
The Use of Selective Serotonin Reuptake Inhibitor (SSRI) Antidepressants in the Treatment of Lung Cancer


Abstract
1. Introduction
2. Classification of Antidepressants
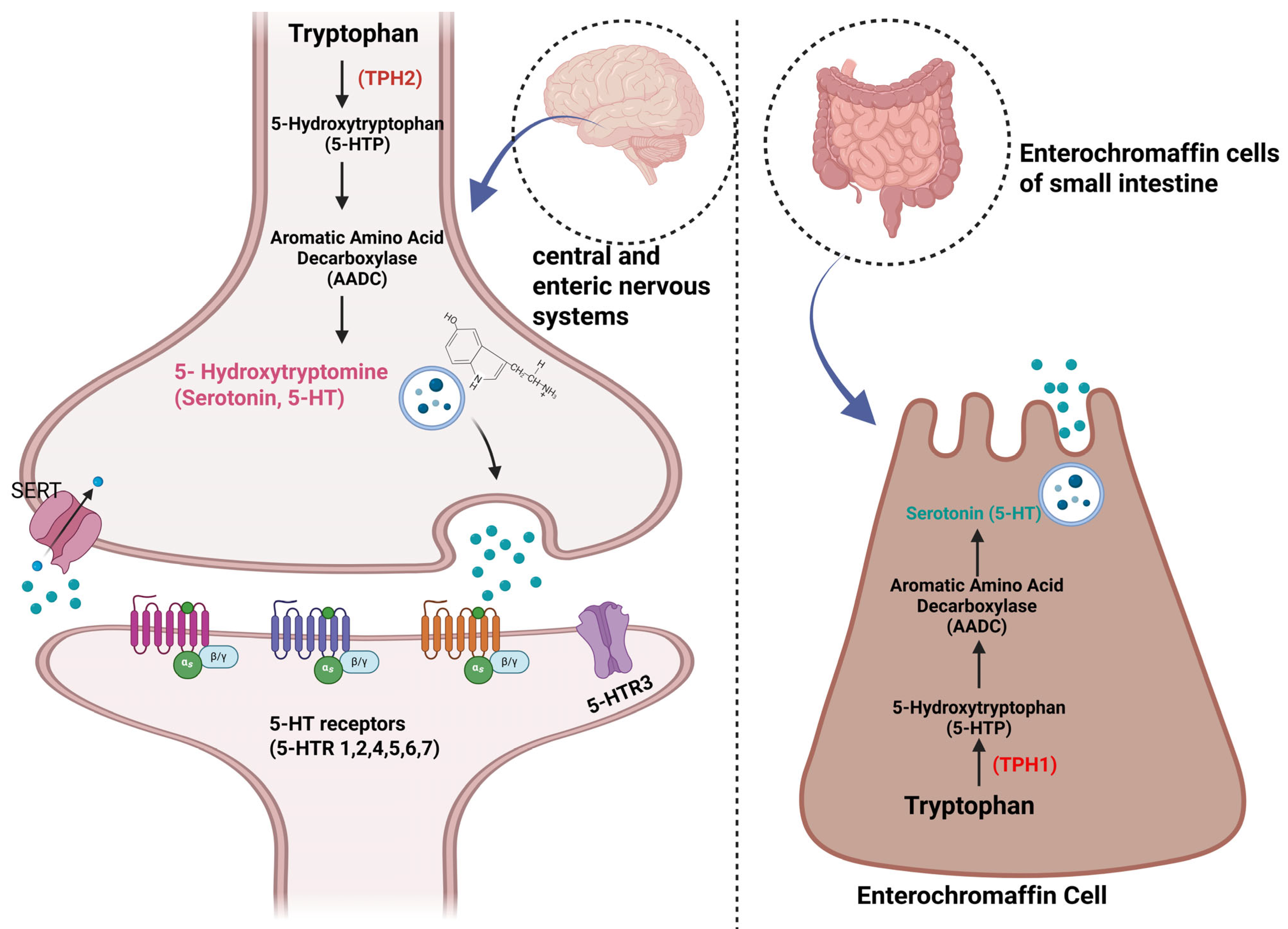
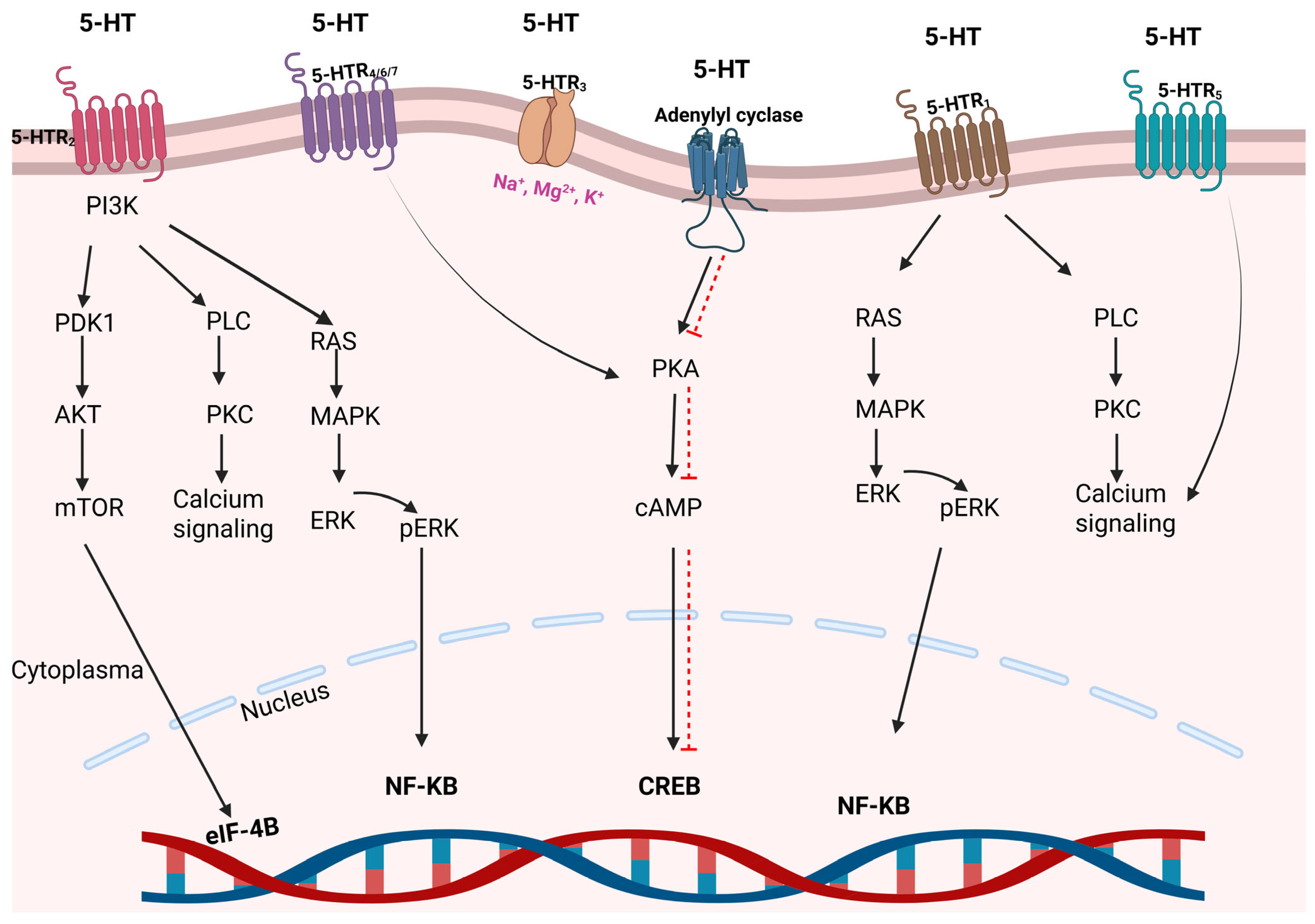
3. Serotonin’s Functions and Its Relationship with Cancer
4. Pharmacokinetics/Pharmacodynamics
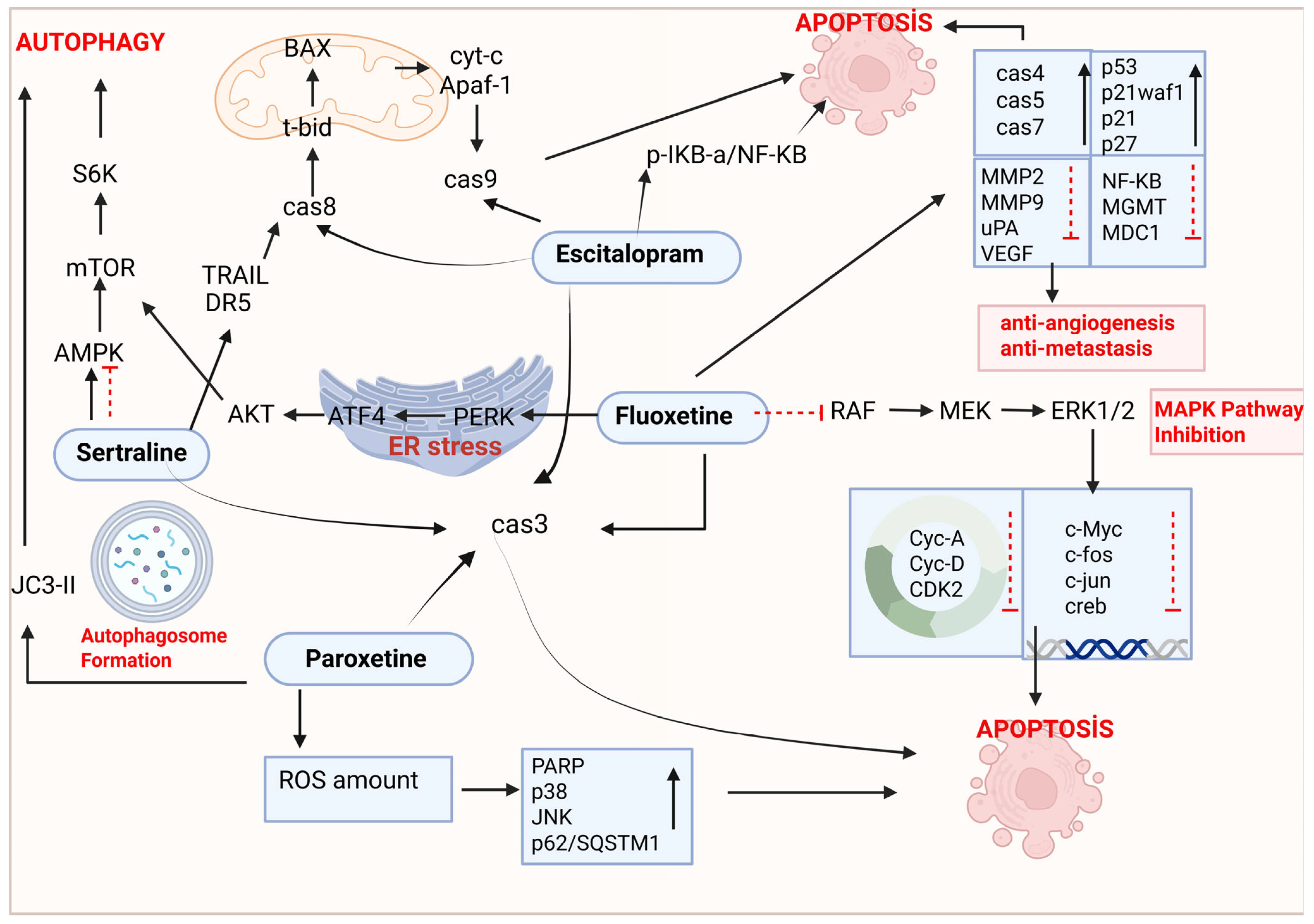
5. The Anticancer Effects of SSRI Antidepressants in the Treatment of Lung Cancer
5.1. Fluoxetine
5.2. Sertraline
5.3. Paroxetine
5.4. Escitalopram

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | Cell Line(s) | Time | Dose Range | Effective Concentration | Mechanism | Reference |
|---|---|---|---|---|---|---|
| FLX | A549 | 24, 48 h | 1–10 μM | 10 μM | Apoptosis | [56] |
| CL1-5-F4 | 48 h | 0, 10, 20, 30, 40 μM | 40 μM | Apoptosis, anti-angiogenesis, and anti-metastatic effect | [57] | |
| CL1-5-F4 | 28 days | dose of 10 mg/kg | Apoptosis, anti-angiogenesis, and anti-metastatic effect | [58] | ||
| A549 and mouse model | 72 h (in vitro) and 7 days (in vivo) | 15 μmol/L and 20 mg/kg (mice) | 15 μmol/L | Apoptosis | [59] | |
| H460 and A549 | 24 h | 0, 5, 10, 20, 30, and 40 μM | 20 μM | Autophagy | [54] | |
| SRT | H522, A549, H1975, PC9, and PC9/R | 72 h | 0, 5 and 10 µM | 10.50, 11.10, 9.40, 4.40, and 9.60 μM | Autophagy | [64] |
| A549 and HCC 15 vs. Calu 3 | 18 h | 0, 2.5, 5 and 10 µM | 10 µM | Extrinsic pathway of the apoptosis | [65] | |
| PRX | ChaGo-K1 and Hude | 24 h | 5, 10, 50, 100 µM | 5 and 10 µM | Apoptotic effect | [72] |
| NCI-H1299 and NCI-H1650 | 24 h | 0, 10, 20, 3 40, 50, 60, 70 µM | 36.97 µM and 45.43 µM | Mitochondria-dependent apoptosis | [73] | |
| H460 and A549 | 24 h | 0, 5, 10, 20, 30, and 40 μM | 20 μM | Antiproliferative effect | [54] | |
| A549 | 48 h | 10, 20, and 30 μg/mL | 30 μg/mL | Increased amount of ROS, mitochondria-dependent apoptosis | [75] | |
| ES | A549 and H460 | 24 h | 0.1, 0.2, 0.4, or 0.5 mM | 0.2 and 0.4 mM | Mitochondria-dependent apoptosis | [7] |
6. The Synergistic Effects of SSRIs in Lung Cancer Treatment
| Drugs | Cancer | Time | Doses | Mechanism | Reference |
|---|---|---|---|---|---|
| PRX/Dakomitinib combination | NSLCL | 10 days | 45 mg | Potential drug–drug interaction and cytotoxic effect | [93] |
| SRT/Erlotinib combination | PC9, PC9/R, A549, H522 and H1975 | 24 h | 5.10 vs. 20 μM | Exhibits a synergistic effect by promoting autophagy through the increase in autophagic markers (LC3) and the development of autophagic lysosomes | [64] |
| SRT/Radiation combination | A549, H1299, H125, H520, H1975, HCC15, and Calu-6 | 24 h | 10 µM SRT, 6 Gy | Reduces NSCLC tumor growth and alters the tumor microenvironment through cytokines linked to natural killer cells | [94] |
| SRT/TRAIL combination | A549 and Calu-3 vs. HCC-15 | 18 h | SRT (0, 2,5, and 5 vs. 10 µM) TRAIL (100 ng/mL) | AMPK inhibits the autophagic flux in TRAIL-resistant cells via TRAIL-mediated apoptosis and extrinsic apoptosis in A549, HCC-15, and Calu-3 cells. | [65] |
| FLX/Paclitaxel combination | MRC-5 | 48 h | 6.12 µM and 2.61 nM | Palmitoyl-protein thioesterase 1 (PPT1) | [29] |
| PRX/Amitriptyline combination | A549 | 48 h | 10, 20, and 30 μg/mL | Inhibits the growth of cancer cells by inducing apoptosis and LDH leakage and inducing oxidative stress | [75] |
| (Metformin/FLX), (Efavirenz/FLX), and (Metformin/Efavirenz/FLX) combination | A549 | 24 h | Metformin (5 mM), Efavirenz (1.5 μM), FLX (0.9 μM) | Increased cellular ROS levels | [98] |
| FLX/Gefitinib or FLX/Erlotinib | 0.1 to 100 μM | Pre-clinical drug–drug interaction (DDI) | [99] | ||
| PRX/Cisplatin combination | H1299 | 24 h | 0, 10, 15, and 20 μM | Increased ROS, JNK, and p38 activation and intrinsic apoptosis | [74] |
| ES/Etoposide | A549, H1299, A54990E | 24 h | ES (5, 10, 25, 50, 100, 200, 250, 400, and 500 μg/mL) and Etoposide (5, 25, 50, 100, 250, and 500 ng/mL) | Triggers cytotoxic and apoptotic activity and reduces resistance to etoposide by reducing the amount of P-gP | [100] |
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| TLA | Three letter acronym |
| LD | Linear dichroism |
References
- Li, Y.; Yan, B.; He, S. Advances and Challenges in the Treatment of Lung Cancer. Biomed. Pharmacother. 2023, 169, 115891. [Google Scholar] [CrossRef] [PubMed]
- Woodman, C.; Vundu, G.; George, A.; Wilson, C.M. Applications and Strategies in Nanodiagnosis and Nanotherapy in Lung Cancer. Semin. Cancer Biol. 2021, 69, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Chen, H.-D.; Yu, Y.-W.; Li, N.; Chen, W.-Q. Changing Profiles of Cancer Burden Worldwide and in China: A Secondary Analysis of the Global Cancer Statistics 2020. Chin. Med. J. 2021, 134, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Leiter, A.; Veluswamy, R.R.; Wisnivesky, J.P. The Global Burden of Lung Cancer: Current Status and Future Trends. Nat. Rev. Clin. Oncol. 2023, 20, 624–639. [Google Scholar] [CrossRef]
- Li, C.; Lei, S.; Ding, L.; Xu, Y.; Wu, X.; Wang, H.; Zhang, Z.; Gao, T.; Zhang, Y.; Li, L. Global Burden and Trends of Lung Cancer Incidence and Mortality. Chin. Med. J. 2023, 136, 1583–1590. [Google Scholar] [CrossRef]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non–Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef]
- Yuan, I.; Horng, C.; Chen, V.; Chen, C.; Chen, L.; Hsu, T.; Tzang, B. Escitalopram Oxalate Inhibits Proliferation and Migration and Induces Apoptosis in Non-Small Cell Lung Cancer Cells. Oncol. Lett. 2017, 15, 3376–3382. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [CrossRef]
- Hammerman, P.S.; Voet, D.; Lawrence, M.S.; Voet, D.; Jing, R.; Cibulskis, K.; Sivachenko, A.; Stojanov, P.; McKenna, A.; Lander, E.S.; et al. Comprehensive Genomic Characterization of Squamous Cell Lung Cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Gazdar, A.F.; Bunn, P.A.; Minna, J.D. Small-Cell Lung Cancer: What We Know, What We Need to Know and the Path Forward. Nat. Rev. Cancer 2017, 17, 725–737. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Ju, D.-T.; Chang, C.-F.; Muralidhar Reddy, P.; Velmurugan, B.K. A Review on the Effects of Current Chemotherapy Drugs and Natural Agents in Treating Non–Small Cell Lung Cancer. Biomedicine 2017, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Mehta, M.; Dhanjal, D.S.; Kaur, S.; Gupta, G.; Singh, H.; Thangavelu, L.; Rajeshkumar, S.; Tambuwala, M.; Bakshi, H.A.; et al. Emerging Trends in the Novel Drug Delivery Approaches for the Treatment of Lung Cancer. Chem. Biol. Interact. 2019, 309, 108720. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Yu, Z.; Feng, L.; Deng, L.; Fang, Z.; Liu, Z.; Li, Y.; Wu, X.; Qin, L.; Guo, R.; et al. Chitosan-Based Nanoparticle Co-Delivery of Docetaxel and Curcumin Ameliorates Anti-Tumor Chemoimmunotherapy in Lung Cancer. Carbohydr. Polym. 2021, 268, 118237. [Google Scholar] [CrossRef]
- Norouzi, M.; Hardy, P. Clinical Applications of Nanomedicines in Lung Cancer Treatment. Acta Biomater. 2021, 121, 134–142. [Google Scholar] [CrossRef]
- Helvacı Çelik, F.; Hocaoğlu, Ç. Major Depresif Bozukluk’ Tanımı, Etyolojisi ve Epidemiyolojisi: Bir Gözden Geçirme. J. Contemp. Med. 2016, 6, 51–66. [Google Scholar] [CrossRef]
- Mete, H.E. Chronic probability and availability. J. Chem. Phys. 2008, 11, 3–18. [Google Scholar]
- Wittchen, H.U.; Jacobi, F. Size and Burden of Mental Disorders in Europe—A Critical Review and Appraisal of 27 Studies. Eur. Neuropsychopharmacol. 2005, 15, 357–376. [Google Scholar] [CrossRef]
- McIntosh, E.; Gillanders, D.; Rodgers, S. Rumination, Goal Linking, Daily Hassles and Life Events in Major Depression. Clin. Psychol. Psychother. 2010, 17, 33–43. [Google Scholar] [CrossRef]
- Horwath, E.; Cohen, R.S.; Weissman, M.M. Epidemiology of Depressive and Anxiety Disorders. In Psychiatry Psychiatric Epidemiology; Wiley: Hoboken, NJ, USA, 2002; pp. 389–426. [Google Scholar]
- Kersting, A.; Reutemann, M.; Ohrmann, P.; Schütt, K.; Wesselmann, U.; Rothermundt, M.; Suslow, T.; Arolt, V. Traumatische Trauer—Ein Eigenständiges Krankheitsbild? Psychotherapeut 2001, 46, 301–308. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Molecular Theory of Cancer. Cancer Biol. Ther. 2005, 4, 621–627. [Google Scholar] [CrossRef]
- Pinquart, M.; Duberstein, P.R. Depression and Cancer Mortality: A Meta-Analysis. Psychol. Med. 2010, 40, 1797–1810. [Google Scholar] [CrossRef]
- Smith, H.R. Depression in Cancer Patients: Pathogenesis, Implications and Treatment (Review). Oncol. Lett. 2015, 9, 1509–1514. [Google Scholar] [CrossRef]
- Fisch, M.J.; Zhao, F.; Manola, J.; Miller, A.H.; Pirl, W.F.; Wagner, L.I. Patterns and Predictors of Antidepressant Use in Ambulatory Cancer Patients with Common Solid Tumors. Psychooncology 2015, 24, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Zheng, L.; Tang, S.; Gu, J.; Jiang, X.; Wang, L. In-Vitro and in Situ Assessment of the Efflux of Five Antidepressants by Breast Cancer Resistance Protein. J. Pharm. Pharmacol. 2019, 71, 1133–1141. [Google Scholar] [CrossRef]
- Debela, D.T.; Muzazu, S.G.; Heraro, K.D.; Ndalama, M.T.; Mesele, B.W.; Haile, D.C.; Kitui, S.K.; Manyazewal, T. New Approaches and Procedures for Cancer Treatment: Current Perspectives. SAGE Open Med. 2021, 9, 20503121211034366. [Google Scholar] [CrossRef] [PubMed]
- Deep, A.; Kumar, D.; Bansal, N.; Narasimhan, B.; Marwaha, R.K.; Sharma, P.C. Understanding Mechanistic Aspects and Therapeutic Potential of Natural Substances as Anticancer Agents. Phytomedicine Plus 2023, 3, 100418. [Google Scholar] [CrossRef]
- Antoszczak, M.; Markowska, A.; Markowska, J.; Huczyński, A. Antidepressants and Antipsychotic Agents as Repurposable Oncological Drug Candidates. Curr. Med. Chem. 2021, 28, 2137–2174. [Google Scholar] [CrossRef] [PubMed]
- Duarte, D.; Nunes, M.; Ricardo, S.; Vale, N. Combination of Antimalarial and CNS Drugs with Antineoplastic Agents in MCF-7 Breast and HT-29 Colon Cancer Cells: Biosafety Evaluation and Mechanism of Action. Biomolecules 2022, 12, 1490. [Google Scholar] [CrossRef]
- Song, Y.; Yang, X.; Yu, B. Repurposing Antidepressants for Anticancer Drug Discovery. Drug Discov. 2022, 27, 1924–1935. [Google Scholar] [CrossRef]
- Edinoff, A.N.; Apgar, T.L.; Rogers, J.J.; Harper, J.D.; Cornett, E.M.; Kaye, A.M.; Kaye, A.D. Attention Deficit Hyperactivity Disorder and Bipolar Disorder: Diagnosis, Treatments, and Clinical Considerations: A Narrative Review. Psychiatry Int. 2021, 3, 17–28. [Google Scholar] [CrossRef]
- Caraci, F.; Crupi, R.; Drago, F.; Spina, E. Metabolic Drug Interactions Between Antidepressants and Anticancer Drugs: Focus on Selective Serotonin Reuptake Inhibitors and Hypericum Extract. Curr. Drug Metab. 2011, 12, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Khushboo; Sharma, B. Antidepressants: Mechanism of Action, Toxicity and Possible Amelioration. J. Appl. Biotechnol. Bioeng. 2017, 3, 437–448. [Google Scholar] [CrossRef]
- Rodin, G.; Lloyd, N.; Katz, M.; Green, E.; Mackay, J.A.; Wong, R.K.S. The Treatment of Depression in Cancer Patients: A Systematic Review. Support. Care Cancer 2007, 15, 123–136. [Google Scholar] [CrossRef]
- Alvano, S.A.; Zieher, L.M. An Updated Classification of Antidepressants: A Proposal to Simplify Treatment. Pers. Med. Psychiatry 2020, 19–20, 100042. [Google Scholar] [CrossRef]
- Sarrouilhe, D.; Mesnil, M. Serotonin and Human Cancer: A Critical View. Biochimie 2019, 161, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Balakrishna, P.; George, S.; Hatoum, H.; Mukherjee, S. Serotonin Pathway in Cancer. Int. J. Mol. Sci. 2021, 22, 1268. [Google Scholar] [CrossRef]
- Chen, L.; Huang, S.; Wu, X.; He, W.; Song, M. Serotonin Signalling in Cancer: Emerging Mechanisms and Therapeutic Opportunities. Clin. Transl. Med. 2024, 14, e1750. [Google Scholar] [CrossRef]
- Karmakar, S.; Lal, G. Role of Serotonin Receptor Signaling in Cancer Cells and Anti-Tumor Immunity. Theranostics 2021, 11, 5296. [Google Scholar] [CrossRef]
- Pourhamzeh, M.; Moravej, F.G.; Arabi, M.; Shahriari, E.; Mehrabi, S.; Ward, R.; Ahadi, R.; Joghataei, M.T. The Roles of Serotonin in Neuropsychiatric Disorders. Cell Mol. Neurobiol. 2021, 42, 1671–1692. [Google Scholar] [CrossRef]
- Xue, W.; Wang, P.; Li, B.; Li, Y.; Xu, X.; Yang, F.; Yao, X.; Chen, Y.Z.; Xu, F.; Zhu, F. Identification of the Inhibitory Mechanism of FDA Approved Selective Serotonin Reuptake Inhibitors: An Insight from Molecular Dynamics Simulation Study. Phys. Chem. Chem. Phys. 2016, 18, 3260–3271. [Google Scholar] [CrossRef]
- Edinoff, A.N.; Akuly, H.A.; Hanna, T.A.; Ochoa, C.O.; Patti, S.J.; Ghaffar, Y.A.; Kaye, A.D.; Viswanath, O.; Urits, I.; Boyer, A.G.; et al. Selective Serotonin Reuptake Inhibitors and Adverse Effects: A Narrative Review. Neurol. Int. 2021, 13, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Niu, M.; Zang, Y.; Zhuang, H.; Jia, F.; Bao, S.; Liu, S. Which Can Predict the Outcome of Antidepressants: Metabolic Genes or Pharmacodynamic Genes? Curr. Drug Metab. 2023, 24, 525–535. [Google Scholar] [CrossRef]
- Özkaya, S.; Aydemir, E. SSRI Grubu Antidepresanlar ve Antikanser Etkileri. Türk Bilimsel Derlemeler Derg. 2021, 14, 37–46. [Google Scholar]
- Frick, L.R.; Rapanelli, M. Antidepressants: Influence on Cancer and Immunity? Life Sci. 2013, 92, 525–532. [Google Scholar] [CrossRef]
- He, L.; Fu, Y.; Tian, Y.; Wang, X.; Zhou, X.; Ding, R.-B.; Qi, X.; Bao, J. Antidepressants as Autophagy Modulators for Cancer Therapy. Molecules 2023, 28, 7594. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kumar, M.; Gautam, A.K.; Sonkar, A.B.; Verma, A.; Singh, A.; Nisha, R.; Kumar, U.; Kumar, D.; Mahata, T.; et al. Ameliorative Effect of Fluvoxamine against Colon Carcinogenesis via COX-2 Blockade with Oxidative and Metabolic Stress Reduction at the Cellular, Molecular and Metabolic Levels. BBA Adv. 2022, 2, 100046. [Google Scholar] [CrossRef]
- Fernández-García, B.; Fraile-Rodriguez, C.; Abasolo-Tamayo, X.; Aquerreta-González Clínica, I.; Cancela-Díez, B.; Gómez-Derueda, F.; Sánchez-Argaiz, M.; Martinez-Casanova, N.; Sanchez-Yañez, E.; Moya-Carmona, I. 5PSQ-149 Drug Interaction in Breast Cancer Triple Negative Therapy: Docetaxel and Fluvoxamine. A Case Report. Eur. J. Hosp. Pharm. 2021, 28, A130. [Google Scholar] [CrossRef]
- Ramasamy, S.; Jeyaram, K.; Narayanan, A.; Arunachalam, S.; Ethiraj, S.; Sankar, M.; Pandian, B. Repurposing Fluvoxamine as an Inhibitor for NUDT5 in Breast Cancer Cell: An in Silico and in Vitro Study. Silico Pharmacol. 2024, 13, 5. [Google Scholar] [CrossRef]
- Suzuki, N.; Ninomiya, M.; Maruta, T.; Hosonuma, S.; Yoshioka, N.; Ohara, T.; Nishigaya, Y.; Kobayashi, Y.; Kiguchi, K.; Ishizuka, B. Clinical Study on the Efficacy of Fluvoxamine for Psychological Distress in Gynecologic Cancer Patients. Int. J. Gynecol. Cancer 2011, 21, 1143–1149. [Google Scholar] [CrossRef]
- Ahmadian, E.; Eftekhari, A.; Babaei, H.; Nayebi, A.M.; Eghbal, M.A. Anti-Cancer Effects of Citalopram on Hepatocellular Carcinoma Cells Occur via Cytochrome C Release and the Activation of NF-KB. Anticancer. Agents Med. Chem. 2017, 17, 1570–1577. [Google Scholar] [CrossRef]
- Dong, F.; He, K.; Zhang, S.; Song, K.; Jiang, L.; Hu, L.-P.; Li, Q.; Zhang, X.-L.; Zhang, N.; Li, B.-T.; et al. SSRI Antidepressant Citalopram Reverses the Warburg Effect to Inhibit Hepatocellular Carcinoma by Directly Targeting GLUT1. Cell Rep. 2024, 43, 114818. [Google Scholar] [CrossRef]
- Sakka, L.; Delétage, N.; Chalus, M.; Aissouni, Y.; Sylvain-Vidal, V.; Gobron, S.; Coll, G. Assessment of Citalopram and Escitalopram on Neuroblastoma Cell Lines: Cell Toxicity and Gene Modulation. Oncotarget 2017, 8, 42789. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Zhuang, X.; Zhang, L.; Qiao, T. Antidepressants Fluoxetine Mediates Endoplasmic Reticulum Stress and Autophagy of Non–Small Cell Lung Cancer Cells Through the ATF4-AKT-MTOR Signaling Pathway. Front. Pharmacol. 2022, 13, 904701. [Google Scholar] [CrossRef]
- Kadasah, S.F.; Alqahtani, A.M.S.; Alkhammash, A.; Radwan, M.O. Beyond Psychotropic: Potential Repurposing of Fluoxetine toward Cancer Therapy. Int. J. Mol. Sci. 2024, 25, 6314. [Google Scholar] [CrossRef] [PubMed]
- Stepulak, A.; Rzeski, W.; Sifringer, M.; Brocke, K.; Gratopp, A.; Kupisz, K.; Turski, L.; Ikonomidou, C. Fluoxetine Inhibits the Extracellular Signal Regulated Kinase Pathway and Suppresses Growth of Cancer Cells. Cancer Biol. Ther. 2008, 7, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Lin, S.S.; Hsu, F.T.; Chung, J.G. Fluoxetine Inhibits DNA Repair and NF-ĸB-Modulated Metastatic Potential in Non-Small Cell Lung Cancer. Anticancer. Res. 2018, 38, 5201–5210. [Google Scholar] [CrossRef]
- Hsu, L.-C.; Tu, H.-F.; Hsu, F.-T.; Yueh, P.-F.; Chiang, I.-T. Beneficial Effect of Fluoxetine on Anti-Tumor Progression on Hepatocellular Carcinoma and Non-Small Cell Lung Cancer Bearing Animal Model. Biomed. Pharmacother. 2020, 126, 110054. [Google Scholar] [CrossRef]
- Yang, Z.; Li, Z.; Guo, Z.; Ren, Y.; Zhou, T.; Xiao, Z.; Duan, J.; Han, C.; Cheng, Y.; Xu, F. Antitumor Effect of Fluoxetine on Chronic Stress-Promoted Lung Cancer Growth via Suppressing Kynurenine Pathway and Enhancing Cellular Immunity. Front. Pharmacol. 2021, 12, 685898. [Google Scholar] [CrossRef]
- He, A.; Wu, M.; Pu, Y.; Li, R.; Zhang, Y.; He, J.; Xia, Y.; Ma, Y. Fluoxetine as a Potential Therapeutic Agent for Inhibiting Melanoma Brain and Lung Metastasis: Induction of Apoptosis, G0/G1 Cell Cycle Arrest, and Disruption of Autophagy Flux. J. Cancer 2024, 15, 3825–3840. [Google Scholar] [CrossRef]
- Huddart, R.; Hicks, J.K.; Ramsey, L.B.; Strawn, J.R.; Smith, D.M.; Bobonis Babilonia, M.; Altman, R.B.; Klein, T.E. PharmGKB Summary: Sertraline Pathway, Pharmacokinetics. Pharmacogenet. Genom. 2020, 30, 26–33. [Google Scholar] [CrossRef]
- Duarte, D.; Vale, N. Antidepressant Drug Sertraline against Human Cancer Cells. Biomolecules 2022, 12, 1513. [Google Scholar] [CrossRef] [PubMed]
- Hiemke, C.; Härtter, S. Pharmacokinetics of Selective Serotonin Reuptake Inhibitors. Pharmacol. Ther. 2000, 85, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Lu, W.; Shen, X.; Wang, Q.; Lv, J.; Liu, M.; Cheng, F.; Zhao, Z.; Pang, X. Repurposing Sertraline Sensitizes Non–Small Cell Lung Cancer Cells to Erlotinib by Inducing Autophagy. JCI Insight 2018, 3, e98921. [Google Scholar] [CrossRef]
- Zinnah, K.; Seol, J.; Park, S. Inhibition of Autophagy Flux by Sertraline Attenuates TRAIL Resistance in Lung Cancer via Death Receptor 5 Upregulation. Int. J. Mol. Med. 2020, 46, 795–805. [Google Scholar] [CrossRef]
- Bourin, M.; Chue, P.; Guillon, Y. Paroxetine: A Review. CNS Drug Rev. 2001, 7, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Nevels, R.M.; Gontkovsky, S.T.; Williams, B.E. Paroxetine-The Antidepressant from Hell? Probably Not, But Caution Required. Psychopharmacol. Bull. 2016, 46, 77–104. [Google Scholar] [PubMed]
- Jang, W.; Jung, S.K.; Vo, T.T.L.; Jeong, C. Anticancer Activity of Paroxetine in Human Colon Cancer Cells: Involvement of MET and ERBB3. J. Cell Mol. Med. 2019, 23, 1106–1115. [Google Scholar] [CrossRef]
- Cabral, V.P.; Rodrigues, D.S.; Barbosa, A.D.; Moreira, L.E.; Sá, L.G.; Silva, C.R.; Neto, J.B.; Silva, J.; Marinho, E.S.; Santos, H.S.; et al. Antibacterial Activity of Paroxetine Against Staphylococcus Aureus and Possible Mechanisms of Action. Future Microbiol. 2023, 18, 415–426. [Google Scholar] [CrossRef]
- Cakil, Y.D.; Ozunal, Z.G.; Kayali, D.G.; Aktas, R.G.; Saglam, E. Anti-Proliferative Effects of Paroxetine Alone or in Combination with Sorafenib in HepG2 Cells. Braz. J. Pharm. 2022, 58, e201148. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, L.; Wang, M.; Yang, Y.; Hu, B.; Li, G.; Wei, F. Paroxetine Promotes Longevity via Ser-7-Dop-4-IIS Axis in Caenorhabditis Elegans. Geroscience 2024, 1–13. [Google Scholar] [CrossRef]
- Rosetti, M.; Frasnelli, M.; Tesei, A.; Zoli, W.; Oncol, M.C.-J.E.T. Cytotoxicity of Different Selective Serotonin Reuptake Inhibitors (SSRIs) against Cancer Cells. J. Exp. Ther. Oncol. 2006, 6, 23–29. [Google Scholar]
- Wang, K.; Chen, B.; Yin, T.; Zhan, Y.; Lu, Y.; Zhang, Y.; Chen, J.; Wu, W.; Zhou, S.; Mao, W.; et al. N-Methylparoxetine Blocked Autophagic Flux and Induced Apoptosis by Activating ROS-MAPK Pathway in Non-Small Cell Lung Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3415. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Gong, Q.; Zhan, Y.; Chen, B.; Yin, T.; Lu, Y.; Zhang, Y.; Wang, H.; Ke, J.; Du, B.; et al. Blockage of Autophagic Flux and Induction of Mitochondria Fragmentation by Paroxetine Hydrochloride in Lung Cancer Cells Promotes Apoptosis via the ROS-MAPK Pathway. Front. Cell Dev. Biol. 2020, 7, 397. [Google Scholar] [CrossRef] [PubMed]
- Motafeghi, F.; Shahsavari, R.; Mortazavi, P.; Shokrzadeh, M. Anticancer Effect of Paroxetine and Amitriptyline on HT29 and A549 Cell Lines. Toxicol. Vitr. 2023, 87, 105532. [Google Scholar] [CrossRef]
- Von Moltke, L.L.; Greenblatt, D.J.; Giancarlo, G.M.; Granda, B.W.; Harmatz, J.S.; Shader, R.I. Escitalopram (S-Citalopram) and Its Metabolites in Vitro: Cytochromes Mediating Biotransformation, Inhibitory Effects, and Comparison to R-Citalopram. Drug Metab. Dispos. 2001, 29, 1102–1109. [Google Scholar]
- Rao, N. The Clinical Pharmacokinetics of Escitalopram. Clin. Pharmacokinet. 2007, 46, 281–290. [Google Scholar] [CrossRef]
- Spina, E.; Trifirò, G.; Caraci, F. Clinically Significant Drug Interactions with Newer Antidepressants. CNS Drugs 2012, 26, 39–67. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Haddjeri, N.; Sánchez, C. Escitalopram, an Antidepressant with an Allosteric Effect at the Serotonin Transporter-a Review of Current Understanding of Its Mechanism of Action. Psychopharmacology 2012, 219, 1–13. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, H.; Wang, Z.; Wu, P.; Gong, W. 5-Hydroxytryptamine1a Receptors on Tumour Cells Induce Immune Evasion in Lung Adenocarcinoma Patients with Depression via Autophagy/PSTAT3. Eur. J. Cancer 2019, 114, 8–24. [Google Scholar] [CrossRef]
- Dikmen, M.; Cantürk, Z.; Öztürk, Y. Escitalopram Oxalate, a Selective Serotonin Reuptake Inhibitor, Exhibits Cytotoxic and Apoptotic Effects in Glioma C6 Cells. Acta Neuropsychiatr. 2011, 23, 173–178. [Google Scholar] [CrossRef]
- Chen, L.J.; Hsu, T.C.; Chan, H.L.; Lin, C.F.; Huang, J.Y.; Stewart, R.; Tzang, B.S.; Chen, V.C.H. Protective Effect of Escitalopram on Hepatocellular Carcinoma by Inducing Autophagy. Int. J. Mol. Sci. 2022, 23, 9247. [Google Scholar] [CrossRef]
- Chen, V.C.H.; Hsieh, Y.H.; Chen, L.J.; Hsu, T.C.; Tzang, B.S. Escitalopram Oxalate Induces Apoptosis in U-87MG Cells and Autophagy in GBM8401 Cells. J. Cell Mol. Med. 2018, 22, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.; Huang, C.-H.; Tzang, J.-Y.; Hsu, B.-S.; Mcintyre, T.-C.; Kim, N.D.; Chen, V.C.-H.; Huang, J.-Y.; Tzang, B.-S.; Hsu, T.-C.; et al. Synergistic Effects of the Combinational Use of Escitalopram Oxalate and 5-Fluorouracil on the Inhibition of Gastric Cancer SNU-1 Cells. Int. J. Mol. Sci. 2022, 23, 16179. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.C.H.; Huang, S.L.; Huang, J.Y.; Hsu, T.C.; Tzang, B.S.; McIntyre, R.S. Combined Administration of Escitalopram Oxalate and Nivolumab Exhibits Synergistic Growth-Inhibitory Effects on Liver Cancer Cells through Inducing Apoptosis. Int. J. Mol. Sci. 2023, 24, 12630. [Google Scholar] [CrossRef] [PubMed]
- Gwynne, W.D.; Hallett, R.M.; Girgis-Gabardo, A.; Bojovic, B.; Dvorkin-Gheva, A.; Aarts, C.; Dias, K.; Bane, A.; Hassell, J.A. Serotonergic System Antagonists Target Breast Tumor Initiating Cells and Synergize with Chemotherapy to Shrink Human Breast Tumor Xenografts. Oncotarget 2017, 8, 32101. [Google Scholar] [CrossRef]
- Zinnah, K.; Park, S.-Y. Sensitizing TRAIL resistant A549 Lung Cancer Cells and Enhancing TRAIL induced Apoptosis with the Antidepressant Amitriptyline. Oncol. Rep. 2021, 46, 144. [Google Scholar] [CrossRef]
- Fatehi, R.; Nouraei, M.; Panahiyan, M.; Rashedinia, M.; Firouzabadi, N. Modulation of ACE2/Ang1-7/Mas and ACE/AngII/AT1 Axes Affects Anticancer Properties of Sertraline in MCF-7 Breast Cancer Cells. Biochem. Biophys. Rep. 2024, 38, 101738. [Google Scholar] [CrossRef]
- Peer, D.; Dekel, Y.; Melikhov, D.; Margalit, R. Fluoxetine Inhibits Multidrug Resistance Extrusion Pumps and Enhances Responses to Chemotherapy in Syngeneic and in Human Xenograft Mouse Tumor Models. Cancer Res. 2004, 64, 7562–7569. [Google Scholar] [CrossRef]
- Liu, B.-H.; Yuan, T.-M.; Huang, C.-J.; Hsu, D.-T.; Chen, S.-W.; Hsiao, N.-W.; Lin, S.-C.; Wu, S.-W.; Lin, Y.-M.J.; Chuang, S.-M. DNA Repair Proteins as the Targets for Paroxetine to Induce Cytotoxicity in Gastric Cancer Cell AGS. Am. J. Cancer Res. 2022, 12, 1465–1483. [Google Scholar]
- Gul, S.O.; Korkut, A.; Aydemir, E. Combined Effect of Sertraline and Capecitabine on Breast Cancer Cell Lines In Vitro and In Silico Evidence for Synergistic Interaction. Sci. Pharm. 2024, 92, 38. [Google Scholar] [CrossRef]
- Huang, Q.; Wu, M.; Pu, Y.; Zhou, J.; Zhang, Y.; Li, R.; Xia, Y.; Zhang, Y.; Ma, Y. Inhibition of TNBC Cell Growth by Paroxetine: Induction of Apoptosis and Blockage of Autophagy Flux. Cancers 2024, 16, 885. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Garcia, A.; Giri, N.; Labadie, R.R.; Ni, G.; Boutros, T.; Richie, N.; Kocinsky, H.S.; Checchio, T.M.; Bello, C.L. A Phase I Open-Label Study to Investigate the Potential Drug–Drug Interaction between Single-Dose Dacomitinib and Steady-State Paroxetine in Healthy Volunteers. J. Clin. Pharmacol. 2014, 54, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Castillo, A.; Heylen, E.; Hounjet, J.; Savelkouls, K.G.; Lieuwes, N.G.; Biemans, R.; Dubois, L.J.; Reynders, K.; Rouschop, K.M.; Vaes, R.D.W.; et al. Targeting Serine/Glycine Metabolism Improves Radiotherapy Response in Non-Small Cell Lung Cancer. Br. J. Cancer 2023, 130, 568–584. [Google Scholar] [CrossRef]
- Torta, R.G.V.; Miniotti, M.; Leombruni, P. Antidepressants in Oncology: Reason and Choice. Neurobiol. Dis. 2012, 1, 179–202. [Google Scholar] [CrossRef]
- Irarrázaval, O.M.E.; Gaete, G.L. Elección Del Mejor Antidepresivo En Pacientes Con Cáncer de Mama En Tratamiento Con Tamoxifeno: Revisión de La Evidencia Básica y Clínica. Rev. Med. Chil. 2016, 144, 1326–1335. [Google Scholar] [CrossRef]
- Zheng, Y.; Chang, X.; Huang, Y.; He, D. The Application of Antidepressant Drugs in Cancer Treatment. Biomed. Pharmacother. 2023, 157, 113985. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.-G.; Shende, M.; Inci, G.; Park, S.-H.; Jung, J.-S.; Kim, S.B.; Kim, J.H.; Mo, Y.W.; Seo, J.-H.; Feng, J.-H.; et al. Combination of Metformin/Efavirenz/Fluoxetine Exhibits Profound Anticancer Activity via a Cancer Cell-Specific ROS Amplification. Cancer Biol. Ther. 2023, 24, 2161803. [Google Scholar] [CrossRef]
- Luong, T.-L.T.; McAnulty, M.J.; Evers, D.L.; Reinhardt, B.J.; Weina, P.J. Pre-Clinical Drug-Drug Interaction (DDI) of Gefitinib or Erlotinib with Cytochrome P450 (CYP) Inhibiting Drugs, Fluoxetine and/or Losartan. Curr. Res. Toxicol. 2021, 2, 217–224. [Google Scholar] [CrossRef]
- Özkaya Gül, S.; Şimşek, B.; Yıldız, F.; Aydemir, E. Cytotoxic Effect of Escitalopram/Etoposide Combination on Etoposide-Resistant Lung Cancer. Pharmaceuticals 2025, 18, 531. [Google Scholar] [CrossRef]

| Category | Drug Class | Mechanism | Generic Names |
|---|---|---|---|
| First generation | Tricyclic antidepressants (TCAs) | İnhibit reuptake of norepinephrine and serotonin into presynaptic terminals | Amiltriptyline, Clomipmine, Desipramine, Doxepin, İmipramine, Nortriptyline, Amoxapine, Protriptyline, Trimipramine |
| Monoamine oxidase inhibitors (MAOIs) | Competitively inhibit monoamine oxidase; agents in the class differ in their reversibility and their activity against MAOOa and MAOb | Tranylcypromine, Phenelzine, Selegiline, Isocarboxazid | |
| Second generation | Selective serotonin reuptake inhibitors (SSRIs) | Selectively inhibit the reuptake of serotonin (5-HT) at the presynaptic neuronal membrane | Fluoxetine, Fluvoxamine, Paroxetine, Sertraline, Citalopram, Escitalopram |
| Selective norepinephrine reuptake inhibitors | İnhibit reuptake of both serotonin and norepinephrine; weakly inhibit dopamine reuptake | Venlafaxine, Mirtazapine, Duloxetine | |
| Serotonin antagonist and reuptake inhibitors (SARIs) | 5-HT2 receptor antagonists | Nefazodone | |
| Dopamine reuptake inhibitors | İnhibit dopamine reuptake with some effect on norepinephrine | Bupropion |
| Drugs | Apoptosis | Autophagy | ROS Production | Anti-Angiogenesis/Anti-Metastasis |
|---|---|---|---|---|
| FLX | + | + | − | + |
| ES | + | − | − | − |
| SRT | + | + | − | − |
| PRX | + | + | + | − |
| FVX | + | − | − | − |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Özkaya Gül, S.; Aydemir, E. The Use of Selective Serotonin Reuptake Inhibitor (SSRI) Antidepressants in the Treatment of Lung Cancer. Int. J. Mol. Sci. 2025, 26, 4546. https://doi.org/10.3390/ijms26104546
Özkaya Gül S, Aydemir E. The Use of Selective Serotonin Reuptake Inhibitor (SSRI) Antidepressants in the Treatment of Lung Cancer. International Journal of Molecular Sciences. 2025; 26(10):4546. https://doi.org/10.3390/ijms26104546
Chicago/Turabian StyleÖzkaya Gül, Serap, and Esra Aydemir. 2025. "The Use of Selective Serotonin Reuptake Inhibitor (SSRI) Antidepressants in the Treatment of Lung Cancer" International Journal of Molecular Sciences 26, no. 10: 4546. https://doi.org/10.3390/ijms26104546
APA StyleÖzkaya Gül, S., & Aydemir, E. (2025). The Use of Selective Serotonin Reuptake Inhibitor (SSRI) Antidepressants in the Treatment of Lung Cancer. International Journal of Molecular Sciences, 26(10), 4546. https://doi.org/10.3390/ijms26104546