
Metabolic Pathways and Molecular Regulatory Mechanisms of Fruit Color Change During Greening Stage of Peppers (Capsicum annuum L.)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
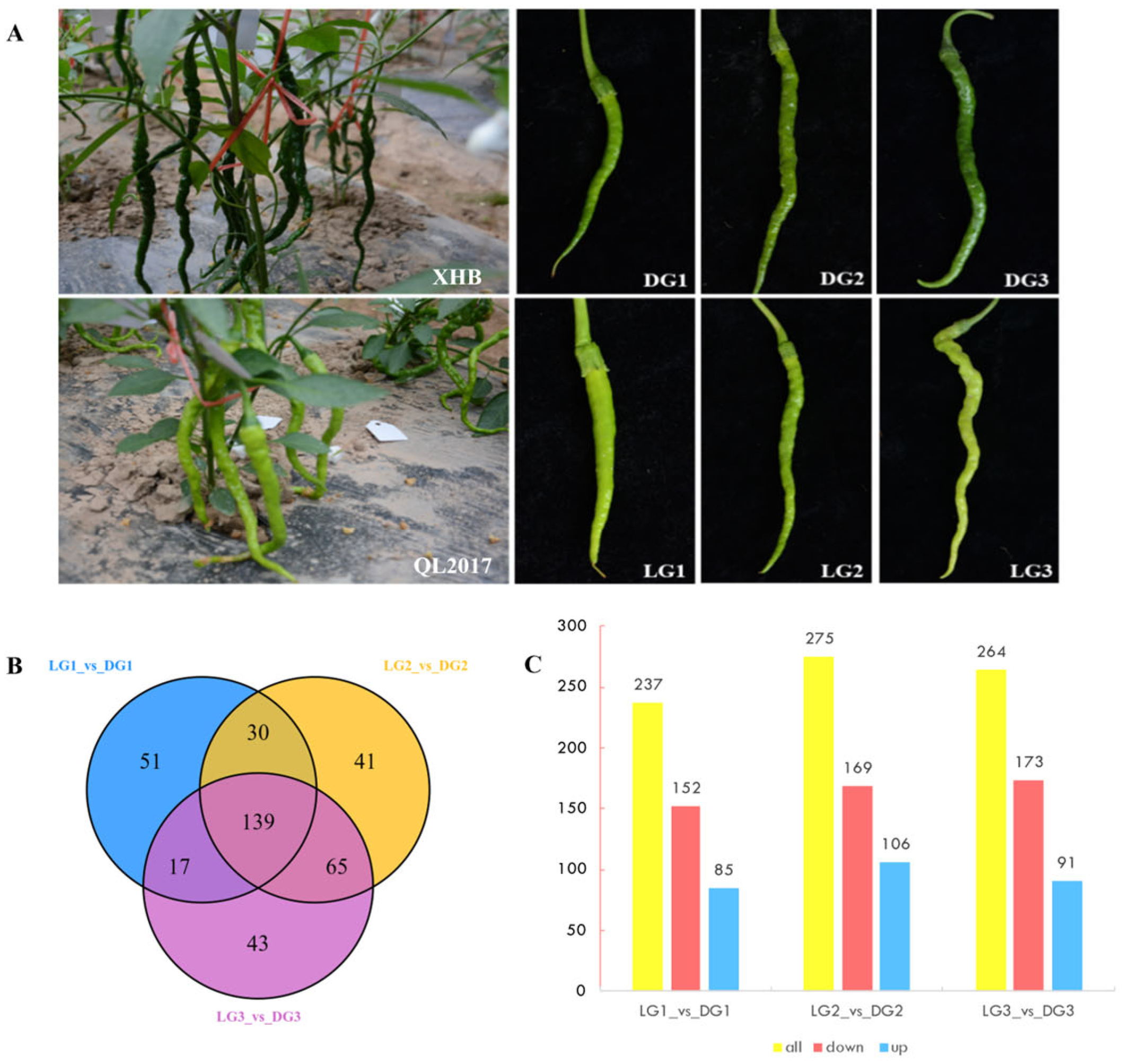
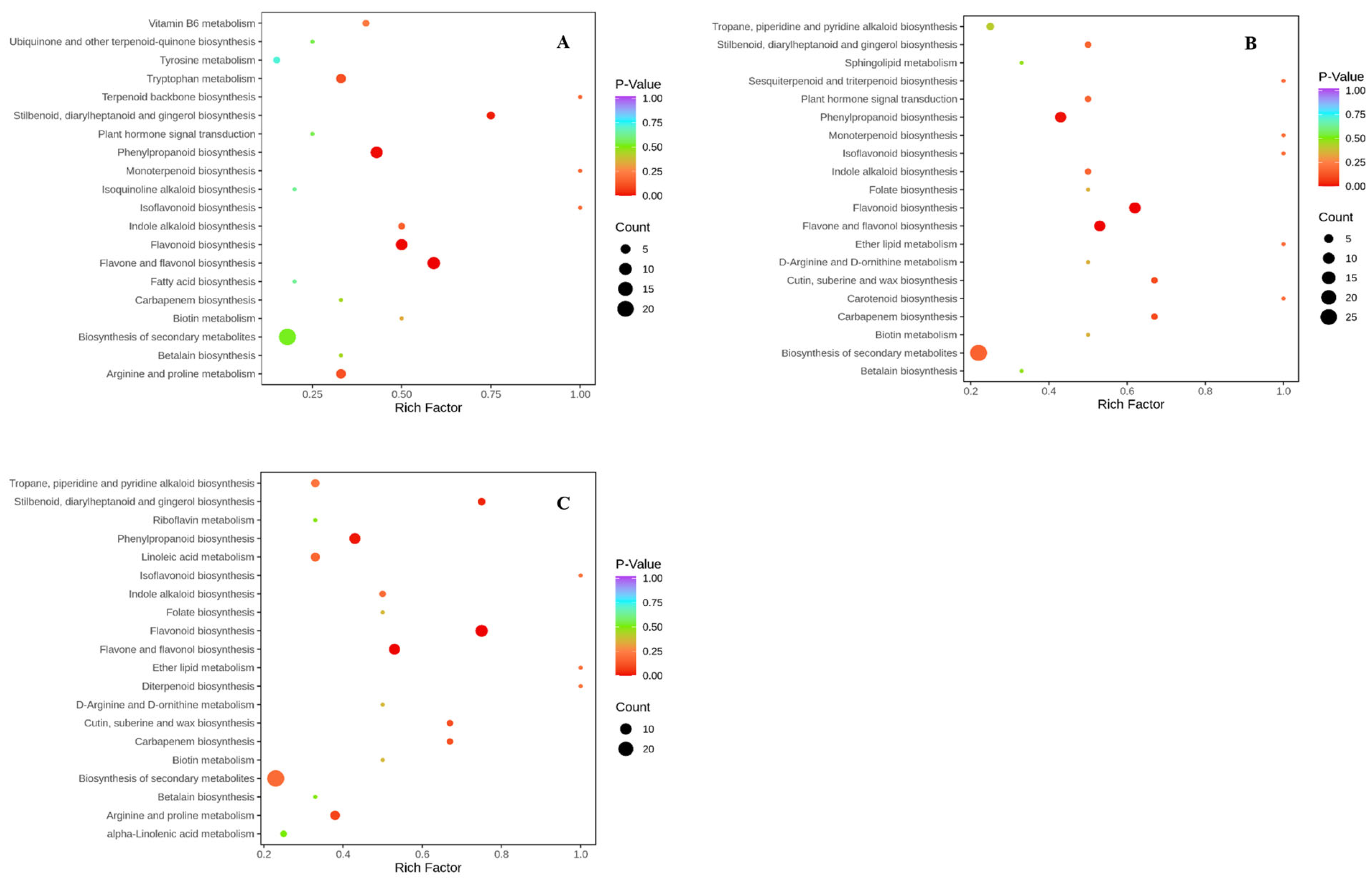
2.1. Metabolite Analysis Results
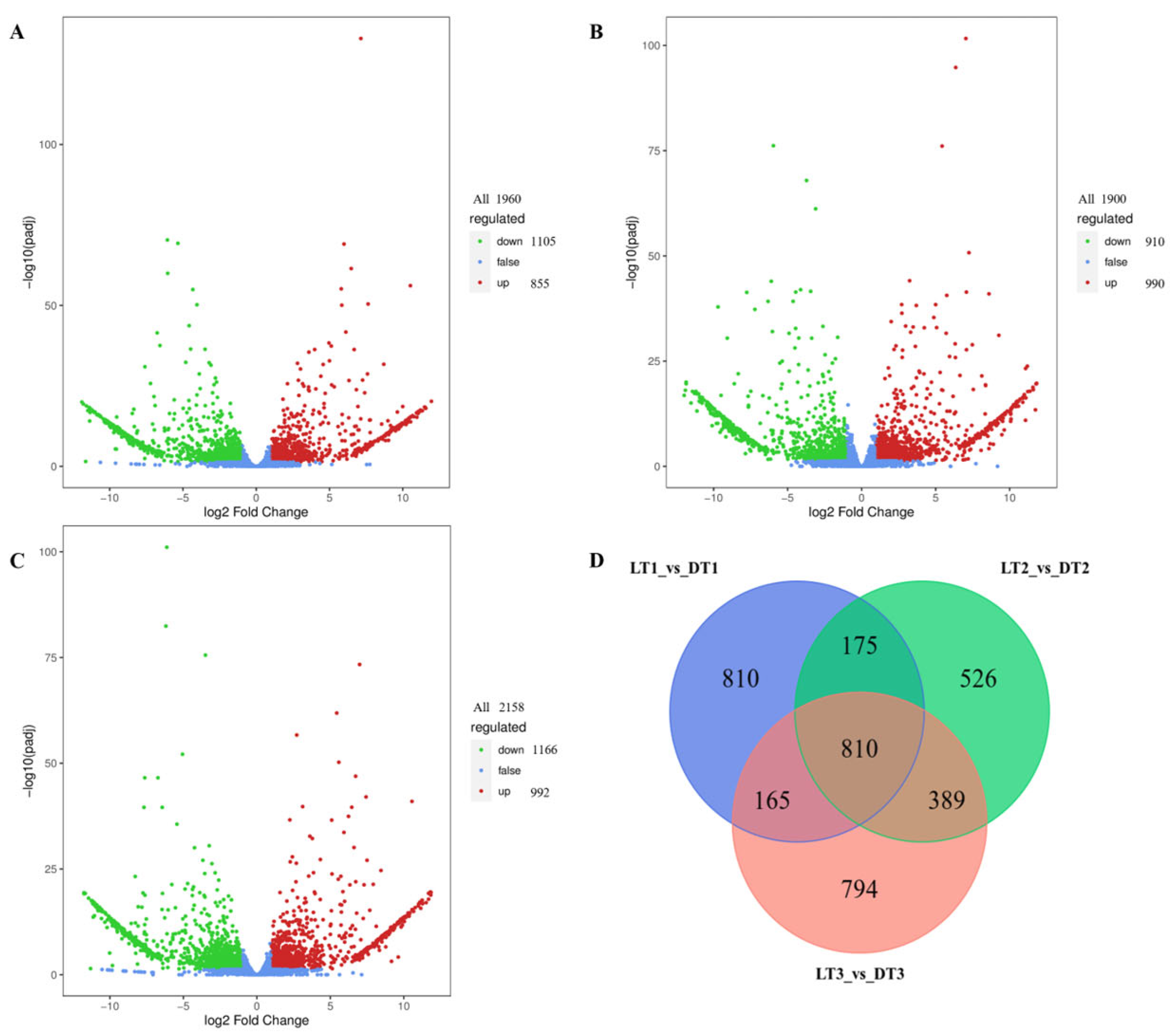
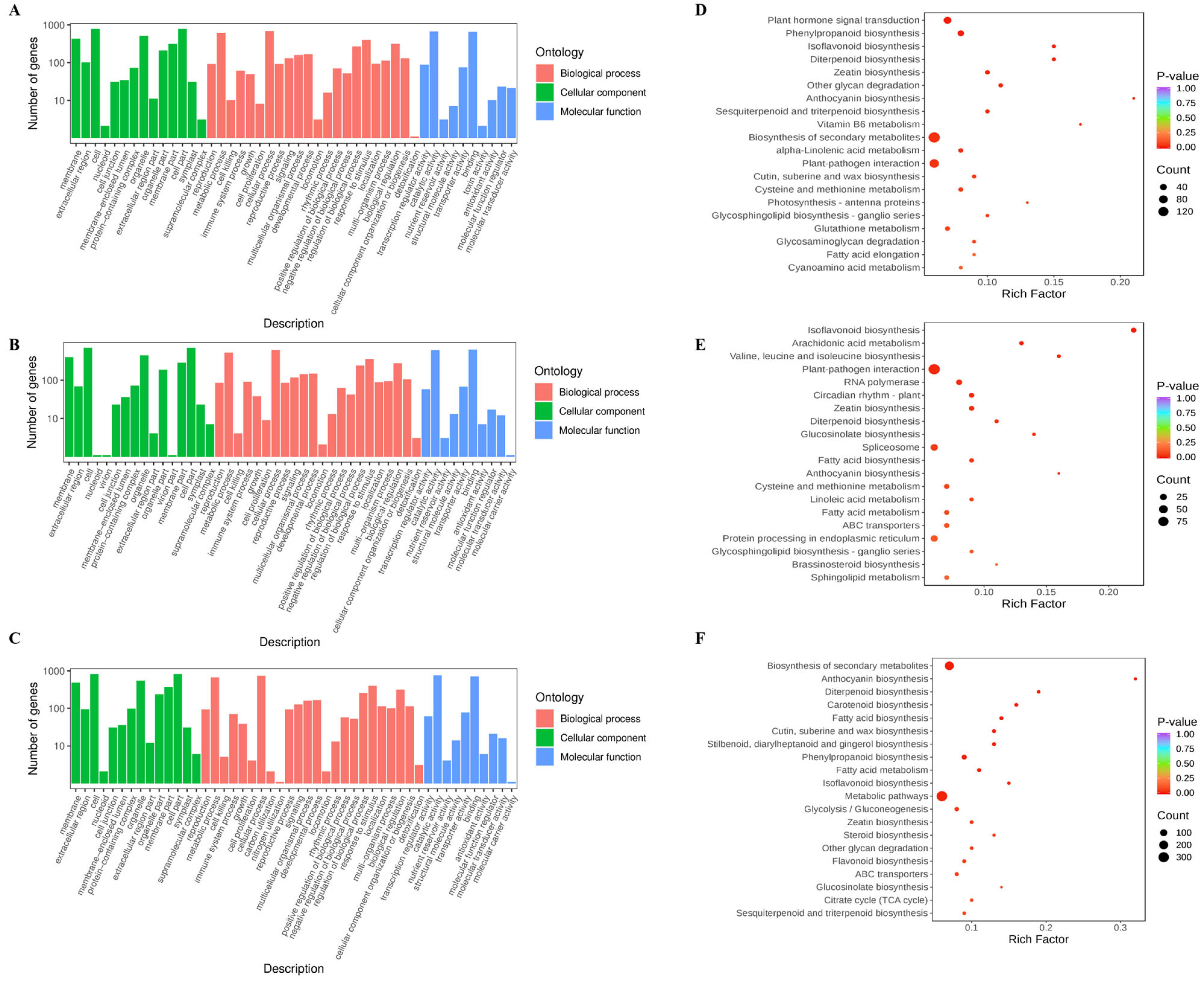
2.2. Transcriptome Analysis Results
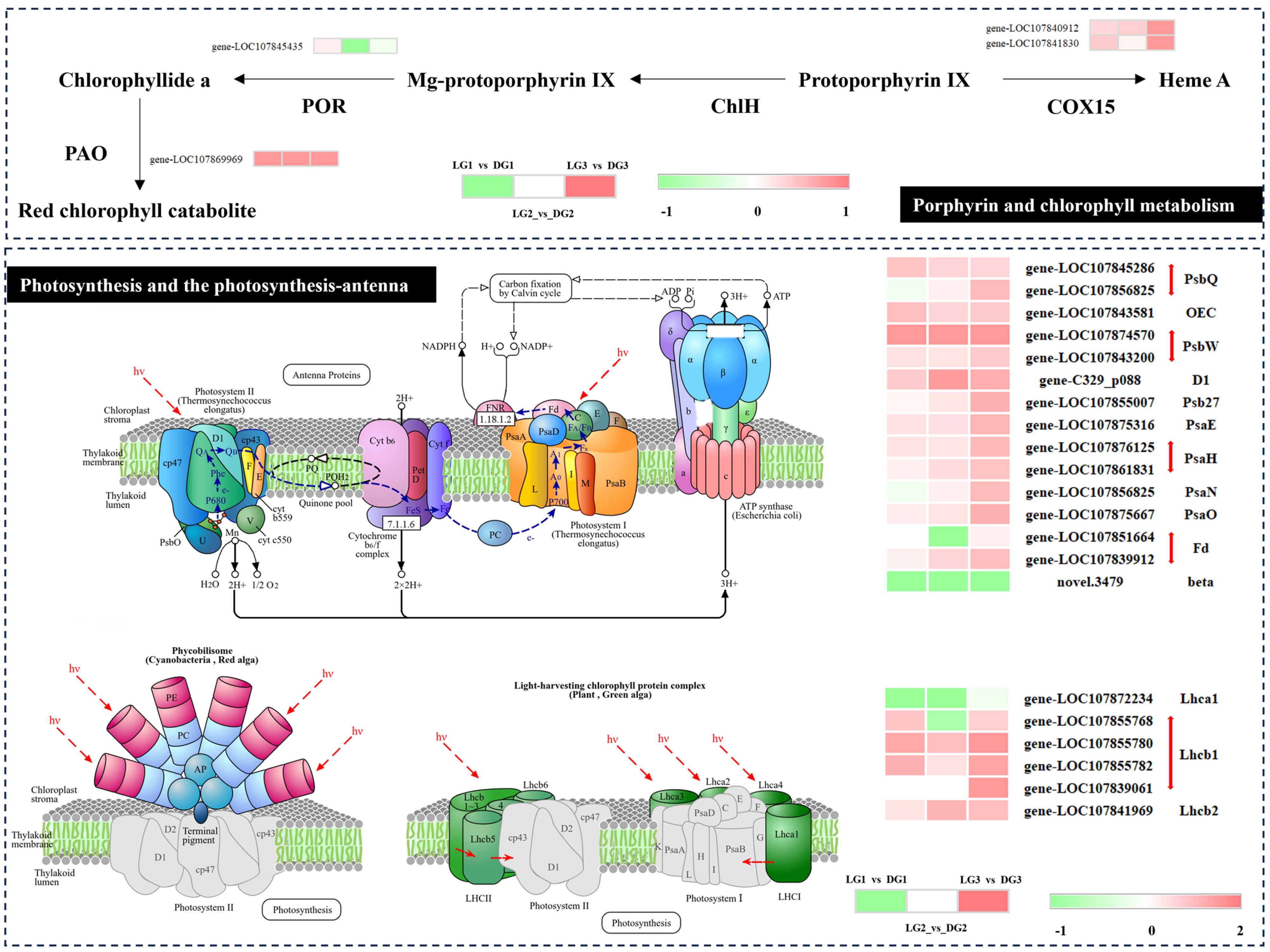
2.3. Chlorophyll Metabolism
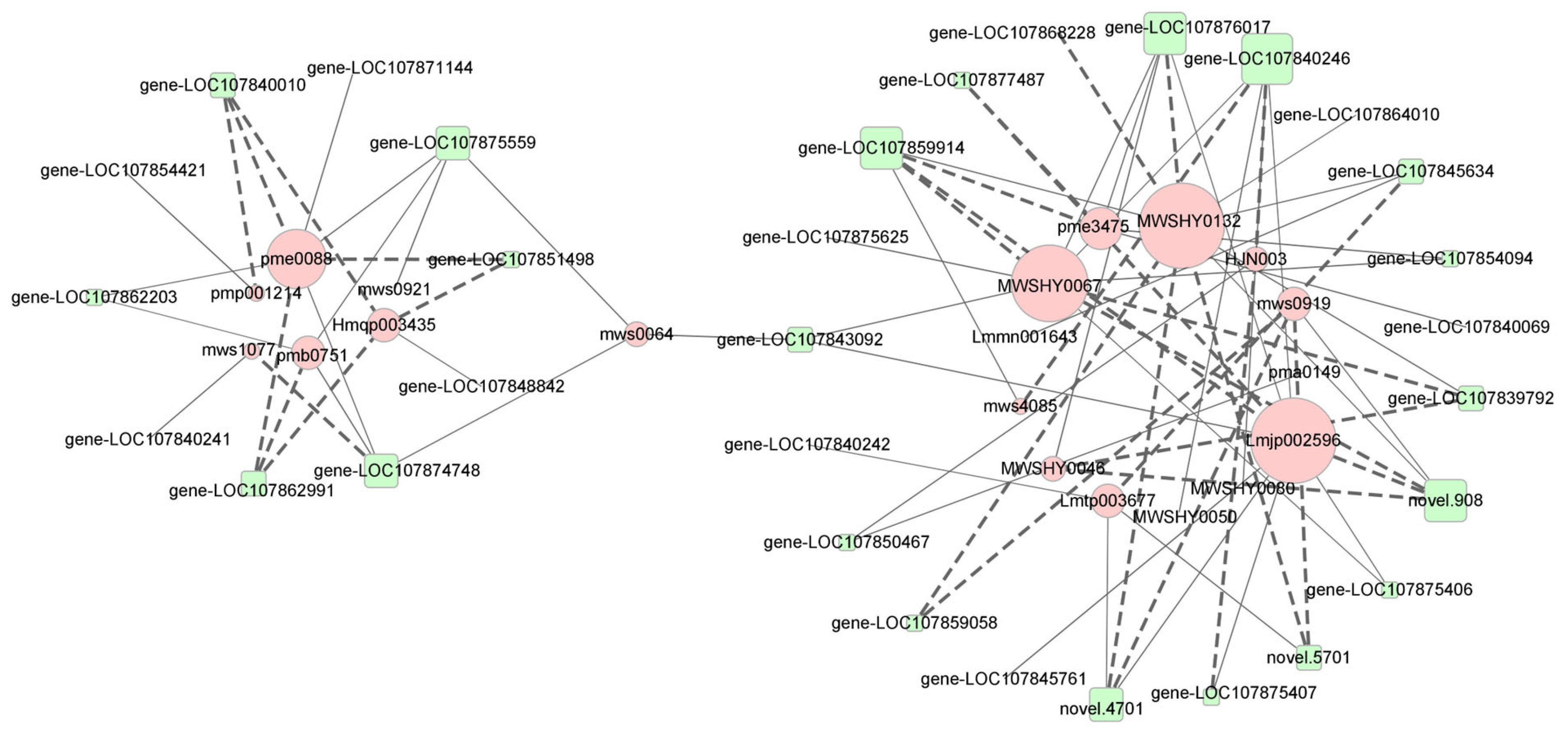
2.4. Correlation Analysis of Flavonoid Metabolism
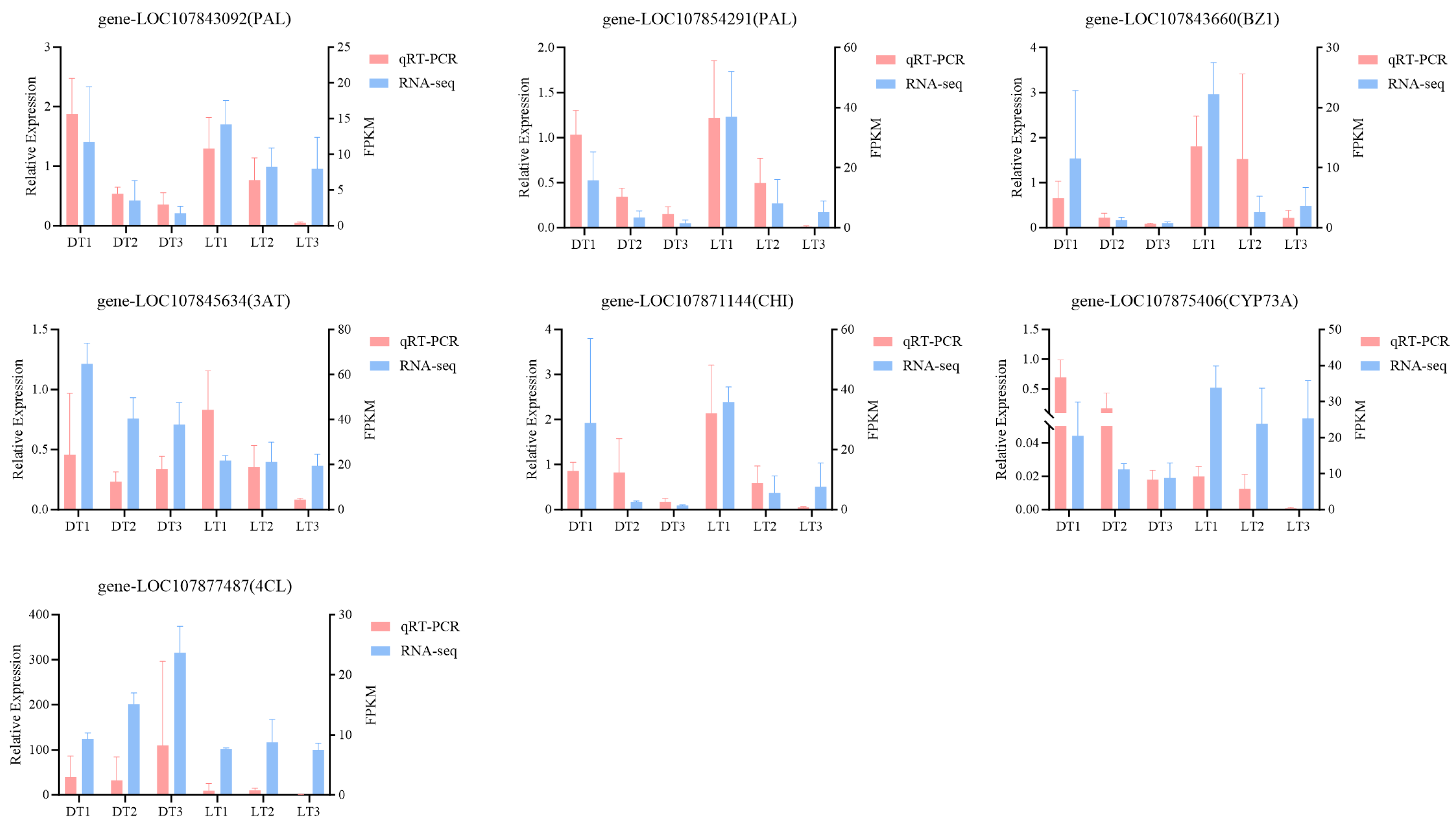
2.5. qRT-PCR Analysis
3. Discussion
4. Materials and Methods
4.1. Experimental Materials and Sampling
4.2. Metabolome Analysis
4.3. Transcriptome Results Analysis
4.4. Relevance Analysis
4.5. qRT-PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Paran, I.; van der Knaap, E. Genetic and molecular regulation of fruit and plant domestication traits in tomato and pepper. J. Exp. Bot. 2007, 58, 3841–3852. [Google Scholar] [CrossRef] [PubMed]
- Timberlake, C.F. Plant pigments for colouring food. Nutr. Bull. 1989, 14, 113–125. [Google Scholar] [CrossRef]
- Wang, L.; Zhong, Y.; Liu, J.; Ma, R.; Miao, Y.; Chen, W.; Zheng, J.; Pang, X.; Wan, H. Pigment Biosynthesis and Molecular Genetics of Fruit Color in Pepper. Plants 2023, 12, 2156. [Google Scholar] [CrossRef] [PubMed]
- Lightbourn, G.J.; Griesbach, R.J.; Novotny, J.A.; Clevidence, B.A.; Rao, D.D.; Stommel, J.R. Effects of Anthocyanin and Carotenoid Combinations on Foliage and Immature Fruit Color of Capsicum annuum L. J. Hered. 2008, 99, 105–111. [Google Scholar] [CrossRef]
- Borovsky, Y.; Paran, I. Chlorophyll breakdown during pepper fruit ripening in the chlorophyll retainer mutation is impaired at the homolog of the senescence-inducible stay-green gene. Theor. Appl. Genet. 2008, 117, 235–240. [Google Scholar] [CrossRef]
- Lai, B.; Hu, B.; Qin, Y.H.; Zhao, J.T.; Wang, H.C.; Hu, G.B. Transcriptomic analysis of Litchi chinensis pericarp during maturation with a focus on chlorophyll degradation and flavonoid biosynthesis. BMC Genom. 2015, 16, 225. [Google Scholar] [CrossRef]
- Wen, C.H.; Lin, S.S.; Chu, F.H. Transcriptome analysis of a subtropical deciduous tree: Autumn leaf senescence gene expression profile of Formosan gum. Plant Cell Physiol. 2015, 56, 163–174. [Google Scholar] [CrossRef]
- Mlodzinska, E. Survey of plant pigments: Molecular and environmental determinants of plant colors. Acta Biol. Cracoviensia Ser. Bot. 2009, 51, 7–16. [Google Scholar]
- Eckhardt, U.; Grimm, B.; Hörtensteiner, S. Recent advances in chlorophyll biosynthesis and breakdown in higher plants. Plant Mol. Biol. 2004, 56, 1–14. [Google Scholar] [CrossRef]
- Luo, F.; Cheng, S.-C.; Cai, J.-H.; Wei, B.-D.; Zhou, X.; Zhou, Q.; Zhao, Y.-B.; Ji, S.-J. Chlorophyll degradation and carotenoid biosynthetic pathways: Gene expression and pigment content in broccoli during yellowing. Food Chem. 2019, 297, 124964. [Google Scholar] [CrossRef]
- Bollivar, D.W. Recent advances in chlorophyll biosynthesis. Photosynth. Res. 2006, 90, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tang, W.; Ma, T.; Niu, D.; Jin, J.B.; Wang, H.; Lin, R. A pair of light signaling factors FHY3 and FAR1 regulates plant immunity by modulating chlorophyll biosynthesis. J. Integr. Plant Biol. 2016, 58, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Gu, T.; Khan, I.; Zada, A.; Jia, T. Research Progress in the Interconversion, Turnover and Degradation of Chlorophyll. Cells 2021, 10, 3134. [Google Scholar] [CrossRef] [PubMed]
- Hotta, Y.; Tanaka, T.; Takaoka, H.; Takeuchi, Y.; Konnai, M. Promotive effects of 5-aminolevulinic acid on the yield of several crops. Plant Growth Regul. 1997, 22, 109–114. [Google Scholar] [CrossRef]
- Rüdiger, W. Biosynthesis of chlorophyll b and the chlorophyll cycle. Photosynth. Res. 2002, 74, 187–193. [Google Scholar] [CrossRef]
- Li, C.; Ran, M.; Liu, J.; Wang, X.; Wu, Q.; Zhang, Q.; Yang, J.; Yi, F.; Zhang, H.; Zhu, J.-K.; et al. Functional analysis of CqPORB in the regulation of chlorophyll biosynthesis in Chenopodium quinoa. Front. Plant Sci. 2022, 13, 1083438. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, Y.; Zhang, L.; Wang, X.; Zhang, K.; Cheng, F.; Chen, S. Genetic Regulation of Chlorophyll Biosynthesis in Pepper Fruit: Roles of CaAPRR2 and CaGLK2. Genes 2025, 16, 219. [Google Scholar] [CrossRef]
- Yuan, M.; Yu-Qing, Z.; Zhong-Wei, Z.; Yang-Er, C.; Chun-Bang, D.; and Yuan, S. Light Regulates Transcription of Chlorophyll Biosynthetic Genes During Chloroplast Biogenesis. Crit. Rev. Plant Sci. 2017, 36, 35–54. [Google Scholar] [CrossRef]
- Santos, C.V. Regulation of chlorophyll biosynthesis and degradation by salt stress in sunflower leaves. Sci. Hortic. 2004, 103, 93–99. [Google Scholar] [CrossRef]
- Winkel-Shirley, B. Molecular genetics and control of anthocyanin expression. In Advances in Botanical Research; Academic Press: Cambridge, MA, USA, 2002; Volume 37, pp. 75–94. [Google Scholar]
- Huang, R.; Zhang, Y.; Shen, S.; Zhi, Z.; Cheng, H.; Chen, S.; Ye, X. Antioxidant and pancreatic lipase inhibitory effects of flavonoids from different citrus peel extracts: An in vitro study. Food Chem. 2020, 326, 126785. [Google Scholar] [CrossRef]
- Goulas, V.; Manganaris, G.A. Exploring the phytochemical content and the antioxidant potential of Citrus fruits grown in Cyprus. Food Chem. 2012, 131, 39–47. [Google Scholar] [CrossRef]
- Li, Y.; Fang, J.; Qi, X.; Lin, M.; Zhong, Y.; Sun, L.; Cui, W. Combined Analysis of the Fruit Metabolome and Transcriptome Reveals Candidate Genes Involved in Flavonoid Biosynthesis in Actinidia arguta. Int. J. Mol. Sci. 2018, 19, 1471. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, M.K.; Murrell, J.R.; Shirley, B.W. Characterization of flavonol synthase and leucoanthocyanidin dioxygenase genes in Arabidopsis. Further evidence for differential regulation of “early” and “late” genes. Plant Physiol. 1997, 113, 1437–1445. [Google Scholar] [CrossRef]
- Falcone Ferreyra, M.L.; Rius, S.P.; Casati, P. Flavonoids: Biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 2012, 3, 222. [Google Scholar] [CrossRef]
- Yonekura-Sakakibara, K.; Higashi, Y.; Nakabayashi, R. The Origin and Evolution of Plant Flavonoid Metabolism. Front. Plant Sci. 2019, 10, 943. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, J.; Yi, C.; Chen, F.; Liu, Y.; Liao, Y.; Zhang, Z.; Liu, W.; Lv, J. Integrative analysis of non-targeted metabolome and transcriptome reveals the mechanism of volatile formation in pepper fruit. Front. Genet. 2023, 14, 1290492. [Google Scholar] [CrossRef]
- Gong, C.; Guo, G.; Pan, B.; Gao, C.; Liu, J.; Wang, S.; Diao, W. Integrating transcriptome and metabolome to explore the formation of fruit aroma in different types of pepper. Food Biosci. 2024, 62, 105157. [Google Scholar] [CrossRef]
- Zhang, Y.; Shu, H.; Mumtaz, M.A.; Hao, Y.; Li, L.; He, Y.; Jin, W.; Li, C.; Zhou, Y.; Lu, X.; et al. Transcriptome and Metabolome Analysis of Color Changes during Fruit Development of Pepper (Capsicum baccatum). Int. J. Mol. Sci. 2022, 23, 12524. [Google Scholar] [CrossRef]
- Liu, Y.; Lv, J.; Liu, Z.; Wang, J.; Yang, B.; Chen, W.; Ou, L.; Dai, X.; Zhang, Z.; Zou, X. Integrative analysis of metabolome and transcriptome reveals the mechanism of color formation in pepper fruit (Capsicum annuum L.). Food Chem. 2020, 306, 125629. [Google Scholar] [CrossRef]
- Lv, J.; Zhang, R.; Mo, Y.; Zhou, H.; Li, M.; Wu, R.; Cheng, H.; Zhang, M.; Wang, H.; Hua, W.; et al. Integrative Metabolome and Transcriptome Analyses Provide Insights into Carotenoid Variation in Different-Colored Peppers. Int. J. Mol. Sci. 2023, 24, 16563. [Google Scholar] [CrossRef]
- Wang, J.; Lv, J.; Liu, Z.; Liu, Y.; Song, J.; Ma, Y.; Ou, L.; Zhang, X.; Liang, C.; Wang, F.; et al. Integration of Transcriptomics and Metabolomics for Pepper (Capsicum annuum L.) in Response to Heat Stress. Int. J. Mol. Sci. 2019, 20, 5042. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Zhou, K.-H.; Chen, X.-J.; Huang, Y.-Q.; Yuan, X.-J.; Li, G.-G.; Xie, Y.-Y.; Fang, R. Transcriptome and metabolome analyses revealed the response mechanism of pepper roots to Phytophthora capsici infection. BMC Genom. 2023, 24, 626. [Google Scholar] [CrossRef] [PubMed]
- Wahyuni, Y.; Ballester, A.-R.; Sudarmonowati, E.; Bino, R.J.; Bovy, A.G. Metabolite biodiversity in pepper (Capsicum) fruits of thirty-two diverse accessions: Variation in health-related compounds and implications for breeding. Phytochemistry 2011, 72, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Matsufuji, H.; Ishikawa, K.; Nunomura, O.; Chino, M.; Takeda, M. Anti-oxidant content of different coloured sweet peppers, white, green, yellow, orange and red (Capsicum annuum L.). Food Sci. Technol. 2007, 42, 1482–1488. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, Z.; Chen, W.; Liang, C.; Li, X.; Liu, Z.; Cui, Q.; Ma, Y.; Zou, X. Fine-mapping and transcriptome analysis of the photosensitive leaf -yellowing gene CaLY1 in pepper (Capsicum annuum L.). Hortic. Plant J. 2023, 9, 122–132. [Google Scholar] [CrossRef]
- Pan, Y.; Bradley, G.; Pyke, K.; Ball, G.; Lu, C.; Fray, R.; Marshall, A.; Jayasuta, S.; Baxter, C.; van Wijk, R.; et al. Network Inference Analysis Identifies an APRR2-Like Gene Linked to Pigment Accumulation in Tomato and Pepper Fruits. Plant Physiol. 2013, 161, 1476–1485. [Google Scholar] [CrossRef]
- Fitter, D.W.; Martin, D.J.; Copley, M.J.; Scotland, R.W.; Langdale, J.A. GLK gene pairs regulate chloroplast development in diverse plant species. Plant J. 2002, 31, 713–727. [Google Scholar] [CrossRef]
- Matile, P.; Hörtensteiner, S.; Thomas, H. Chlorophyll degradation. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1999, 50, 67–95. [Google Scholar] [CrossRef]
- Pruzinská, A.; Tanner, G.; Aubry, S.; Anders, I.; Moser, S.; Muller, T.; Ongania, K.-H.; Kräutler, B.; Youn, J.-Y.; Liljegren, S.J.; et al. Chlorophyll Breakdown in Senescent Arabidopsis Leaves. Characterization of Chlorophyll Catabolites and of Chlorophyll Catabolic Enzymes Involved in the Degreening Reaction. Plant Physiol. 2005, 139, 52–63. [Google Scholar] [CrossRef]
- Jansson, S. A guide to the Lhc genes and their relatives in Arabidopsis. Trends Plant Sci. 1999, 4, 236–240. [Google Scholar] [CrossRef]
- Jansson, S. The light-harvesting chlorophyll ab-binding proteins. Biochim. Biophys. Acta (BBA)—Bioenerg. 1994, 1184, 1–19. [Google Scholar] [CrossRef]
- Liu, Z.F.; Yan, H.C.; Wang, K.B.; Kuang, T.Y.; Zhang, J.P.; Gui, L.L.; An, X.M.; Chang, W.R. Crystal structure of spinach major light-harvesting complex at 2.72 A resolution. Nature 2004, 428, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Han, S.A.; Xie, H.; Wang, M.; Zhang, J.G.; Xu, Y.H.; Zhu, X.H.; Caikasimu, A.; Zhou, X.W.; Mai, S.L.; Pan, M.Q.; et al. Transcriptome and metabolome reveal the effects of three canopy types on the flavonoids and phenolic acids in ‘Merlot’ (Vitis vinifera L.) berry pericarp. Food Res. Int. 2023, 163, 112196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tian, C.P.; Zhang, Y.; Li, C.Z.Y.; Li, X.; Yu, Q.; Wang, S.; Wang, X.Y.; Chen, X.S.; Feng, S.Q. Transcriptomic and metabolomic analysis provides insights into anthocyanin and procyanidin accumulation in pear. BMC Plant Biol. 2020, 20, 129. [Google Scholar] [CrossRef]
- Wang, Z.R.; Cui, Y.Y.; Vainstein, A.; Chen, S.W.; Ma, H.Q. Regulation of Fig (Ficus carica L.) Fruit Color: Metabolomic and Transcriptomic Analyses of the Flavonoid Biosynthetic Pathway. Front. Plant Sci. 2017, 8, 1990. [Google Scholar] [CrossRef]
- Dwibedi, V.; Jain, S.; Singhal, D.; Mittal, A.; Rath, S.K.; Saxena, S. Inhibitory activities of grape bioactive compounds against enzymes linked with human diseases. Appl. Microbiol. Biotechnol. 2022, 106, 1399–1417. [Google Scholar] [CrossRef]
- Wang, F.; Huang, Y.; Wu, W.; Zhu, C.; Zhang, R.; Chen, J.; Zeng, J. Metabolomics Analysis of the Peels of Different Colored Citrus Fruits (Citrus reticulata cv. ‘Shatangju’) During the Maturation Period Based on UHPLC-QQQ-MS. Molecules 2020, 25, 396. [Google Scholar] [CrossRef]
- Zhang, A.; Zheng, J.; Chen, X.; Shi, X.; Wang, H.; Fu, Q. Comprehensive Analysis of Transcriptome and Metabolome Reveals the Flavonoid Metabolic Pathway Is Associated with Fruit Peel Coloration of Melon. Molecules 2021, 26, 2830. [Google Scholar] [CrossRef]
- Wang, M.; Chen, L.; Liang, Z.; He, X.; Liu, W.; Jiang, B.; Yan, J.; Sun, P.; Cao, Z.; Peng, Q.; et al. Metabolome and transcriptome analyses reveal chlorophyll and anthocyanin metabolism pathway associated with cucumber fruit skin color. BMC Plant biology 2020, 20, 386. [Google Scholar] [CrossRef]
- Hildreth, S.B.; Littleton, E.S.; Clark, L.C.; Puller, G.C.; Kojima, S.; Winkel, B.S.J. Mutations that alter Arabidopsis flavonoid metabolism affect the circadian clock. Plant J. 2022, 110, 932–945. [Google Scholar] [CrossRef]
- Ma, X.; Yu, Y.-N.; Jia, J.-H.; Li, Q.-H.; Gong, Z.-H. The pepper MYB transcription factor CaMYB306 accelerates fruit coloration and negatively regulates cold resistance. Sci. Hortic. 2022, 295, 110892. [Google Scholar] [CrossRef]
- Singh, P.; Arif, Y.; Bajguz, A.; Hayat, S. The role of quercetin in plants. Plant Physiol. Biochem. 2021, 166, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.; Mestre, T.C.; Rubio, F.; Girones-Vilaplana, A.; Moreno, D.A.; Mittler, R.; Rivero, R.M. Accumulation of Flavonols over Hydroxycinnamic Acids Favors Oxidative Damage Protection under Abiotic Stress. Front. Plant Sci. 2016, 7, 838. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, M.; Wen, J.; Ren, Y.; Shao, D.; Wang, Y.; Li, J.; Li, Q. Metabolic Pathways and Molecular Regulatory Mechanisms of Fruit Color Change During Greening Stage of Peppers (Capsicum annuum L.). Int. J. Mol. Sci. 2025, 26, 4508. https://doi.org/10.3390/ijms26104508
Wei M, Wen J, Ren Y, Shao D, Wang Y, Li J, Li Q. Metabolic Pathways and Molecular Regulatory Mechanisms of Fruit Color Change During Greening Stage of Peppers (Capsicum annuum L.). International Journal of Molecular Sciences. 2025; 26(10):4508. https://doi.org/10.3390/ijms26104508
Chicago/Turabian StyleWei, Mengyuan, Junqin Wen, Yanjing Ren, Dengkui Shao, Yayi Wang, Jiang Li, and Quanhui Li. 2025. "Metabolic Pathways and Molecular Regulatory Mechanisms of Fruit Color Change During Greening Stage of Peppers (Capsicum annuum L.)" International Journal of Molecular Sciences 26, no. 10: 4508. https://doi.org/10.3390/ijms26104508
APA StyleWei, M., Wen, J., Ren, Y., Shao, D., Wang, Y., Li, J., & Li, Q. (2025). Metabolic Pathways and Molecular Regulatory Mechanisms of Fruit Color Change During Greening Stage of Peppers (Capsicum annuum L.). International Journal of Molecular Sciences, 26(10), 4508. https://doi.org/10.3390/ijms26104508