
Enhanced Cytotoxicity and Receptor Modulation by SMA-WIN 55,212-2 Micelles in Glioblastoma Cells

 , , ,
, , ,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Results
2.1. Baseline Receptor Expression
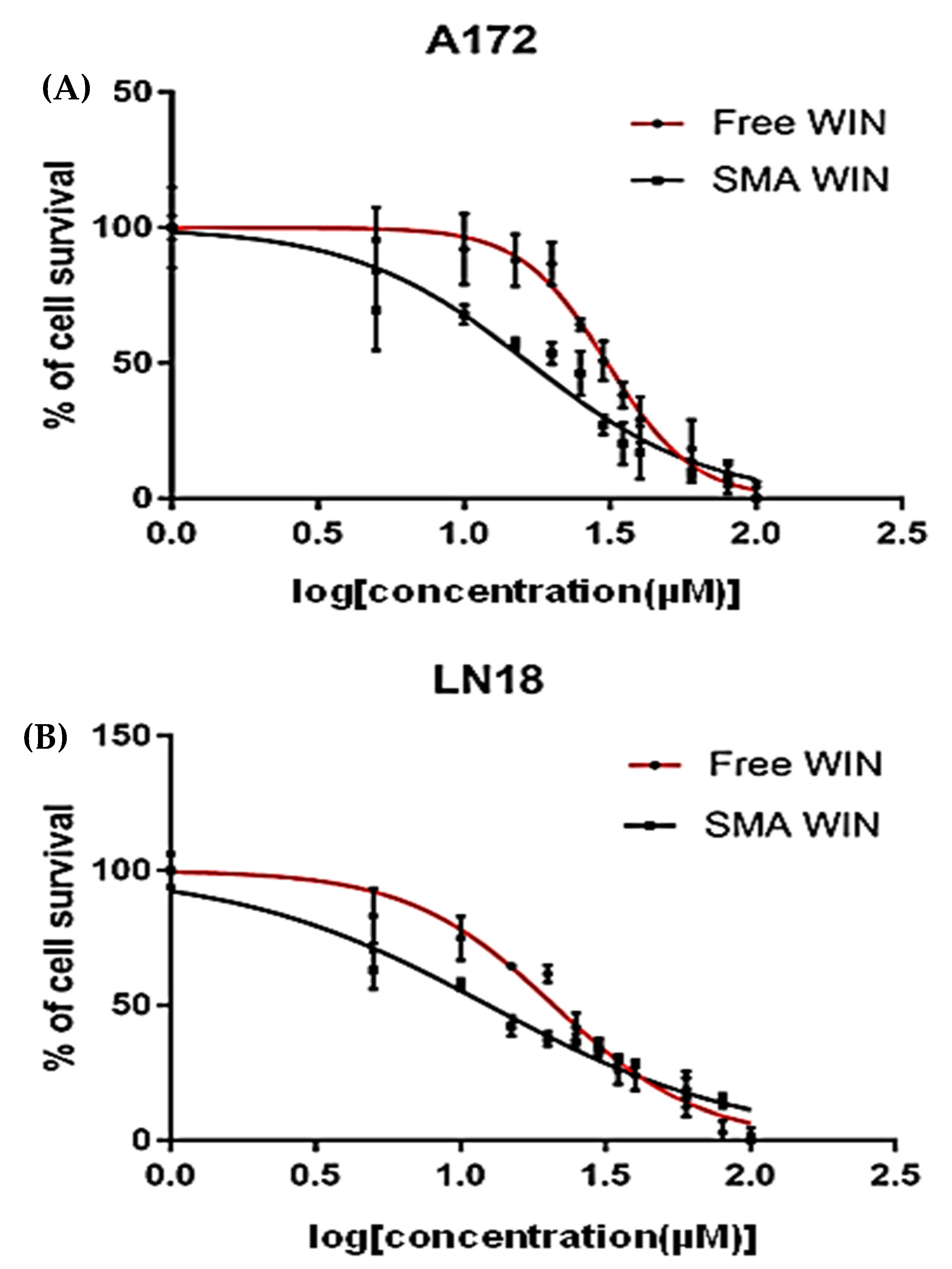
2.2. Cytotoxicity of Free WIN and SMA-WIN
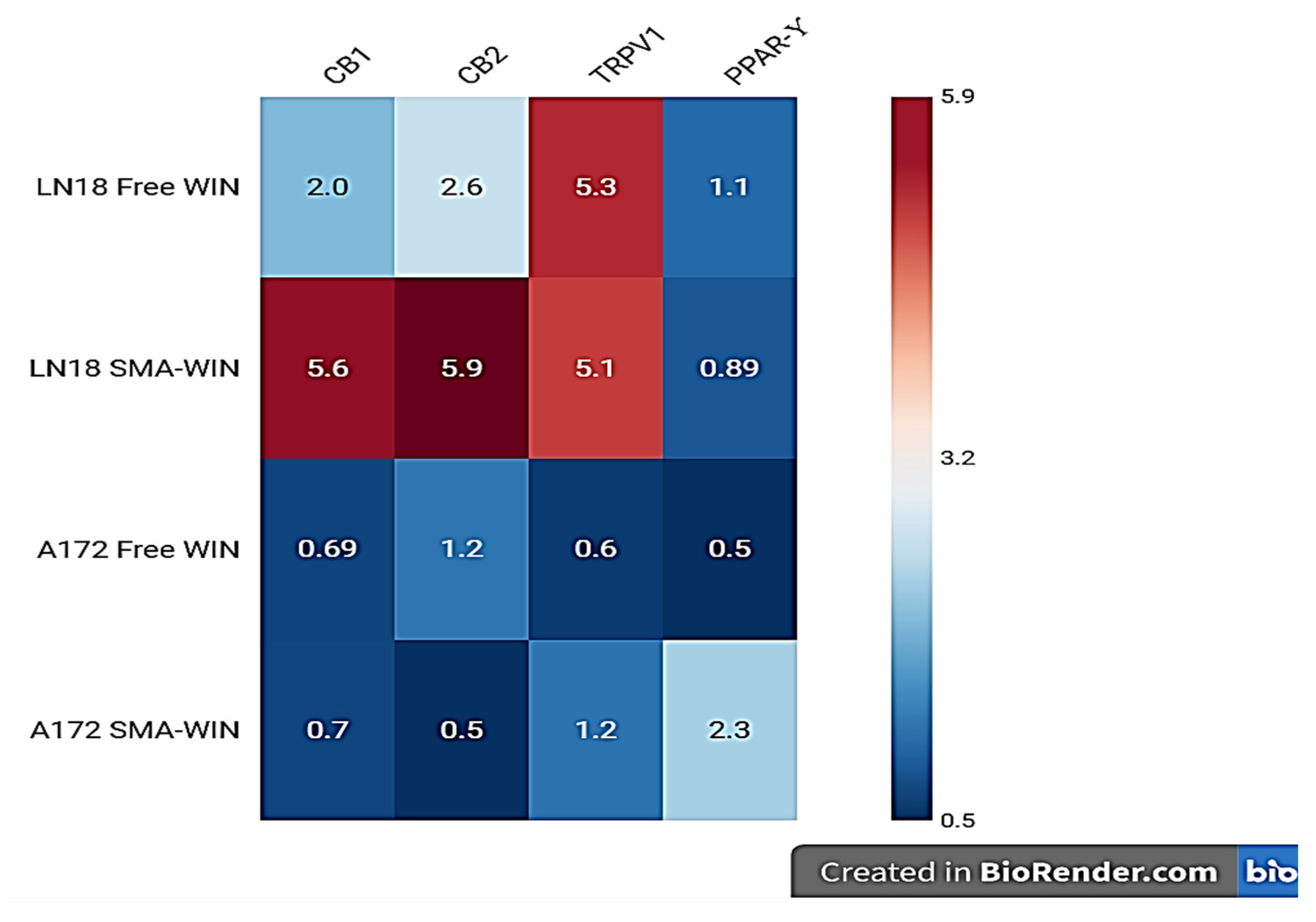
2.3. Receptor Expression Post-Treatment
3. Discussion
- In Vitro to In Vivo Translation: Orthotopic GBM Mouse Models: Validate SMA-WIN’s blood–brain barrier penetration and anti-tumor efficacy, building on these in vitro findings.Intracranial GBM Xenografts: Assess SMA-WIN’s ability to cross the blood–brain barrier (BBB) and induce tumor regression using bioluminescence imaging and survival analysis.
- Receptor Dependency: CB1/CB2 antagonists (rimonabant, AM630) and PPAR-γ inhibitors (e.g., GW9662) to confirm mechanistic pathways.
- Additional Receptors: GPR55 agonists/antagonists to explore its role in SMA-WIN’s effects.
- Molecular Pathways: Western blots for STAT3 phosphorylation, caspase-3 activation, and Bax/Bcl-2 ratios to elucidate apoptosis mechanisms.
- Combination Therapies: Pairing SMA-WIN with TGF-β inhibitors to target EMT in mesenchymal GBM, potentially reversing resistance.
4. Materials and Methods
4.1. Cell Culture
4.2. SMA-WIN Micelle Preparation
4.3. Baseline Receptor Expression Analysis
4.4. Cytotoxicity Assay
4.5. RNA Extraction and RT-PCR Post-Treatment
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tamimi, A.F.; Juweid, M. Epidemiology and outcome of glioblastoma. In Codon Publications eBooks; Codon Publications: Brisbane, Australia, 2017; pp. 143–153. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016–2020. Neuro Oncol. 2023, 25 (Suppl. 4), iv1–iv99. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Manfioletti, G. Proneural-Mesenchymal Transition: Phenotypic Plasticity to Acquire Multitherapy Resistance in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 2746. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Dehouck, M.; Jolliet-Riant, P.; Brée, F.; Fruchart, J.; Cecchelli, R.; Tillement, J. Drug transfer across the Blood-Brain Barrier: Correlation between in vitro and in vivo models. J. Neurochem. 1992, 58, 1790–1797. [Google Scholar] [CrossRef]
- Shi, W.; Cui, X.; Shi, J.; Chen, J.; Wang, Y. Overcoming the blood–brain barrier for glioma-targeted therapy based on an interleukin-6 receptor-mediated micelle system. RSC Adv. 2017, 7, 27162–27169. [Google Scholar] [CrossRef]
- Pertwee, R.G. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol. Ther. 1997, 74, 129–180. [Google Scholar] [CrossRef]
- Console-Bram, L.; Brailoiu, E.; Brailoiu, G.C.; Sharir, H.; Abood, M.E. Activation of GPR18 by cannabinoid compounds: A tale of biased agonism. Br. J. Pharmacol. 2012, 171, 3908–3917. [Google Scholar] [CrossRef]
- Glass, M. Cannabinoid receptor binding and mRNA expression in human brain tissue. Adv. Exp. Med. Biol. 1998, 437, 85–90. [Google Scholar]
- Ellert-Miklaszewska, A.; Ciechomska, I.A.; Kaminska, B. Synthetic cannabinoids induce autophagy and mitochondrial apoptotic pathways in human glioblastoma cells independently of deficiency in TP53 or PTEN tumor suppressors. Cancers 2021, 13, 419. [Google Scholar] [CrossRef] [PubMed]
- Kargbo, R.B. TRPV1 modulators for the treatment of pain and inflammation. ACS Med. Chem. Lett. 2019, 10, 143–144. [Google Scholar] [CrossRef]
- Hua, T.N.M.; Oh, J.; Kim, S.; Antonio, J.M.; Vo, V.T.A.; Om, J.; Choi, J.; Kim, J.; Jung, C.; Park, M.; et al. Peroxisome proliferator-activated receptor gamma as a theragnostic target for mesenchymal-type glioblastoma patients. Exp. Mol. Med. 2020, 52, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, M. Cannabinoids: Potential anticancer agents. Nat. Rev. Cancer. 2003, 3, 745–755. [Google Scholar] [CrossRef]
- Velasco, G.; Hernández-Tiedra, S.; Dávila, D.; Lorente, M. The use of cannabinoids as anticancer agents. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 259–266. [Google Scholar] [CrossRef]
- Grotenhermen, F.; Müller-Vahl, K. The therapeutic potential of cannabis and cannabinoids. Dtsch. Arztebl. Int. 2012, 109, 495–501. [Google Scholar] [CrossRef]
- Galve-Roperh, I.; Sánchez, C.; Cortés, M.L.; Izquierdo, M.; Guzmán, M. Anti-tumoral action of cannabinoids: Involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat. Med. 2000, 6, 313–319. [Google Scholar] [CrossRef]
- Devane, W.A.; Hanuš, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Vučković, S.; Srebro, D.; Vujović, K.S.; Vučetić, Č.; Prostran, M. Cannabinoids and pain: New insights from old molecules. Front. Pharmacol. 2018, 9, 1259. [Google Scholar] [CrossRef]
- Greish, K.; Sawa, T.; Fang, J.; Akaike, T.; Maeda, H. SMA-doxorubicin, a new polymeric micellar drug for effective targeting to solid tumors. J. Control Release 2005, 101, 189–201. [Google Scholar]
- Linsell, O.; Brownjohn, P.W.; Nehoff, H.; Greish, K.; Ashton, J.C. Effect of styrene maleic acid WIN55,212-2 micelles on neuropathic pain in a rat model. J. Drug Target. 2015, 23, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Xian, S.; Parayath, N.N.; Nehoff, H.; Giles, N.M.; Greish, K. The use of styrene maleic acid nanomicelles encapsulating the synthetic cannabinoid analog WIN55,212-2 for the treatment of cancer. Anticancer Res. 2015, 35, 4707–4712. [Google Scholar]
- De Jesús, M.L.; Hostalot, C.; Garibi, J.M.; Sallés, J.; Meana, J.J.; Callado, L.F. Opposite changes in cannabinoid CB1 and CB2 receptor expression in human gliomas. Neurochem. Int. 2010, 56, 829–833. [Google Scholar] [CrossRef]
- Sánchez, C.; de Ceballos, M.L.; del Pulgar, T.G.; Rueda, D.; Corbacho, C.; Velasco, G.; Galve-Roperh, I.; Huffman, J.W.; Ramón y Cajal, S.; Guzmán, M. Inhibition of glioma growth in vivo by selective activation of the CB2 cannabinoid receptor. Cancer Res. 2001, 61, 5784–5789. [Google Scholar]
- Lenman, A.; Fowler, C.J. Interaction of ligands for the peroxisome proliferator-activated receptor γ with the endocannabinoid system. Br. J. Pharmacol. 2007, 151, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Ellert-Miklaszewska, A.; Grajkowska, W.; Gabrusiewicz, K.; Kaminska, B.; Konarska, L. Distinctive pattern of cannabinoid receptor type II (CB2) expression in adult and pediatric brain tumors. Brain Res. 2006, 1137, 161–169. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Grommes, C.; Landreth, G.E.; Sastre, M.; Beck, M.; Feinstein, D.L.; Jacobs, A.H.; Schlegel, U.; Heneka, M.T. Inhibition of in Vivo Glioma Growth and Invasion by Peroxisome Proliferator-Activated Receptor γ Agonist Treatment. Mol. Pharmacol. 2006, 70, 1524–1533. [Google Scholar] [CrossRef]
- Sheu, M.; Pan, L.; Hu, H.; Su, H.; Sheehan, J.; Tsou, H.; Pan, H. Potential therapeutic effects of thiazolidinedione on malignant glioma. Int. J. Mol. Sci. 2022, 23, 13510. [Google Scholar] [CrossRef]
- Andradas, C.; Caffarel, M.M.; Pérez-Gómez, E.; Salazar, M.; Lorente, M.; Velasco, G.; Guzmán, M.; Sánchez, C. The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene 2010, 30, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Damani, M.; Nilawar, N.; Momin, M.; Ningthoujham, R.S.; Khan, T. Nanoparticles assisted drug delivery for effective management of Glioblastoma. Next Nanotechnol. 2025, 7, 100137. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′-3′) | Reverse Primer (5′-3′) | Amplicon Size (bp) |
|---|---|---|---|
| CB1 | AAGACCCTCATCACCATCCT | GTTGATGAGGCCTTCGGGAA | 100 |
| CB2 | CGGAGCTCATGCTGTCTTTA | TCAGGAAGGTCCAGGTCATC | 101 |
| TRPV1 | AGCCATTGAGCATGGCATAG | GTGATGTCCTTGGTGTCCAG | 170 |
| PPAR-γ | GAGATCACAGAGTATGCCAA | CTGTCATCTAATTCCAGTGC | 148 |
| GAPDH | GGAGCGAGATCCCTCCAAAA | GGCTGTTGTCATACTTCTCA | 218 |
| Cell Line | Maximum Inhibition (% ± SEM) | p-Value | IC50 (µM ± SEM) | p-Value | ||
|---|---|---|---|---|---|---|
| Free WIN | SMA-WIN | Free WIN | SMA-WIN | |||
| LN18 | 71.62 ± 0.032 | 80.49 ± 0.018 | 0.03 | 20.97 ± 0.080 | 12.48 ± 0.11 | 4.40 × 10−8 |
| A172 | 54.17 ± 0.058 | 58.29 ± 0.041 | 0.05 | 30.9 ± 0.120 | 16.72 ± 0.098 | 8.94 × 10−9 |
| Cell Line | Treatment | Cell Survival at 20 µM (% ± SEM) | Cell Survival at 100 µM (% ± SEM) | IC50 (µM ± SEM) |
|---|---|---|---|---|
| LN18 | Free WIN | 52.0 ± 1.5 | 10.0 ± 0.8 | 20.97 ± 0.08 |
| SMA-WIN | 30.0 ± 1.3 | 5.0 ± 0.5 | 12.48 ± 0.11 | |
| A172 | Free WIN | 60.0 ± 1.7 | 12.0 ± 0.9 | 30.9 ± 0.12 |
| SMA-WIN | 35.0 ± 1.5 | 6.0 ± 0.6 | 16.72 ± 0.09 |
| Receptor | LN-18 | A-172 | ||||||
|---|---|---|---|---|---|---|---|---|
| Free WIN | SMA-WIN | Free WIN | SMA-WIN | |||||
| FC | p | FC | p | FC | p | FC | p | |
| CB1 | 2.01 ↑ | 0.0001 *** | 2.60 ↑ | 0.0001 *** | 1.10 ↑ | 0.0039 ** | 5.30 ↑ | 0.040 * |
| CB2 | 2.6 ↑ | 0.00002 *** | 5.90 ↑ | 0.00003 *** | 0.89 ↓ | 0.0002 *** | 5.10 ↑ | 0.083 ns |
| TRPV1 | 0.69 ↓ | 0.0014 ** | 1.22 ↑ | 0.0207 * | 0.60 ↓ | 0.003 ** | 0.50 ↓ | 0.001 *** |
| PPARγ | 0.70 ↓ | 0.001 *** | 0.50 ↓ | 0.0003 *** | 1.20 ↑ | 0.020 * | 2.30 ↑ | 0.001 *** |
| Parameter | Value | Method |
|---|---|---|
| Size (nm) | 120 ± 20 | Dynamic Light Scattering |
| Zeta Potential (mV) | −30 ± 5 | Electrophoretic Mobility |
| Drug Loading (%) | 90 ± 3 | UV-Vis at 320 nm |
| Polydispersity Index | 0.2 ± 0.05 | Dynamic Light Scattering |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taha, S.; Aljishi, M.; Sultan, A.; Sridharan, K.; Taurin, S.; Greish, K.; Bakhiet, M. Enhanced Cytotoxicity and Receptor Modulation by SMA-WIN 55,212-2 Micelles in Glioblastoma Cells. Int. J. Mol. Sci. 2025, 26, 4544. https://doi.org/10.3390/ijms26104544
Taha S, Aljishi M, Sultan A, Sridharan K, Taurin S, Greish K, Bakhiet M. Enhanced Cytotoxicity and Receptor Modulation by SMA-WIN 55,212-2 Micelles in Glioblastoma Cells. International Journal of Molecular Sciences. 2025; 26(10):4544. https://doi.org/10.3390/ijms26104544
Chicago/Turabian StyleTaha, Safa, Muna Aljishi, Ameera Sultan, Kannan Sridharan, Sebastien Taurin, Khaled Greish, and Moiz Bakhiet. 2025. "Enhanced Cytotoxicity and Receptor Modulation by SMA-WIN 55,212-2 Micelles in Glioblastoma Cells" International Journal of Molecular Sciences 26, no. 10: 4544. https://doi.org/10.3390/ijms26104544
APA StyleTaha, S., Aljishi, M., Sultan, A., Sridharan, K., Taurin, S., Greish, K., & Bakhiet, M. (2025). Enhanced Cytotoxicity and Receptor Modulation by SMA-WIN 55,212-2 Micelles in Glioblastoma Cells. International Journal of Molecular Sciences, 26(10), 4544. https://doi.org/10.3390/ijms26104544