
Mediating Role of the ANGPTL3/TFPI Protein Ratio in Regulating T-Cell Surface Glycoprotein CD5 Levels on Knee Osteoarthritis (KOA): A Mendelian Randomization Study




Abstract
1. Introduction
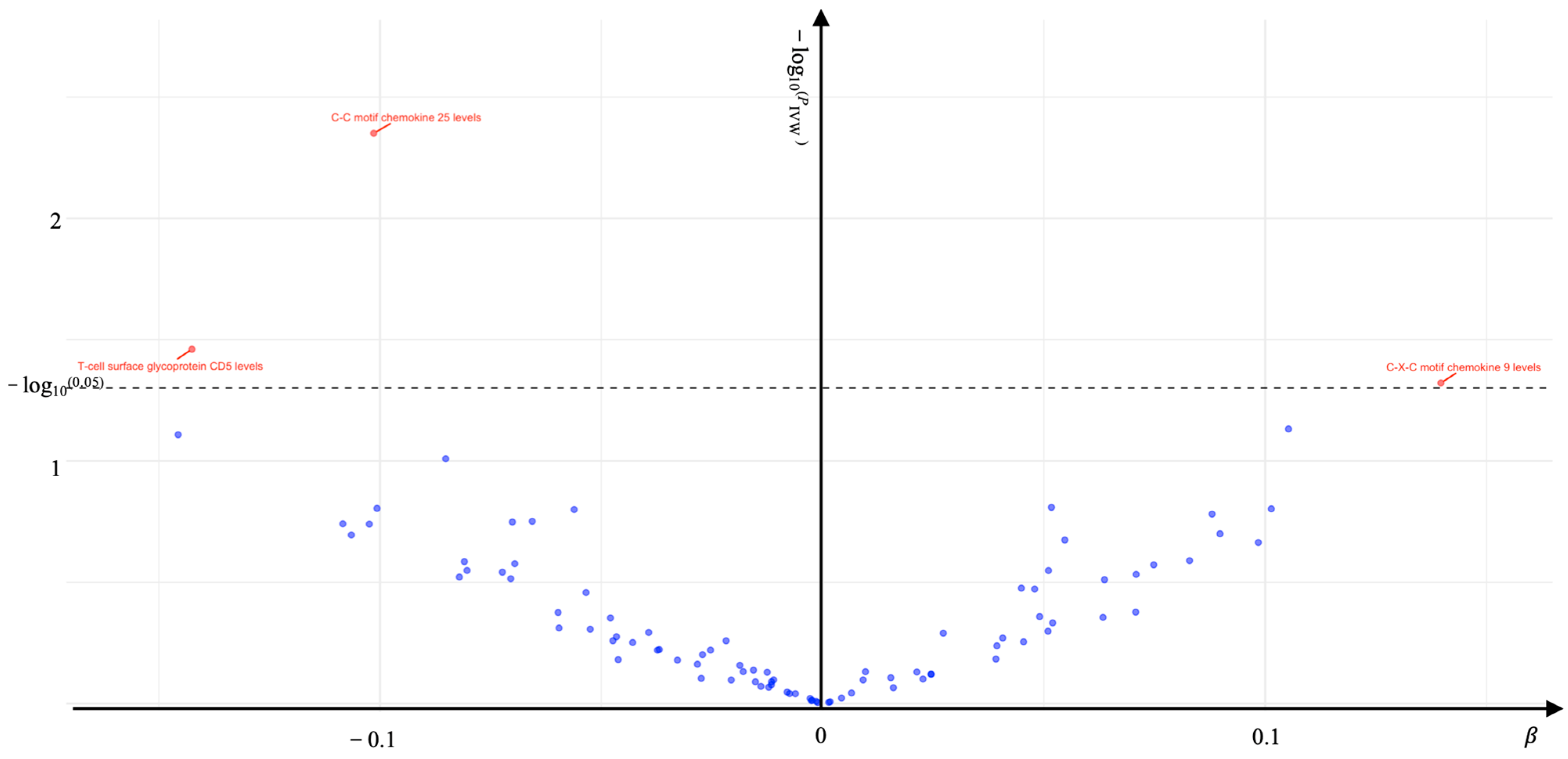
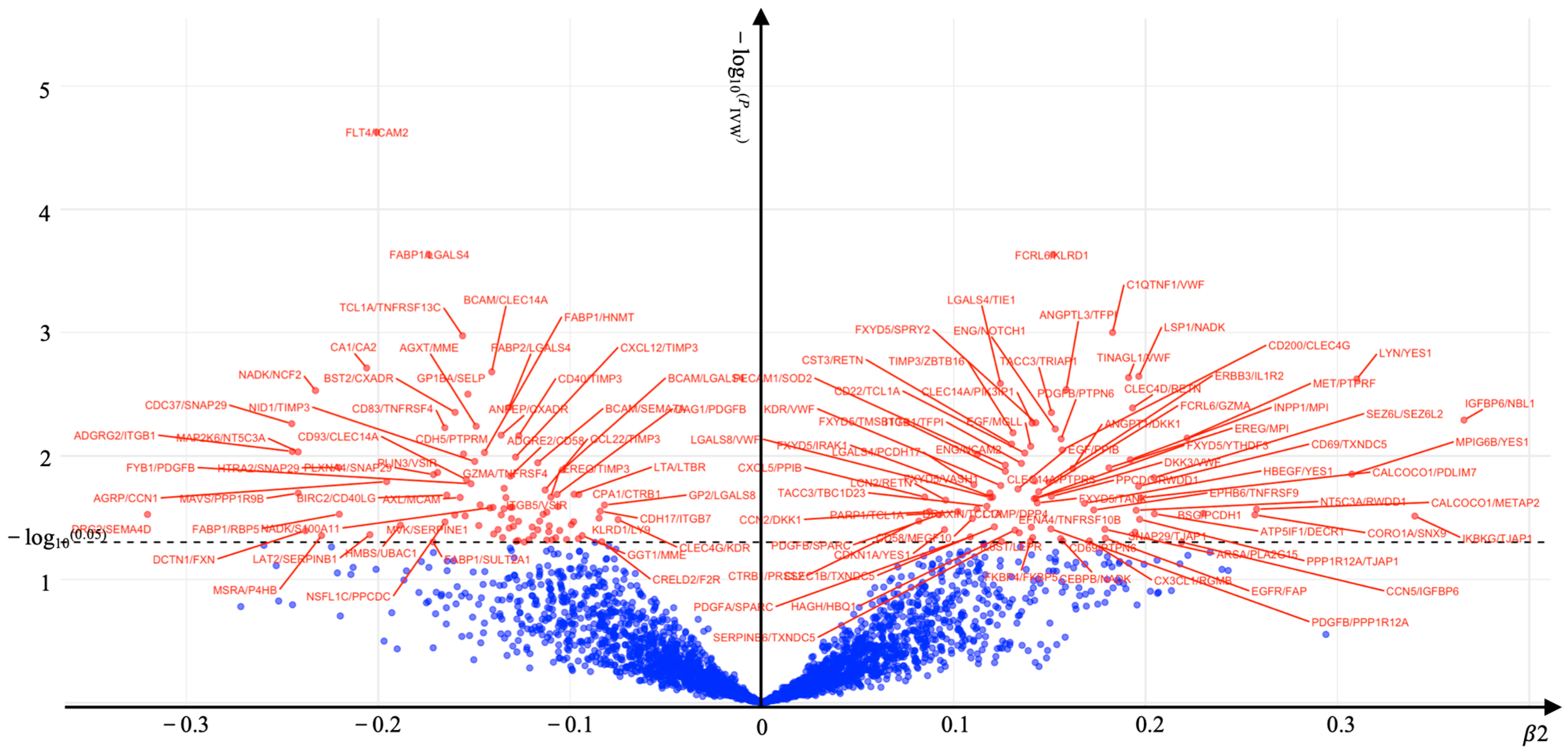
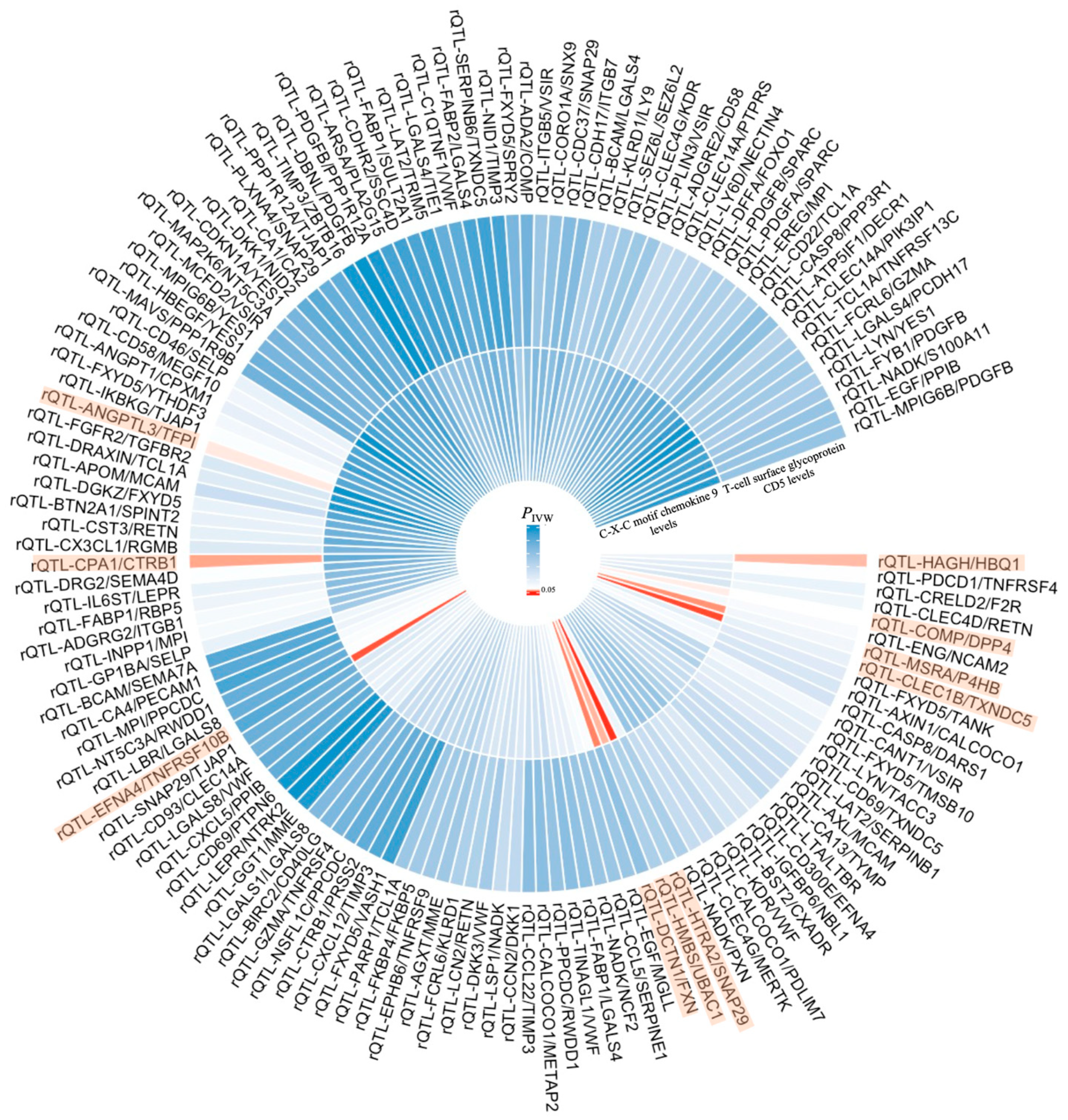
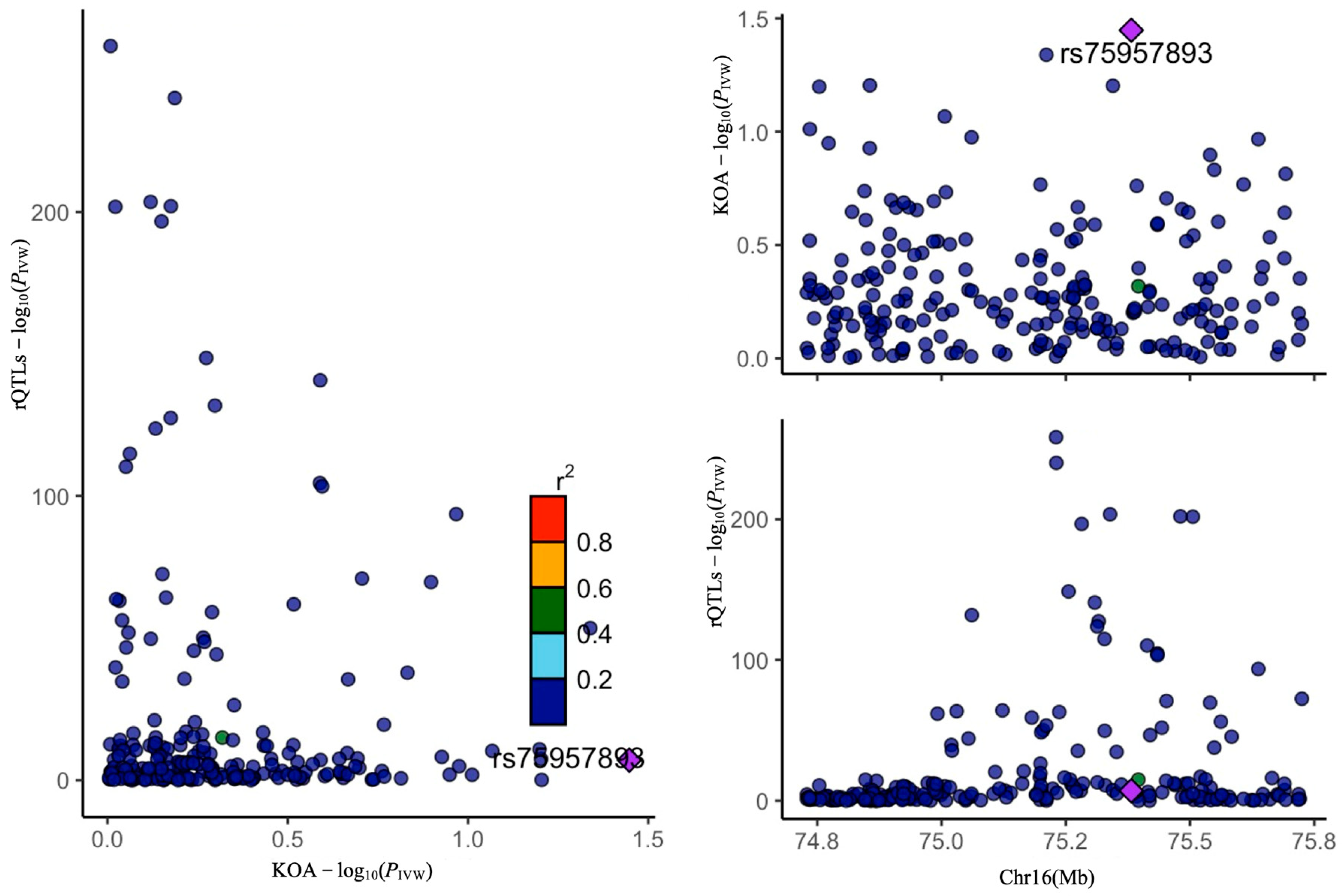
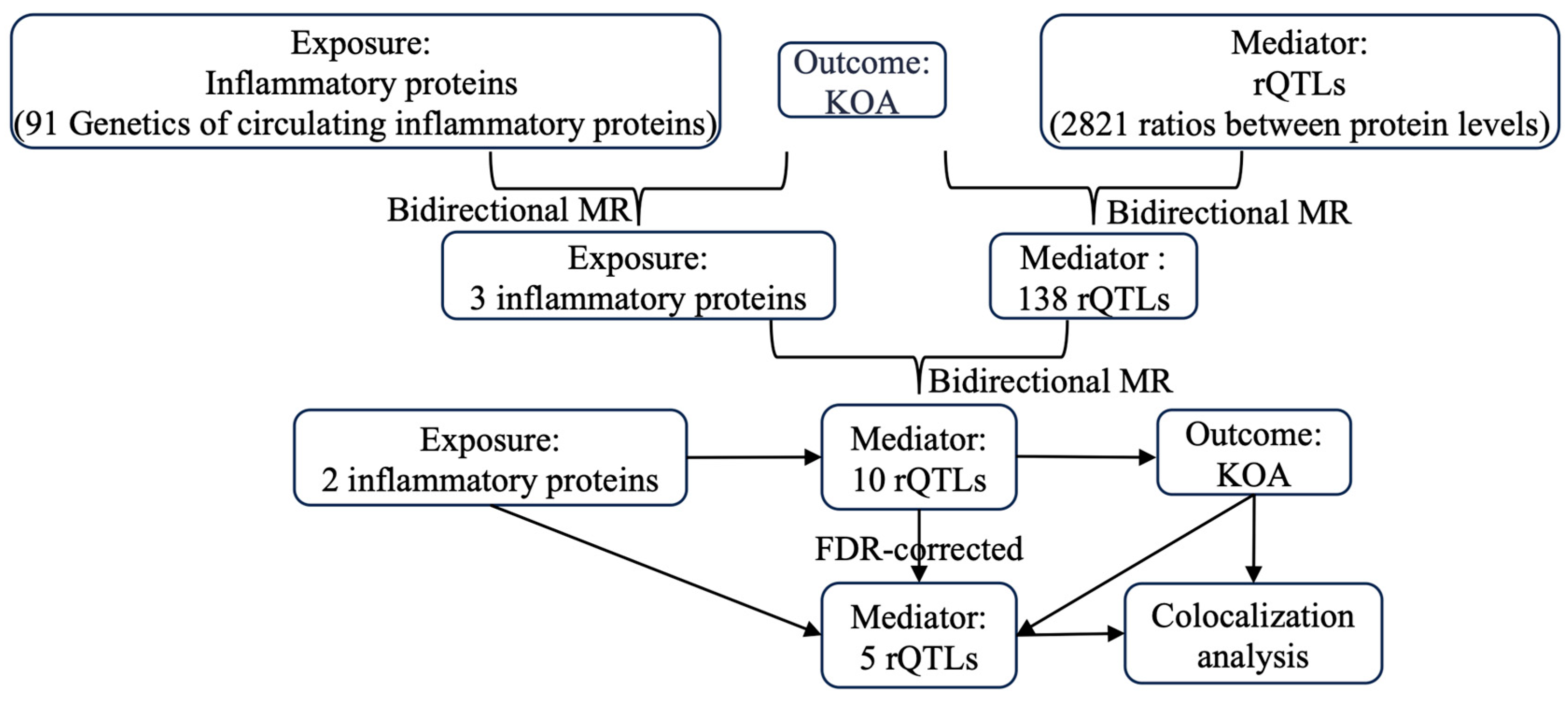
2. Results
3. Discussion
3.1. The Impact of T-Cell Surface Glycoprotein CD5 Levels on KOA
3.1.1. The Total Effect (T-Cell Surface Glycoprotein CD5 Levels Were Negatively Correlated with KOA (β = −0.143))
3.1.2. The Mediating Effect of rQTL-ANGPTL3/TFPI
3.1.3. The Mediating Effect of rQTL-CPA1/CTRB1
3.1.4. The Mediating Effect of rQTL-HAGH/HBQ1
3.2. The Impact of C-X-C Motif Chemokine 9 Levels on KOA
3.2.1. The Total Effect (C-X-C Motif Chemokine 9 Levels Were Positively Correlated with KOA (β = 0.140))
3.2.2. The Mediating Effect of rQTL-MSRA/P4HB
3.2.3. The Mediating Effect of rQTL-COMP/DPP4
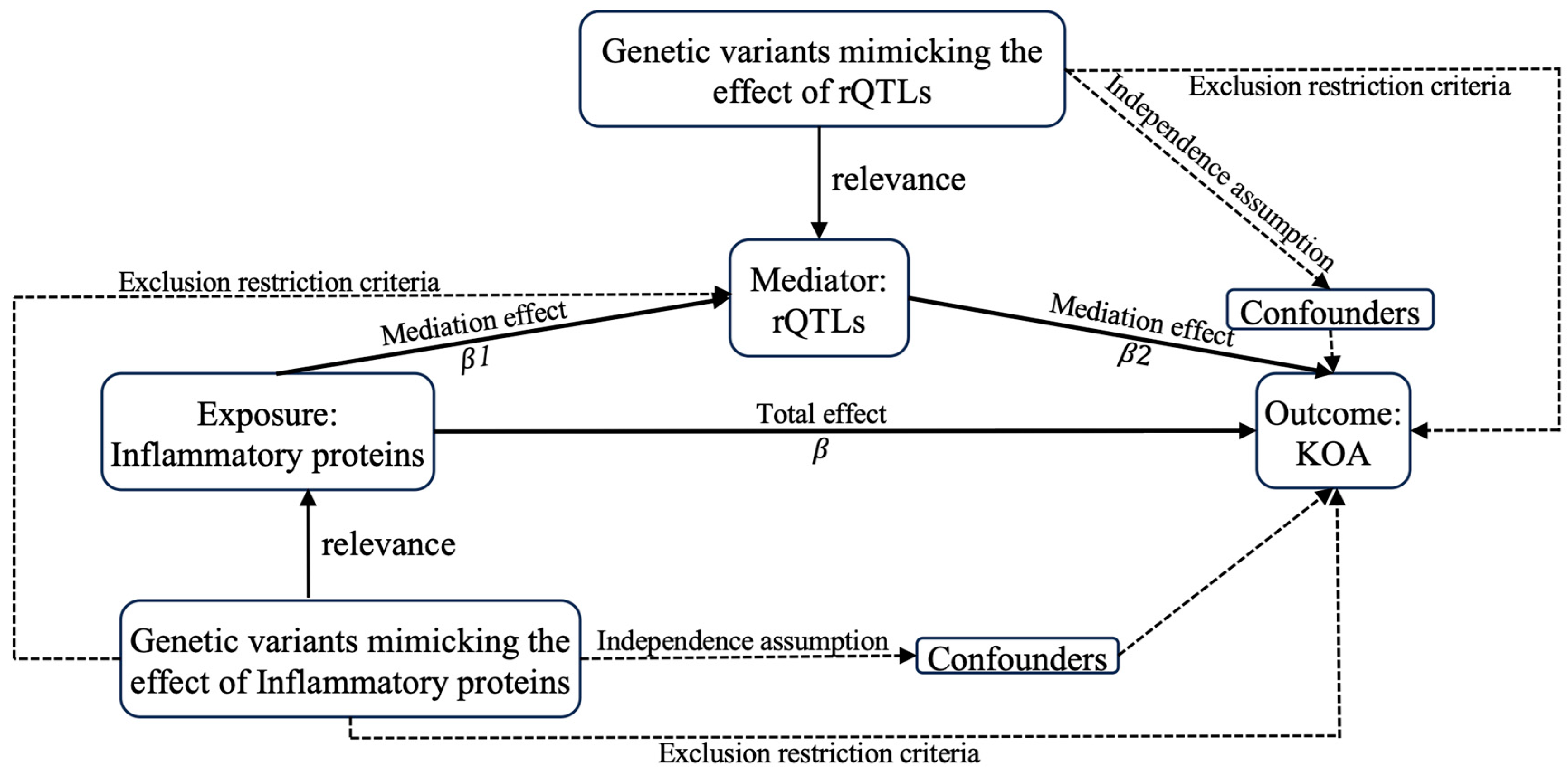
4. Methods and Materials
4.1. Data
4.1.1. Exposure Data
4.1.2. Mediator Data
4.1.3. Outcome Data
4.2. Statistics
4.2.1. MR Analysis
Forward MR Analysis
Reverse MR Analysis
4.2.2. Colocalization Analysis
4.2.3. False Discovery Rate Corrected (FDR-Corrected)
4.2.4. Mediation Analysis
5. Conclusions
Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, Z.; Alexander, P.G.; Ocasio-Nieves, B.D.; Yocum, L.; Lin, H.; Tuan, R.S. Pathogenesis of osteoarthritis: Risk factors, regulatory pathways in chondrocytes, and experimental models. Biology 2020, 9, 194. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Liu, L.; Li, J.; Zhao, G.; Cai, Y.; Dong, Y.; Wang, J.; Wu, S. Prevalence and factors associated with knee osteoarthritis among middle-aged and elderly individuals in rural Tianjin: A population-based cross-sectional study. J. Orthop. Surg. Res. 2023, 18, 266. [Google Scholar] [CrossRef] [PubMed]
- Cui, A.; Li, H.; Wang, D.; Zhong, J.; Chen, Y.; Lu, H. Global, regional prevalence, incidence and risk factors of knee osteoarthritis in population-based studies. eClinicalMedicine 2020, 29, 100587. [Google Scholar] [CrossRef]
- Vos, T.; Flaxman, A.D.; Naghavi, M.; Lozano, R.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; Aboyans, V. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2163–2196. [Google Scholar] [CrossRef]
- Krakowski, P.; Rejniak, A.; Sobczyk, J.; Karpiński, R. In Cartilage integrity: A review of mechanical and frictional properties and repair approaches in osteoarthritis. Healthcare 2024, 12, 1648. [Google Scholar] [CrossRef]
- Sharma, L. Osteoarthritis of the knee. N. Engl. J. Med. 2021, 384, 51–59. [Google Scholar] [CrossRef]
- Eckstein, F.; Maschek, S.; Wirth, W.; Hudelmaier, M.; Hitzl, W.; Wyman, B.; Nevitt, M.; Le Graverand, M.H.; Group, O.I. One year change of knee cartilage morphology in the first release of participants from the Osteoarthritis Initiative progression subcohort: Association with sex, body mass index, symptoms and radiographic osteoarthritis status. Ann. Rheum. Dis. 2009, 68, 674–679. [Google Scholar] [CrossRef]
- Lv, Z.; Yang, Y.X.; Li, J.; Fei, Y.; Guo, H.; Sun, Z.; Lu, J.; Xu, X.; Jiang, Q.; Ikegawa, S. Molecular classification of knee osteoarthritis. Front. Cell Dev. Biol. 2021, 9, 725568. [Google Scholar] [CrossRef]
- Chen, X.; Gong, W.; Shao, X.; Shi, T.; Zhang, L.; Dong, J.; Shi, Y.; Shen, S.; Qin, J.; Jiang, Q. METTL3-mediated m6A modification of ATG7 regulates autophagy-GATA4 axis to promote cellular senescence and osteoarthritis progression. Ann. Rheum. Dis. 2022, 81, 85–97. [Google Scholar] [CrossRef]
- Cuollo, L.; Antonangeli, F.; Santoni, A.; Soriani, A. The senescence-associated secretory phenotype (SASP) in the challenging future of cancer therapy and age-related diseases. Biology 2020, 9, 485. [Google Scholar] [CrossRef]
- Geng, N.; Xian, M.; Deng, L.; Kuang, B.; Pan, Y.; Liu, K.; Ye, Y.; Fan, M.; Bai, Z.; Guo, F. Targeting the senescence-related genes MAPK12 and FOS to alleviate osteoarthritis. J. Orthop. Transl. 2024, 47, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Chen, T.; Qiu, J.; Gao, W.; Qiu, X.; Zhu, Y.; Wang, X.; Chen, Y.; Zhou, H.; Deng, Z. Inhibition of nuclear receptor RORα attenuates cartilage damage in osteoarthritis by modulating IL-6/STAT3 pathway. Cell Death Dis. 2021, 12, 886. [Google Scholar] [CrossRef] [PubMed]
- Vermeij, E.A.; Broeren, M.G.; Bennink, M.B.; Arntz, O.J.; Gjertsson, I.; van Lent, P.L.; van den Berg, W.B.; Koenders, M.I.; van de Loo, F.A. Disease-regulated local IL-10 gene therapy diminishes synovitis and cartilage proteoglycan depletion in experimental arthritis. Ann. Rheum. Dis. 2015, 74, 2084–2091. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Long, H.; Chen, F.; Yu, Y. Oxoglaucine mediates Ca2+ influx and activates autophagy to alleviate osteoarthritis through the TRPV5/calmodulin/CAMK-II pathway. Br. J. Pharmacol. 2021, 178, 2931–2947. [Google Scholar] [CrossRef]
- Fan, A.; Wu, G.; Wang, J.; Lu, L.; Wang, J.; Wei, H.; Sun, Y.; Xu, Y.; Mo, C.; Zhang, X. Inhibition of fibroblast activation protein ameliorates cartilage matrix degradation and osteoarthritis progression. Bone Res. 2023, 11, 3. [Google Scholar] [CrossRef]
- Lyu, Y.; Deng, H.; Qu, C.; Qiao, L.; Liu, X.; Xiao, X.; Liu, J.; Guo, Z.; Zhao, Y.; Han, J. Identification of proteins and N-glycosylation sites of knee cartilage in Kashin-Beck disease compared with osteoarthritis. Int. J. Biol. Macromol. 2022, 210, 128–138. [Google Scholar] [CrossRef]
- Yu, H.; Ye, W.B.; Zhong, Z.M.; Ding, R.T.; Chen, J.T. Effect of advanced oxidation protein products on articular cartilage and synovium in a rabbit osteoarthritis model. Orthop. Surg. 2015, 7, 161–167. [Google Scholar] [CrossRef]
- Wen, Z.-Q.; Liu, D.; Zhang, Y.; Cai, Z.-J.; Xiao, W.-F.; Li, Y.-S. G protein-coupled receptors in osteoarthritis: A novel perspective on pathogenesis and treatment. Front. Cell Dev. Biol. 2021, 9, 758220. [Google Scholar] [CrossRef]
- Sheinenzon, A.; Shehadeh, M.; Michelis, R.; Shaoul, E.; Ronen, O. Serum albumin levels and inflammation. Int. J. Biol. Macromol. 2021, 184, 857–862. [Google Scholar] [CrossRef]
- Sedik, A.A.; Salama, M.; Fathy, K.; Salama, A. Cold plasma approach fortifies the topical application of thymoquinone intended for wound healing via up-regulating the levels of TGF-ß, VEGF, and α-SMA in rats. Int. Immunopharmacol. 2023, 122, 110634. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wei, J.; Li, R.; Fu, R.; Han, P.; Wang, H.; Zhang, G.; Li, S.; Chen, S.; Liu, Z. Tyrosine kinase receptor B attenuates liver fibrosis by inhibiting TGF-β/SMAD signaling. Hepatology 2023, 78, 1433–1447. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-S.; Luo, W.; Zhu, S.-A.; Lei, G.-H. T cells in osteoarthritis: Alterations and beyond. Front. Immunol. 2017, 8, 356. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Qiu, L.; Ye, Z.; Tan, X.; Xu, X.; Lu, M.; Kuang, G. The role of Th/Treg immune cells in osteoarthritis. Front. Immunol. 2024, 15, 1393418. [Google Scholar] [CrossRef]
- Wang, L.; Liu, G.; Zheng, L.; Long, H.; Liu, Y. A new era of gene and cell therapy for cancer: A narrative review. Ann. Transl. Med. 2023, 11, 321. [Google Scholar] [CrossRef]
- Balakrishnan, L.; Bhattacharjee, M.; Ahmad, S.; Nirujogi, R.S.; Renuse, S.; Subbannayya, Y.; Marimuthu, A.; Srikanth, S.M.; Raju, R.; Dhillon, M. Differential proteomic analysis of synovial fluid from rheumatoid arthritis and osteoarthritis patients. Clin. Proteom. 2014, 11, 1. [Google Scholar] [CrossRef]
- He, M.; Roussak, K.; Ma, F.; Borcherding, N.; Garin, V.; White, M.; Schutt, C.; Jensen, T.I.; Zhao, Y.; Iberg, C.A. CD5 expression by dendritic cells directs T cell immunity and sustains immunotherapy responses. Science 2023, 379, eabg2752. [Google Scholar] [CrossRef]
- Taher, T.E.; Bystrom, J.; Mignen, O.; Pers, J.-O.; Renaudineau, Y.; Mageed, R.A. CD5 and B lymphocyte responses: Multifaceted effects through multitudes of pathways and channels. Cell. Mol. Immunol. 2020, 17, 1201–1203. [Google Scholar] [CrossRef]
- Rossi, I.; Milani, I.; Lupo, M.; Ferri, N. ANGPTL3 induces a proinflammatory response in THP-1 derived macrophages. Atherosclerosis 2022, 355, 30. [Google Scholar] [CrossRef]
- Lupo, M.G.; Ferri, N. Angiopoietin-like 3 (ANGPTL3) and atherosclerosis: Lipid and non-lipid related effects. J. Cardiovasc. Dev. Dis. 2018, 5, 39. [Google Scholar] [CrossRef]
- Sampath, S.J.P.; Venkatesan, V.; Ghosh, S.; Kotikalapudi, N. Obesity, metabolic syndrome, and osteoarthritis—An updated review. Curr. Obes. Rep. 2023, 12, 308–331. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Huynh, H.; Umikawa, M.; Silvany, R.; Zhang, C.C. Angiopoietin-like protein 3 supports the activity of hematopoietic stem cells in the bone marrow niche. Blood J. Am. Soc. Hematol. 2011, 117, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Wittig, J.; Drekolia, M.-K.; Kyselova, A.; Lagos, F.D.; Bochenek, M.L.; Hu, J.; Schäfer, K.; Fleming, I.; Bibli, S.-I. Endothelial-dependent S-Sulfhydration of tissue factor pathway inhibitor regulates blood coagulation. Redox Biol. 2023, 62, 102694. [Google Scholar] [CrossRef]
- Neubauer, K.; Zieger, B. Endothelial cells and coagulation. Cell Tissue Res. 2022, 387, 391–398. [Google Scholar] [CrossRef]
- Asayag, K.; Peled, E.; Assalia, M.; Crispel, Y.; Yanovich, C.; Cohen, H.; Keren-Politansky, A.; Nadir, Y. Steroids and Malignancy Increase Local Heparanase and Decrease Markers of Osteoblast Activity in Bone Tissue Microcirculation. Biomolecules 2024, 14, 1506. [Google Scholar] [CrossRef]
- So, A.; Varisco, P.; Kemkes-Matthes, B.; Herkenne-Morard, C.; Chobaz-Peclat, V.; Gerster, J.; Busso, N. Arthritis is linked to local and systemic activation of coagulation and fibrinolysis pathways. J. Thromb. Haemost. 2003, 1, 2510–2515. [Google Scholar] [CrossRef]
- Zhao, C.-W.; Song, W.-X.; Liu, B.; Gao, Y.-H.; Ding, L.; Huang, Y.-F.; Qi, X. Resistin induces chemokine and matrix metalloproteinase production via CAP1 receptor and activation of p38-MAPK and NF-κB signalling pathways in human chondrocytes. Clin. Exp. Rheumatol. 2022, 40, 501–513. [Google Scholar] [CrossRef]
- Lee, J.; Ort, T.; Ma, K.; Picha, K.; Carton, J.; Marsters, P.; Lohmander, L.; Baribaud, F.; Song, X.-Y.; Blake, S. Resistin is elevated following traumatic joint injury and causes matrix degradation and release of inflammatory cytokines from articular cartilage in vitro. Osteoarthr. Cartil. 2009, 17, 613–620. [Google Scholar] [CrossRef]
- Yazdanpanah, N.; Yazdanpanah, M.; Wang, Y.; Forgetta, V.; Pollak, M.; Polychronakos, C.; Richards, J.B.; Manousaki, D. Clinically relevant circulating protein biomarkers for type 1 diabetes: Evidence from a two-sample Mendelian randomization study. Diabetes Care 2022, 45, 169–177. [Google Scholar] [CrossRef]
- Marik, W.; Nemec, S.F.; Zbýn, Š.; Zalaudek, M.; Ludvik, B.; Riegler, G.; Karner, M.; Trattnig, S. Changes in cartilage and tendon composition of patients with type I diabetes mellitus: Identification by quantitative sodium magnetic resonance imaging at 7 T. Investig. Radiol. 2016, 51, 266–272. [Google Scholar] [CrossRef]
- Zhou, S.; Tao, B.; Guo, Y.; Gu, J.; Li, H.; Zou, C.; Tang, S.; Jiang, S.; Fu, D.; Li, J. Integrating plasma protein-centric multi-omics to identify potential therapeutic targets for pancreatic cancer. J. Transl. Med. 2024, 22, 557. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Talesa, V.N. Glyoxalases in urological malignancies. Int. J. Mol. Sci. 2018, 19, 415. [Google Scholar] [CrossRef] [PubMed]
- Finn, A.V.; Nakano, M.; Polavarapu, R.; Karmali, V.; Saeed, O.; Zhao, X.; Yazdani, S.; Otsuka, F.; Davis, T.; Habib, A. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J. Am. Coll. Cardiol. 2012, 59, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, U.S.; Tan, B.W.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Xiong, Z.; Zhao, L.; Mei, Y.; Qiu, D.; Li, X.; Zhang, P.; Zhang, M.; Cao, J.; Wang, Y. Proteome-wide Mendelian randomization identified potential drug targets for migraine. J. Headache Pain 2024, 25, 148. [Google Scholar] [CrossRef]
- Edderkaoui, B. Chemokines in cartilage regeneration and degradation: New insights. Int. J. Mol. Sci. 2023, 25, 381. [Google Scholar] [CrossRef]
- Yu, X.; Song, Z.; Rao, L.; Tu, Q.; Zhou, J.; Yin, Y.; Chen, D. Synergistic induction of CCL5, CXCL9 and CXCL10 by IFN-γ and NLRs ligands on human fibroblast-like synoviocytes—A potential immunopathological mechanism for joint inflammation in rheumatoid arthritis. Int. Immunopharmacol. 2020, 82, 106356. [Google Scholar] [CrossRef]
- Dillemans, L.; De Somer, L.; Neerinckx, B.; Proost, P. A review of the pleiotropic actions of the IFN-inducible CXC chemokine receptor 3 ligands in the synovial microenvironment. Cell. Mol. Life Sci. 2023, 80, 78. [Google Scholar] [CrossRef]
- Alder, K.D.; Lee, I.; Munger, A.M.; Kwon, H.-K.; Morris, M.T.; Cahill, S.V.; Back, J.; Kristin, E.Y.; Lee, F.Y. Intracellular Staphylococcus aureus in bone and joint infections: A mechanism of disease recurrence, inflammation, and bone and cartilage destruction. Bone 2020, 141, 115568. [Google Scholar] [CrossRef]
- Xu, Y.; Li, F.; Zhao, X.; Tan, C.; Wang, B.; Chen, Y.; Cao, J.; Wu, D.; Yu, H. Methionine sulfoxide reductase A attenuates atherosclerosis via repairing dysfunctional HDL in scavenger receptor class B type I deficient mice. FASEB J. 2020, 34, 3805–3819. [Google Scholar] [CrossRef]
- Szopa, A.; Bogatko, K.; Herbet, M.; Serefko, A.; Ostrowska, M.; Wośko, S.; Świąder, K.; Szewczyk, B.; Wlaź, A.; Skałecki, P. The interaction of selective A1 and A2A adenosine receptor antagonists with magnesium and zinc ions in mice: Behavioural, biochemical and molecular studies. Int. J. Mol. Sci. 2021, 22, 1840. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Li, D.; Guan, X.; Yang, Y.; Yan, J.; Shi, J.; Ma, R.; Shu, Q. MsrA suppresses inflammatory activation of microglia and oxidative stress to prevent demyelination via inhibition of the NOX2-MAPKs/NF-κB signaling pathway. Drug Des. Dev. Ther. 2020, 14, 1377–1389. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Oses-Prieto, J.A.; Pope, L.E.; Burlingame, A.L.; Dixon, S.J.; Renslo, A.R. Reactivity-based probe of the iron (II)-dependent interactome identifies new cellular modulators of ferroptosis. J. Am. Chem. Soc. 2020, 142, 19085–19093. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Chen, Y.; Xue, F.; Liu, K.; Zhu, B.; Gao, J.; Yin, J.; Zhang, C.; Li, G. Contribution of ferroptosis and GPX4’s dual functions to osteoarthritis progression. EBioMedicine 2022, 76, 103847. [Google Scholar] [CrossRef]
- Yao, X.; Sun, K.; Yu, S.; Luo, J.; Guo, J.; Lin, J.; Wang, G.; Guo, Z.; Ye, Y.; Guo, F. Chondrocyte ferroptosis contribute to the progression of osteoarthritis. J. Orthop. Transl. 2021, 27, 33–43. [Google Scholar] [CrossRef]
- Feng, D.; Li, L.; Li, D.; Wu, R.; Zhu, W.; Wang, J.; Ye, L.; Han, P. Prolyl 4-hydroxylase subunit beta (P4HB) could serve as a prognostic and radiosensitivity biomarker for prostate cancer patients. Eur. J. Med. Res. 2023, 28, 245. [Google Scholar] [CrossRef]
- Müller, G.; Michel, A.; Altenburg, E. COMP (cartilage oligomeric matrix protein) is synthesized in ligament, tendon, meniscus, and articular cartilage. Connect. Tissue Res. 1998, 39, 233–244. [Google Scholar] [CrossRef]
- Di Cesare, P.E.; Chen, F.S.; Moergelin, M.; Carlson, C.S.; Leslie, M.P.; Perris, R.; Fang, C. Matrix–matrix interaction of cartilage oligomeric matrix protein and fibronectin. Matrix Biol. 2002, 21, 461–470. [Google Scholar] [CrossRef]
- Maly, K.; Andres Sastre, E.; Farrell, E.; Meurer, A.; Zaucke, F. COMP and TSP-4: Functional roles in articular cartilage and relevance in osteoarthritis. Int. J. Mol. Sci. 2021, 22, 2242. [Google Scholar] [CrossRef]
- Zhou, H.; Mu, Y.; Ma, C.; Zhang, Z.; Tao, C.; Wang, D.-A. Rejuvenating Hyaline Cartilaginous Phenotype of Dedifferentiated Chondrocytes in Collagen II Scaffolds: A Mechanism Study Using Chondrocyte Membrane Nanoaggregates as Antagonists. ACS Nano 2024, 18, 2077–2090. [Google Scholar] [CrossRef]
- Koelling, S.; Clauditz, T.S.; Kaste, M.; Miosge, N. Cartilage oligomeric matrix protein is involved in human limb development and in the pathogenesis of osteoarthritis. Arthritis Res. Ther. 2006, 8, R56. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Xu, Q.; Yu, X.; Pan, R.; Chen, Y. Dipeptidyl peptidase 4 inhibitors and their potential immune modulatory functions. Pharmacol. Ther. 2020, 209, 107503. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Matsui, T.; Maeda, S.; Higashimoto, Y.; Yamagishi, S.-I. Advanced glycation end products evoke endothelial cell damage by stimulating soluble dipeptidyl peptidase-4 production and its interaction with mannose 6-phosphate/insulin-like growth factor II receptor. Cardiovasc. Diabetol. 2013, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Ro, D.H.; Cho, G.H.; Kim, J.Y.; Min, S.K.; Yang, H.R.; Park, H.J.; Wang, S.Y.; Kim, Y.J.; Lee, M.C.; Bae, H.C. Selective targeting of dipeptidyl-peptidase 4 (DPP-4) positive senescent chondrocyte ameliorates osteoarthritis progression. Aging Cell 2024, 23, e14161. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, E.; Davey Smith, G.; Windmeijer, F.; Bowden, J. An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int. J. Epidemiol. 2019, 48, 713–727. [Google Scholar] [CrossRef]
- Verbanck, M.; Chen, C.-Y.; Neale, B.; Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef]
- Hartwig, F.P.; Davey Smith, G.; Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 2017, 46, 1985–1998. [Google Scholar] [CrossRef]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef]
- Carter, A.R.; Sanderson, E.; Hammerton, G.; Richmond, R.C.; Davey Smith, G.; Heron, J.; Taylor, A.E.; Davies, N.M.; Howe, L.D. Mendelian randomisation for mediation analysis: Current methods and challenges for implementation. Eur. J. Epidemiol. 2021, 36, 465–478. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exposure | Outcome | SNPs | OR (95%CI) | PIVW | PHeterogeneity | PPleiotropy | Reverse MR PIVW |
|---|---|---|---|---|---|---|---|
| C-C motif chemokine 25 levels | KOA | 35 | 0.904 (0.843–0.969) | 0.004 | 0.387 | 0.288 | 0.557 |
| T-cell surface glycoprotein CD5 levels | KOA | 31 | 0.867 (0.760–0.990) | 0.035 | 0.226 | 0.761 | 0.858 |
| C-X-C motif chemokine 9 levels | KOA | 40 | 1.150 (1.001–1.320) | 0.048 | 0.128 | 0.506 | 0.346 |
| Exposure | Outcome | SNPs | OR (95%CI) | PIVW |
|---|---|---|---|---|
| KOA | rQTL-CASP3/PDGFB | 4 | 1.061 (1.000–1.126) | 0.049 |
| KOA | rQTL-CD40/TIMP3 | 3 | 1.161 (1.081–1.247) | 0.00004 |
| KOA | rQTL-CEBPB/NADK | 4 | 1.068 (1.006–1.135) | 0.031 |
| KOA | rQTL-DAG1/PDGFB | 4 | 1.069 (1.008–1.134) | 0.027 |
| KOA | rQTL-EREG/TIMP3 | 3 | 1.129 (1.050–1.214) | 0.001 |
| KOA | rQTL-F2R/PDGFB | 4 | 1.065 (1.004–1.130) | 0.037 |
| KOA | rQTL-FXYD5/IRAK1 | 4 | 1.074 (1.012–1.140) | 0.019 |
| KOA | rQTL-ITGB1/TFPI | 4 | 0.937 (0.884–0.992) | 0.026 |
| KOA | rQTL-MVK/SERPINE1 | 4 | 1.063 (1.002–1.129) | 0.043 |
| Exposure | Mediator | Outcome | Mediation Effect β1 | Mediation Effect β2 | Total Effect β | Mediated Proportion β1 × β2/β |
|---|---|---|---|---|---|---|
| T-cell surface glycoprotein CD5 levels | rQTL-ANGPTL3/TFPI | KOA | −0.084 | 0.159 | −0.143 | 9.340% |
| rQTL-CPA1/CTRB1 | KOA | −0.178 | −0.095 | −0.143 | −11.825% | |
| rQTL-HAGH/HBQ1 | KOA | 0.097 | 0.132 | −0.143 | −8.954% | |
| C-X-C motif chemokine 9 levels | rQTL-COMP/DPP4 | KOA | −0.208 | 0.134 | 0.140 | −19.909% |
| rQTL-MSRA/P4HB | KOA | −0.177 | −0.230 | 0.140 | 29.079% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Liao, X.; Yu, X.; Xiao, Y.; Tao, X.; Zhong, T. Mediating Role of the ANGPTL3/TFPI Protein Ratio in Regulating T-Cell Surface Glycoprotein CD5 Levels on Knee Osteoarthritis (KOA): A Mendelian Randomization Study. Int. J. Mol. Sci. 2025, 26, 4471. https://doi.org/10.3390/ijms26104471
Li Y, Liao X, Yu X, Xiao Y, Tao X, Zhong T. Mediating Role of the ANGPTL3/TFPI Protein Ratio in Regulating T-Cell Surface Glycoprotein CD5 Levels on Knee Osteoarthritis (KOA): A Mendelian Randomization Study. International Journal of Molecular Sciences. 2025; 26(10):4471. https://doi.org/10.3390/ijms26104471
Chicago/Turabian StyleLi, Yongwei, Xi Liao, Xi Yu, Ying Xiao, Xiaoyu Tao, and Tian Zhong. 2025. "Mediating Role of the ANGPTL3/TFPI Protein Ratio in Regulating T-Cell Surface Glycoprotein CD5 Levels on Knee Osteoarthritis (KOA): A Mendelian Randomization Study" International Journal of Molecular Sciences 26, no. 10: 4471. https://doi.org/10.3390/ijms26104471
APA StyleLi, Y., Liao, X., Yu, X., Xiao, Y., Tao, X., & Zhong, T. (2025). Mediating Role of the ANGPTL3/TFPI Protein Ratio in Regulating T-Cell Surface Glycoprotein CD5 Levels on Knee Osteoarthritis (KOA): A Mendelian Randomization Study. International Journal of Molecular Sciences, 26(10), 4471. https://doi.org/10.3390/ijms26104471