
- Tsukamoto, A.S.; Grosschedl, R.; Guzman, R.C.; Parslow, T.; Varmus, H.E. Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell 1988, 55, 619–625. [Google Scholar] [CrossRef]
- Halter, S.A.; Dempsey, P.; Matsui, Y.; Stokes, M.K.; Graves-Deal, R.; Hogan, B.L.; Coffey, R.J. Distinctive patterns of hyperplasia in transgenic mice with mouse mammary tumor virus transforming growth factor-alpha. Characterization of mammary gland and skin proliferations. Am. J. Pathol. 1992, 140, 1131–1146. [Google Scholar] [PubMed]
- Matsui, Y.; Halter, S.A.; Holt, J.T.; Hogan, B.L.; Coffey, R.J. Development of mammary hyperplasia and neoplasia in MMTV-TGF alpha transgenic mice. Cell 1990, 61, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
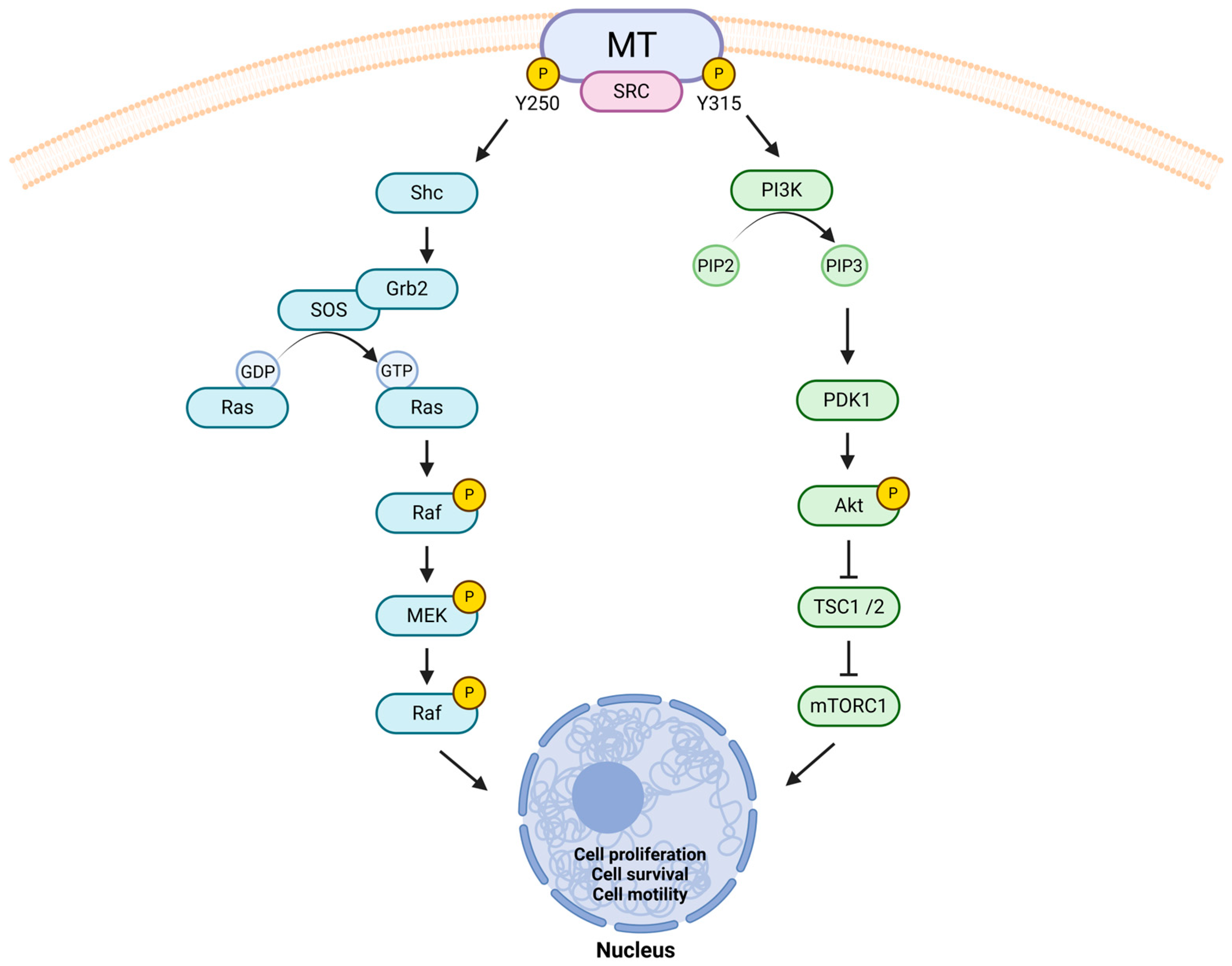
- Dunant, N.M.; Senften, M.; Ballmer-Hofer, K. Polyomavirus middle-T antigen associates with the kinase domain of Src-related tyrosine kinases. J. Virol. 1996, 70, 1323–1330. [Google Scholar] [CrossRef]
- Campbell, K.S.; Ogris, E.; Burke, B.; Su, W.; Auger, K.R.; Druker, B.J.; Schaffhausen, B.S.; Roberts, T.M.; Pallas, D.C. Polyoma middle tumor antigen interacts with SHC protein via the NPTY (Asn-Pro-Thr-Tyr) motif in middle tumor antigen. Proc. Natl. Acad. Sci. USA 1994, 91, 6344–6348. [Google Scholar] [CrossRef]
- Whitman, M.; Kaplan, D.R.; Schaffhausen, B.; Cantley, L.; Roberts, T.M. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature 1985, 315, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Jones, J.G.; Li, P.; Zhu, L.; Whitney, K.D.; Muller, W.J.; Pollard, J.W. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol. 2003, 163, 2113–2126. [Google Scholar] [CrossRef]
- Qiu, T.H.; Chandramouli, G.V.R.; Hunter, K.W.; Alkharouf, N.W.; Green, J.E.; Liu, E.T. Global expression profiling identifies signatures of tumor virulence in MMTV-PyMT-transgenic mice: Correlation to human disease. Cancer Res. 2004, 64, 5973–5981. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Ross, K.N.; Lander, E.S.; Golub, T.R. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 2003, 33, 49–54. [Google Scholar] [CrossRef]
- Attalla, S.; Taifour, T.; Bui, T.; Muller, W. Insights from transgenic mouse models of PyMT-induced breast cancer: Recapitulating human breast cancer progression in vivo. Oncogene 2021, 40, 475–491. [Google Scholar] [CrossRef]
- Müller, V.; Bartsch, R.; Lin, N.U.; Montemurro, F.; Pegram, M.D.; Tolaney, S.M. Epidemiology, clinical outcomes, and unmet needs of patients with human epidermal growth factor receptor 2-positive breast cancer and brain metastases: A systematic literature review. Cancer Treat. Rev. 2023, 115, 102527. [Google Scholar] [CrossRef]
- Bouchard, L.; Lamarre, L.; Tremblay, P.J.; Jolicoeur, P. Stochastic appearance of mammary tumors in transgenic mice carrying the MMTV/c-neu oncogene. Cell 1989, 57, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Ursini-Siegel, J.; Schade, B.; Cardiff, R.D.; Muller, W.J. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat. Rev. Cancer 2007, 7, 389–397. [Google Scholar] [CrossRef]
- Moody, S.E.; Sarkisian, C.J.; Hahn, K.T.; Gunther, E.J.; Pickup, S.; Dugan, K.D.; Innocent, N.; Cardiff, R.D.; Schnall, M.D.; Chodosh, L.A. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell 2002, 2, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Paulson, A.; Iovino, F.; Wicha, M.S. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 2008, 27, 6120–6130. [Google Scholar] [CrossRef]
- Izrailit, J.; Reedijk, M. Developmental pathways in breast cancer and breast tumor-initiating cells: Therapeutic implications. Cancer Lett. 2012, 317, 115–126. [Google Scholar] [CrossRef]
- Liu, J.C.; Deng, T.; Lehal, R.S.; Kim, J.; Zacksenhaus, E. Identification of tumorsphere- and tumor-initiating cells in HER2/Neu-induced mammary tumors. Cancer Res. 2007, 67, 8671–8681. [Google Scholar] [CrossRef]
- Lo, P.-K.; Kanojia, D.; Liu, X.; Singh, U.P.; Berger, F.G.; Wang, Q.; Chen, H. CD49f and CD61 identify Her2/neu-induced mammary tumor-initiating cells that are potentially derived from luminal progenitors and maintained by the integrin–TGFβ signaling. Oncogene 2012, 31, 2614–2626. [Google Scholar] [CrossRef] [PubMed]
- Usary, J.; Zhao, W.; Darr, D.; Roberts, P.J.; Liu, M.; Balletta, L.; Karginova, O.; Jordan, J.; Combest, A.; Bridges, A.; et al. Predicting drug responsiveness in human cancers using genetically engineered mice. Clin. Cancer Res. 2013, 19, 4889–4899. [Google Scholar] [CrossRef]
- Yang, Y.; Leonard, M.; Luo, Z.; Yeo, S.; Bick, G.; Hao, M.; Cai, C.; Charif, M.; Wang, J.; Guan, J.-L.; et al. Functional cooperation between co-amplified genes promotes aggressive phenotypes of HER2-positive breast cancer. Cell Rep. 2021, 34, 108822. [Google Scholar] [CrossRef]
- Liu, Q.; Borcherding, N.C.; Shao, P.; Maina, P.K.; Zhang, W.; Qi, H.H. Contribution of synergism between PHF8 and HER2 signalling to breast cancer development and drug resistance. EBioMedicine 2020, 51, 102612. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, A.D.; Herschkowitz, J.I.; Usary, J.; Harrell, J.C.; Spike, B.T.; Adams, J.R.; Torres-Arzayus, M.I.; Brown, M.; Egan, S.E.; Wahl, G.M.; et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol. 2013, 14, R125. [Google Scholar] [CrossRef]
- Pfefferle, A.D.; Darr, D.B.; Calhoun, B.C.; Mott, K.R.; Rosen, J.M.; Perou, C.M. The MMTV-Wnt1 murine model produces two phenotypically distinct subtypes of mammary tumors with unique therapeutic responses to an EGFR inhibitor. Dis. Model. Mech. 2019, 12, dmm037192. [Google Scholar] [CrossRef]
- Rashid, N.S.; Boyd, D.C.; Olex, A.L.; Grible, J.M.; Duong, A.K.; Alzubi, M.A.; Altman, J.E.; Leftwich, T.J.; Valentine, A.D.; Hairr, N.S.; et al. Transcriptomic changes underlying EGFR inhibitor resistance in human and mouse models of basal-like breast cancer. Sci. Rep. 2022, 12, 21248. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R. Transforming growth factor-alpha. Mol. Reprod. Dev. 1990, 27, 3–9. [Google Scholar] [CrossRef]
- Dogan, S.; Hu, X.; Zhang, Y.; Maihle, N.J.; Grande, J.P.; Cleary, M.P. Effects of high-fat diet and/or body weight on mammary tumor leptin and apoptosis signaling pathways in MMTV-TGF-alpha mice. Breast Cancer Res. 2007, 9, R91. [Google Scholar] [CrossRef] [PubMed]
- Cicekdal, M.B.; Tuna, B.G.; Charehsaz, M.; Cleary, M.P.; Aydin, A.; Dogan, S. Effects of long-term intermittent versus chronic calorie restriction on oxidative stress in a mouse cancer model. IUBMB Life 2019, 71, 1973–1985. [Google Scholar] [CrossRef]
- Lobe, C.G.; Nagy, A. Conditional genome alteration in mice. Bioessays 1998, 20, 200–208. [Google Scholar] [CrossRef]
- Kim, H.; Kim, M.; Im, S.-K.; Fang, S. Mouse Cre-LoxP system: General principles to determine tissue-specific roles of target genes. Lab. Anim. Res. 2018, 34, 147–159. [Google Scholar] [CrossRef]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Lin, S.-C.J.; Lee, K.-F.; Nikitin, A.Y.; Hilsenbeck, S.G.; Cardiff, R.D.; Li, A.; Kang, K.-W.; Frank, S.A.; Lee, W.-H.; Lee, E.Y.-H.P. Somatic mutation of p53 leads to estrogen receptor alpha-positive and -negative mouse mammary tumors with high frequency of metastasis. Cancer Res. 2004, 64, 3525–3532. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.D.; Mo, Q.; Xu, Y.; Qin, L.; Li, Y.; Xu, J. Common genomic aberrations in mouse and human breast cancers with concurrent P53 deficiency and activated PTEN-PI3K-AKT pathway. Int. J. Biol. Sci. 2022, 18, 229–241. [Google Scholar] [CrossRef]
- Wang, S.; Liu, J.C.; Kim, D.; Datti, A.; Zacksenhaus, E. Targeted Pten deletion plus p53-R270H mutation in mouse mammary epithelium induces aggressive claudin-low and basal-like breast cancer. Breast Cancer Res. 2016, 18, 9. [Google Scholar] [CrossRef]
- Hakem, R.; de la Pompa, J.L.; Sirard, C.; Mo, R.; Woo, M.; Hakem, A.; Wakeham, A.; Potter, J.; Reitmair, A.; Billia, F.; et al. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell 1996, 85, 1009–1023. [Google Scholar] [CrossRef]
- Xu, X.; Wagner, K.-U.; Larson, D.; Weaver, Z.; Li, C.; Ried, T.; Hennighausen, L.; Wynshaw-Boris, A.; Deng, C.-X. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat. Genet. 1999, 22, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Hollern, D.P.; Contreras, C.M.; Dance-Barnes, S.; Silva, G.O.; Pfefferle, A.D.; Xiong, J.; Darr, D.B.; Usary, J.; Mott, K.R.; Perou, C.M. A mouse model featuring tissue-specific deletion of p53 and Brca1 gives rise to mammary tumors with genomic and transcriptomic similarities to human basal-like breast cancer. Breast Cancer Res. Treat. 2019, 174, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, T.; Fisher, P.; Murty, V.; Efstratiadis, A. Development of mammary adenocarcinomas by tissue-specific knockout of Brca2 in mice. Oncogene 2001, 20, 3937–3948. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, J.; Meuwissen, R.; van der Gulden, H.; Peterse, H.; van der Valk, M.; Berns, A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 2001, 29, 418–425. [Google Scholar] [CrossRef]
- van’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.M.; Mao, M.; Peterse, H.L.; Van Der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef]
- Thompson, D.; Easton, D.F.; Breast Cancer Linkage Consortium. Cancer Incidence in BRCA1 mutation carriers. J. Natl. Cancer Inst. 2002, 94, 1358–1365. [Google Scholar] [CrossRef]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, S.; Kas, S.M.; Nethe, M.; Yücel, H.; Del Bravo, J.; Pritchard, C.; Bin Ali, R.; van Gerwen, B.; Siteur, B.; Drenth, A.P.; et al. Modeling invasive lobular breast carcinoma by CRISPR/Cas9-mediated somatic genome editing of the mammary gland. Genes Dev. 2016, 30, 1470–1480. [Google Scholar] [CrossRef]
- Annunziato, S.; de Ruiter, J.R.; Henneman, L.; Brambillasca, C.S.; Lutz, C.; Vaillant, F.; Ferrante, F.; Drenth, A.P.; van der Burg, E.; Siteur, B.; et al. Comparative oncogenomics identifies combinations of driver genes and drug targets in BRCA1-mutated breast cancer. Nat. Commun. 2019, 10, 397. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ma, X.; Gao, F.; Guo, Y. Off-target effects in CRISPR/Cas9 gene editing. Front. Bioeng. Biotechnol. 2023, 11, 1143157. [Google Scholar] [CrossRef] [PubMed]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.U.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef]
- Lewis, G.D.; Li, G.; Guo, J.; Yu, S.-F.; Fields, C.T.; Lee, G.; Zhang, D.; Dragovich, P.S.; Pillow, T.; Wei, B.; et al. The HER2-directed antibody-drug conjugate DHES0815A in advanced and/or metastatic breast cancer: Preclinical characterization and phase 1 trial results. Nat. Commun. 2024, 15, 466. [Google Scholar] [CrossRef]
- Rennhack, J.P.; To, B.; Swiatnicki, M.; Dulak, C.; Ogrodzinski, M.P.; Zhang, Y.; Li, C.; Bylett, E.; Ross, C.; Szczepanek, K.; et al. Integrated analyses of murine breast cancer models reveal critical parallels with human disease. Nat. Commun. 2019, 10, 3261. [Google Scholar] [CrossRef]
- Jiang, G.; Tu, J.; Zhou, L.; Dong, M.; Fan, J.; Chang, Z.; Zhang, L.; Bian, X.; Liu, S. Single-cell transcriptomics reveal the heterogeneity and dynamic of cancer stem-like cells during breast tumor progression. Cell Death Dis. 2021, 12, 979. [Google Scholar] [CrossRef]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef]
- Ruggeri, B.A.; Camp, F.; Miknyoczki, S. Animal models of disease: Pre-clinical animal models of cancer and their applications and utility in drug discovery. Biochem. Pharmacol. 2014, 87, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Press, D.J.; Miller, M.E.; Liederbach, E.; Yao, K.; Huo, D. De novo metastasis in breast cancer: Occurrence and overall survival stratified by molecular subtype. Clin. Exp. Metastasis 2017, 34, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Leclercq, G. Relevance of breast cancer cell lines as models for breast tumours: An update. Breast Cancer Res. Treat. 2004, 83, 249–289. [Google Scholar] [CrossRef]
- Chavez, K.J.; Garimella, S.V.; Lipkowitz, S. Triple negative breast cancer cell lines: One tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2010, 32, 35–48. [Google Scholar] [CrossRef]
- Liu, K.; Newbury, P.A.; Glicksberg, B.S.; Zeng, W.Z.D.; Paithankar, S.; Andrechek, E.R.; Chen, B. Evaluating cell lines as models for metastatic breast cancer through integrative analysis of genomic data. Nat. Commun. 2019, 10, 2138. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.J.; Gupta, G.P.; Siegel, P.M.; Bos, P.D.; Shu, W.; Giri, D.D.; Viale, A.; Olshen, A.B.; Gerald, W.L.; Massagué, J. Genes that mediate breast cancer metastasis to lung. Nature 2005, 436, 518–524. [Google Scholar] [CrossRef]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
- Bos, P.D.; Zhang, X.H.F.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar] [CrossRef]
- Jin, X.; Demere, Z.; Nair, K.; Ali, A.; Ferraro, G.B.; Natoli, T.; Deik, A.; Petronio, L.; Tang, A.A.; Zhu, C.; et al. A metastasis map of human cancer cell lines. Nature 2020, 588, 331–336. [Google Scholar] [CrossRef]
- Gawrzak, S.; Rinaldi, L.; Gregorio, S.; Arenas, E.J.; Salvador, F.; Urosevic, J.; Figueras-Puig, C.; Rojo, F.; Del Barco Barrantes, I.; Cejalvo, J.M.; et al. MSK1 regulates luminal cell differentiation and metastatic dormancy in ER+ breast cancer. Nat. Cell Biol. 2018, 20, 211–221, Erratum in Nat. Cell Biol. 2018, 20, 990. [Google Scholar] [CrossRef]
- Caceres, S.; Alonso-Diez, A.; Crespo, B.; Peña, L.; Illera, M.J.; Silvan, G.; de Andres, P.J.; Illera, J.C. Tumor growth progression in ectopic and orthotopic xenografts from inflammatory breast cancer cell lines. Vet. Sci. 2021, 8, 194. [Google Scholar] [CrossRef]
- Gengenbacher, N.; Singhal, M.; Augustin, H.G. Preclinical mouse solid tumour models: Status quo, challenges and perspectives. Nat. Rev. Cancer 2017, 17, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Vaeteewoottacharn, K.; Kariya, R. Application of highly immunocompromised mice for the establishment of patient-derived xenograft (PDX) models. Cells 2019, 8, 889. [Google Scholar] [CrossRef]
- PDX Portal. Available online: https://www.pdxnetwork.org/ (accessed on 1 October 2024).
- Europdx. Available online: https://europdx.eu/ (accessed on 1 October 2024).
- Meehan, T.F.; Conte, N.; Goldstein, T.; Inghirami, G.; Murakami, M.A.; Brabetz, S.; Gu, Z.; Wiser, J.A.; Dunn, P.; Begley, D.A.; et al. PDX-MI: Minimal information for patient-derived tumor xenograft models. Cancer Res. 2017, 77, e62–e66. [Google Scholar] [CrossRef] [PubMed]
- DeRose, Y.S.; Wang, G.; Lin, Y.-C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.W.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Dobrolecki, L.E.; Airhart, S.D.; Alferez, D.G.; Aparicio, S.; Behbod, F.; Bentires-Alj, M.; Brisken, C.; Bult, C.J.; Cai, S.; Clarke, R.B.; et al. Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Rev. 2016, 35, 547–573. [Google Scholar] [CrossRef]
- Guillen, K.P.; Fujita, M.; Butterfield, A.J.; Scherer, S.D.; Bailey, M.H.; Chu, Z.; DeRose, Y.S.; Zhao, L.; Cortes-Sanchez, E.; Yang, C.-H.; et al. A human breast cancer-derived xenograft and organoid platform for drug discovery and precision oncology. Nat. Cancer 2022, 3, 232–250. [Google Scholar] [CrossRef]
- Kabraji, S.; Ni, J.; Sammons, S.; Li, T.; Van Swearingen, A.E.D.; Wang, Y.; Pereslete, A.; Hsu, L.; DiPiro, P.J.; Lascola, C.; et al. Preclinical and clinical efficacy of trastuzumab deruxtecan in breast cancer brain metastases. Clin. Cancer Res. 2023, 29, 174–182. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Serrano-Carbajal, E.A.; Espinal-Enríquez, J.; Hernández-Lemus, E. Targeting metabolic deregulation landscapes in breast cancer subtypes. Front. Oncol. 2020, 10, 97. [Google Scholar] [CrossRef]
- Liao, C.; Glodowski, C.R.; Fan, C.; Liu, J.; Mott, K.R.; Kaushik, A.; Vu, H.; Locasale, J.W.; McBrayer, S.K.; DeBerardinis, R.J.; et al. Integrated metabolic profiling and transcriptional analysis reveals therapeutic modalities for targeting rapidly proliferating breast cancers. Cancer Res. 2022, 82, 665–680. [Google Scholar] [CrossRef]
- Breast Cancer Metabolomics and Gene Expression Explorer. Available online: http://brcametab.org/ (accessed on 1 October 2024).
- Zhang, X.; Claerhout, S.; Prat, A.; Dobrolecki, L.E.; Petrovic, I.; Lai, Q.; Landis, M.D.; Wiechmann, L.; Schiff, R.; Giuliano, M.; et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013, 73, 4885–4897. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.-T.; Du, W.-L.; Lan, H.-R.; Liu, Y.-Y.; Mao, C.-S.; Du, J.-L.; Mou, X.-Z. Development of humanized mouse with patient-derived xenografts for cancer immunotherapy studies: A comprehensive review. Cancer Sci. 2021, 112, 2592–2606. [Google Scholar] [CrossRef]
- Rosato, R.R.; Dávila-González, D.; Choi, D.S.; Qian, W.; Chen, W.; Kozielski, A.J.; Wong, H.; Dave, B.; Chang, J.C. Evaluation of anti-PD-1-based therapy against triple-negative breast cancer patient-derived xenograft tumors engrafted in humanized mouse models. Breast Cancer Res. 2018, 20, 108. [Google Scholar] [CrossRef]
- Scherer, S.D.; Riggio, A.I.; Haroun, F.; DeRose, Y.S.; Ekiz, H.A.; Fujita, M.; Toner, J.; Zhao, L.; Li, Z.; Oesterreich, S.; et al. An immune-humanized patient-derived xenograft model of estrogen-independent, hormone receptor positive metastatic breast cancer. Breast Cancer Res. 2021, 23, 100. [Google Scholar] [CrossRef] [PubMed]
- Burlion, A.; Ramos, R.N.; Kc, P.; Sendeyo, K.; Corneau, A.; Ménétrier-Caux, C.; Piaggio, E.; Olive, D.; Caux, C.; Marodon, G. A novel combination of chemotherapy and immunotherapy controls tumor growth in mice with a human immune system. OncoImmunology 2019, 8, e1596005. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Wu, L.; Chen, J.; Na, N.; Lou, G. Neoadjuvant nivolumab plus bevacizumab therapy improves the prognosis of triple-negative breast cancer in humanized mouse models. Breast Cancer 2024, 31, 371–381. [Google Scholar] [CrossRef]
- Greenblatt, M.B.; Vbranac, V.; Tivey, T.; Tsang, K.; Tager, A.M.; Aliprantis, A.O. Graft versus host disease in the bone marrow, liver and thymus humanized mouse model. PLoS ONE 2012, 7, e44664, Erratum in PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Kumari, R.; Feuer, G.; Bourré, L. Humanized Mouse Models for Immuno-oncology Drug Discovery. Curr. Protoc. 2023, 3, e852. [Google Scholar] [CrossRef]
- Huntington, N.D.; Legrand, N.; Alves, N.L.; Jaron, B.; Weijer, K.; Plet, A.; Corcuff, E.; Mortier, E.; Jacques, Y.; Spits, H.; et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J. Exp. Med. 2009, 206, 25–34. [Google Scholar] [CrossRef]
- Billerbeck, E.; Barry, W.T.; Mu, K.; Dorner, M.; Rice, C.M.; Ploss, A. Development of human CD4+FoxP3+ regulatory T cells in human stem cell factor–, granulocyte-macrophage colony-stimulating factor–, and interleukin-3–expressing NOD-SCID IL2Rγnull humanized mice. Blood 2011, 117, 3076–3086. [Google Scholar] [CrossRef]
- Yoshihara, S.; Li, Y.; Xia, J.; Danzl, N.; Sykes, M.; Yang, Y.-G. Posttransplant Hemophagocytic Lymphohistiocytosis Driven by Myeloid Cytokines and Vicious Cycles of T-Cell and Macrophage Activation in Humanized Mice. Front. Immunol. 2019, 10, 186. [Google Scholar] [CrossRef]
- Stripecke, R.; Münz, C.; Schuringa, J.J.; Bissig, K.; Soper, B.; Meeham, T.; Yao, L.; Di Santo, J.P.; Brehm, M.; Rodriguez, E.; et al. Innovations, challenges, and minimal information for standardization of humanized mice. EMBO Mol. Med. 2020, 12, e8662. [Google Scholar] [CrossRef]
- Council of Europe. European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes. 1991. Available online: https://www.coe.int/en/web/conventions/full-list?module=treaty-detail&treatynum=123 (accessed on 10 February 2025).
- Russell, W.M.S.; Burch, R.L. The Principles of Humane Experimental Technique; Universities Federation for Animal Welfare: Wheathampstead, UK, 1959. [Google Scholar]
- Blyth, K.; Carter, P.; Morrissey, B.; Chelala, C.; Jones, L.; Holen, I.; Speirs, V. SEARCHBreast: A new resource to locate and share surplus archival material from breast cancer animal models to help address the 3Rs. Breast Cancer Res Treat 2016, 156, 447–452. [Google Scholar] [CrossRef]
- Harrison, R.K. Phase II and phase III failures: 2013–2015. Nat. Rev. Drug Discov. 2016, 15, 817–818. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Cell line | ER # | PR # | HER2 # | TP53 Status $ | BRCA1 Status $ | BRCA2 Status $ | Additional Oncogenic Mutated Genes $ | Talazoparib IC50 (μM) * |
|---|---|---|---|---|---|---|---|---|
| Luminal cell lines | ||||||||
| BT474 | + | + | + | SNV | WT | SNV | PIK3CA, RIT1, RHOA | 670.8 |
| CAMA-1 | + | +/− | - | SNV | WT | WT | PTEN | 61.2 |
| MCF-7 | + | + | - | WT | WT | WT | PIK3CA | 77.5 |
| MDA-MB-361 | + | +/− | + | SNV | WT | SNV | PIK3CA | 182.2 |
| T-47-D | + | + | - | SNV | WT | WT | PIK3CA | 70.6 |
| ZR-75-1 | + | +/− | - | WT | WT | WT | PTEN | NA |
| ZR-75-30 | + | - | + | WT | WT | SNV | / | 463.1 |
| HER2-positive cell lines | ||||||||
| AU565 | - | - | + | SNV | WT | WT | / | 16.4 |
| HCC1954 | - | - | + | SNV | WT | WT | PIK3CA, GAB1 | 87.3 |
| MDA-MB-453 | - | - | + | WT | WT | WT | PIK3CA, FGFR4 | 186.5 |
| SKBR-3 | - | - | + | SNV | WT | WT | / | NA |
| Basal A cell lines | ||||||||
| BT20 | - | - | - | SNV | WT | WT | PIK3CA | 127.9 |
| HCC1143 | - | - | - | SNV | WT | WT | FGFR2 | 44.6 |
| HCC1806 | - | - | - | insertion | WT | WT | / | NA |
| HCC1937 | - | - | - | SNV | insertion | WT | / | 118.9 |
| HCC70 | - | - | - | SNV | WT | WT | / | 32.6 |
| MDA-MB-436 | - | - | - | insertion | SNV | WT | / | 49.2 |
| MDA-MB-468 | - | - | - | SNV | WT | SNV | / | 27 |
| Basal B cell lines | ||||||||
| BT549 | - | - | - | SNV | WT | WT | PTPRT | 42 |
| CAL-51 | - | - | - | WT | WT | WT | RRAS2, PIK3CA | 0.8 |
| HCC38 | - | - | - | SNV | WT | WT | / | 39.8 |
| HCC1395 | - | - | - | SNV | SNV | SNV | / | 11.9 |
| HS578T | - | - | - | SNV | WT | WT | HRAS | 97.3 |
| MDA-MB-157 | - | - | - | deletion | WT | WT | RAC1 | 122.9 |
| MDA-MB-231 | - | - | - | SNV | WT | WT | KRAS | 35.5 |
| SUM-149-PT | - | - | - | SNV | deletion | WT | / | NA |
| SUM-159-PT | - | - | - | insertion | WT | WT | HRAS, PIK3CA | NA |
| Transgene | Mean Tumor Latency (Days) | Tumor Penetrance | Metastatic | Reference | Citations on PubMed * |
|---|---|---|---|---|---|
| PyMT | 53–92 | 100% | Yes | [71,72] | 165 |
| Neu | 90 | 100% | Yes | [73] | 28 |
| Wnt-1 | 35–406 | 80% | No | [74] | 16 |
| TGFα | 480 | 30–40% | No | [75,76] | 7 |
| GEMMs | Advantages | Limitations |
|---|---|---|
| MMTV-PyMT |
|
|
| MMTV-NEU |
|
|
| Murine Model | Advantages | Limitations |
| Carcinogen-induced mouse models |
|
|
| CDXs |
|
|
| PDXs |
|
|
| Humanized models |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciringione, A.; Rizzi, F. Facing the Challenge to Mimic Breast Cancer Heterogeneity: Established and Emerging Experimental Preclinical Models Integrated with Omics Technologies. Int. J. Mol. Sci. 2025, 26, 4572. https://doi.org/10.3390/ijms26104572
Ciringione A, Rizzi F. Facing the Challenge to Mimic Breast Cancer Heterogeneity: Established and Emerging Experimental Preclinical Models Integrated with Omics Technologies. International Journal of Molecular Sciences. 2025; 26(10):4572. https://doi.org/10.3390/ijms26104572
Chicago/Turabian StyleCiringione, Alessia, and Federica Rizzi. 2025. "Facing the Challenge to Mimic Breast Cancer Heterogeneity: Established and Emerging Experimental Preclinical Models Integrated with Omics Technologies" International Journal of Molecular Sciences 26, no. 10: 4572. https://doi.org/10.3390/ijms26104572
APA StyleCiringione, A., & Rizzi, F. (2025). Facing the Challenge to Mimic Breast Cancer Heterogeneity: Established and Emerging Experimental Preclinical Models Integrated with Omics Technologies. International Journal of Molecular Sciences, 26(10), 4572. https://doi.org/10.3390/ijms26104572