
A New Strategy in Modulating the Protease-Activated Receptor 2 (Par2) in Autoimmune Diseases


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
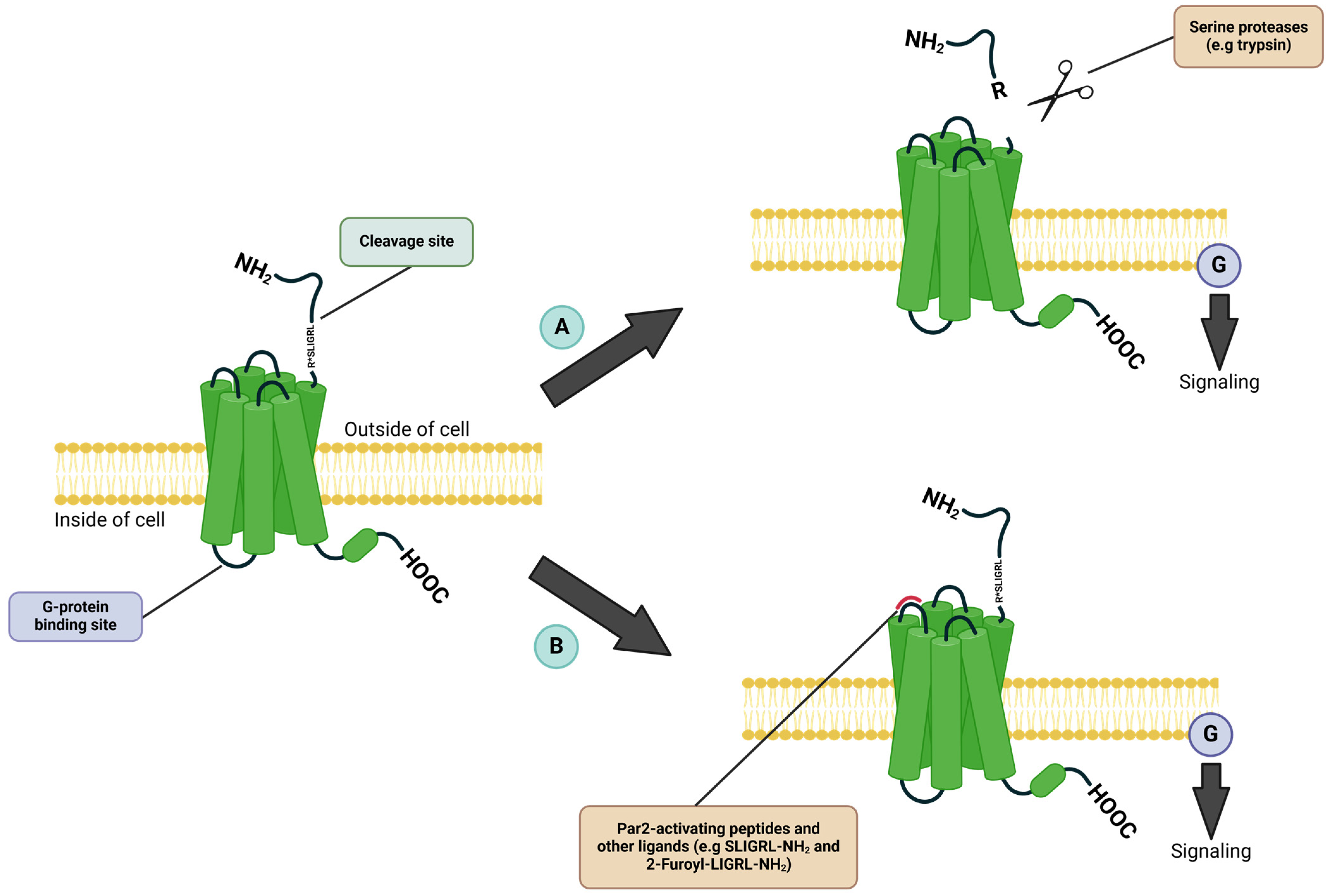
1. Introduction
2. Par2 in Autoimmune Diseases
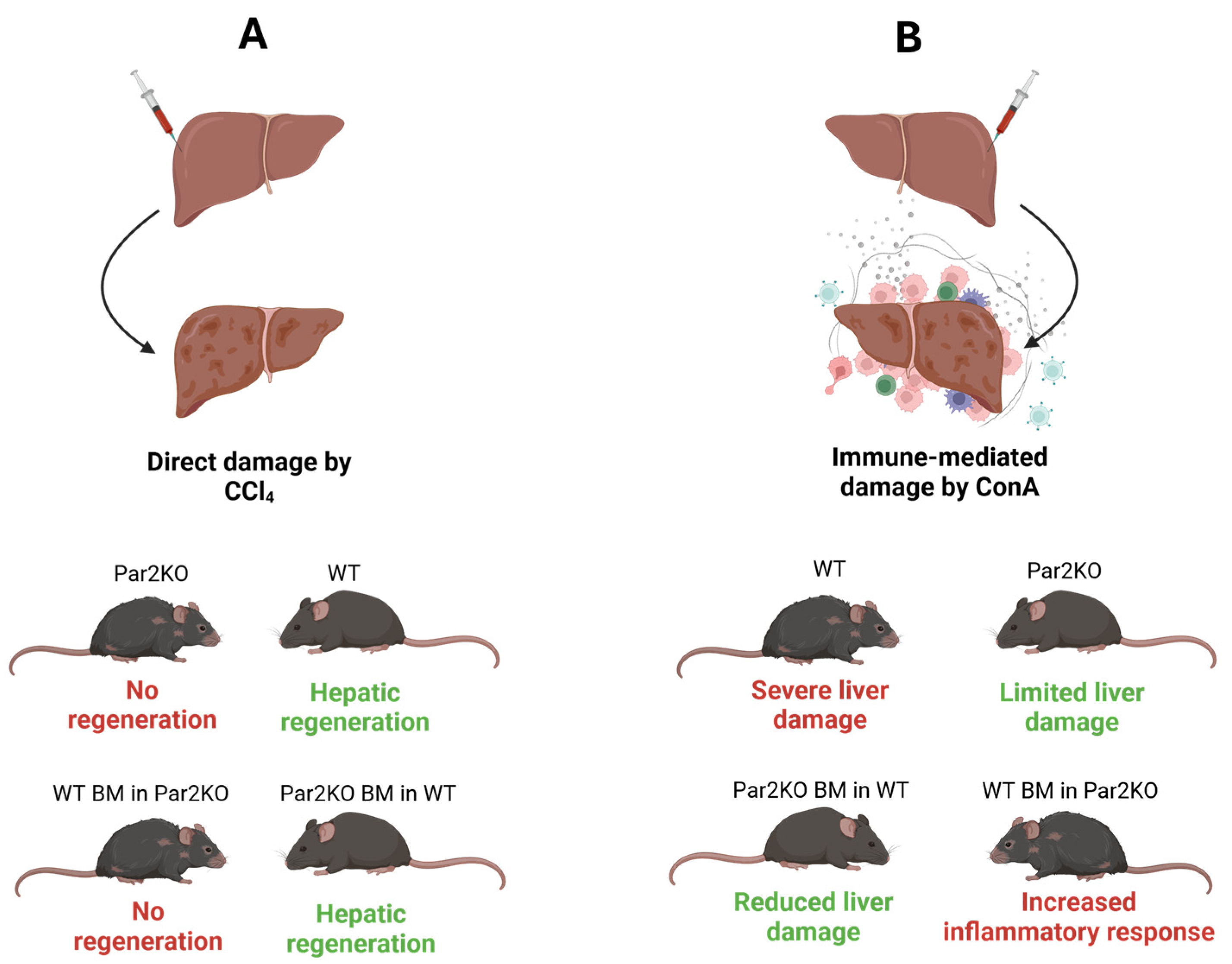
2.1. Autoimmune Hepatitis
2.2. Type 1 Diabetes
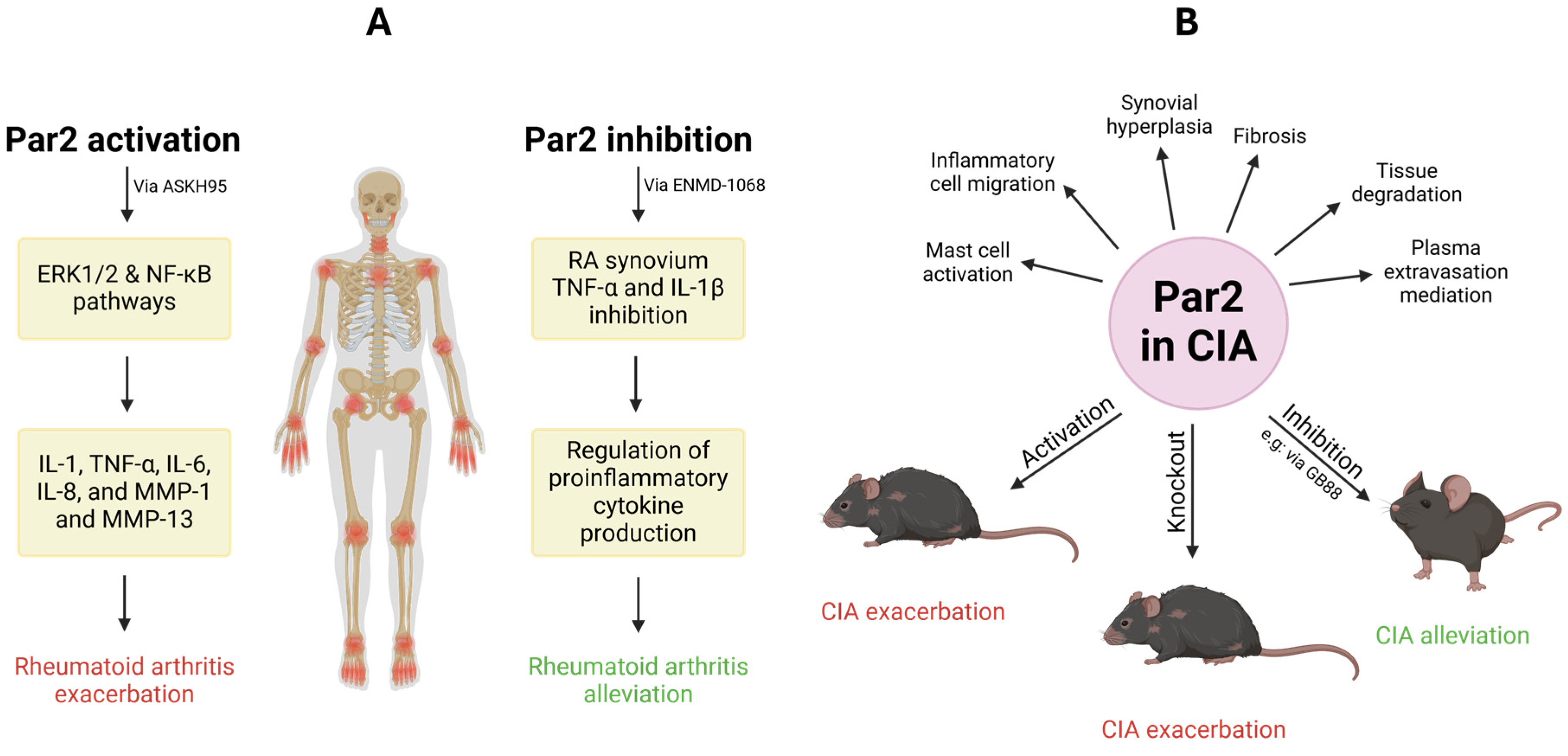
2.3. Rheumatoid Arthritis
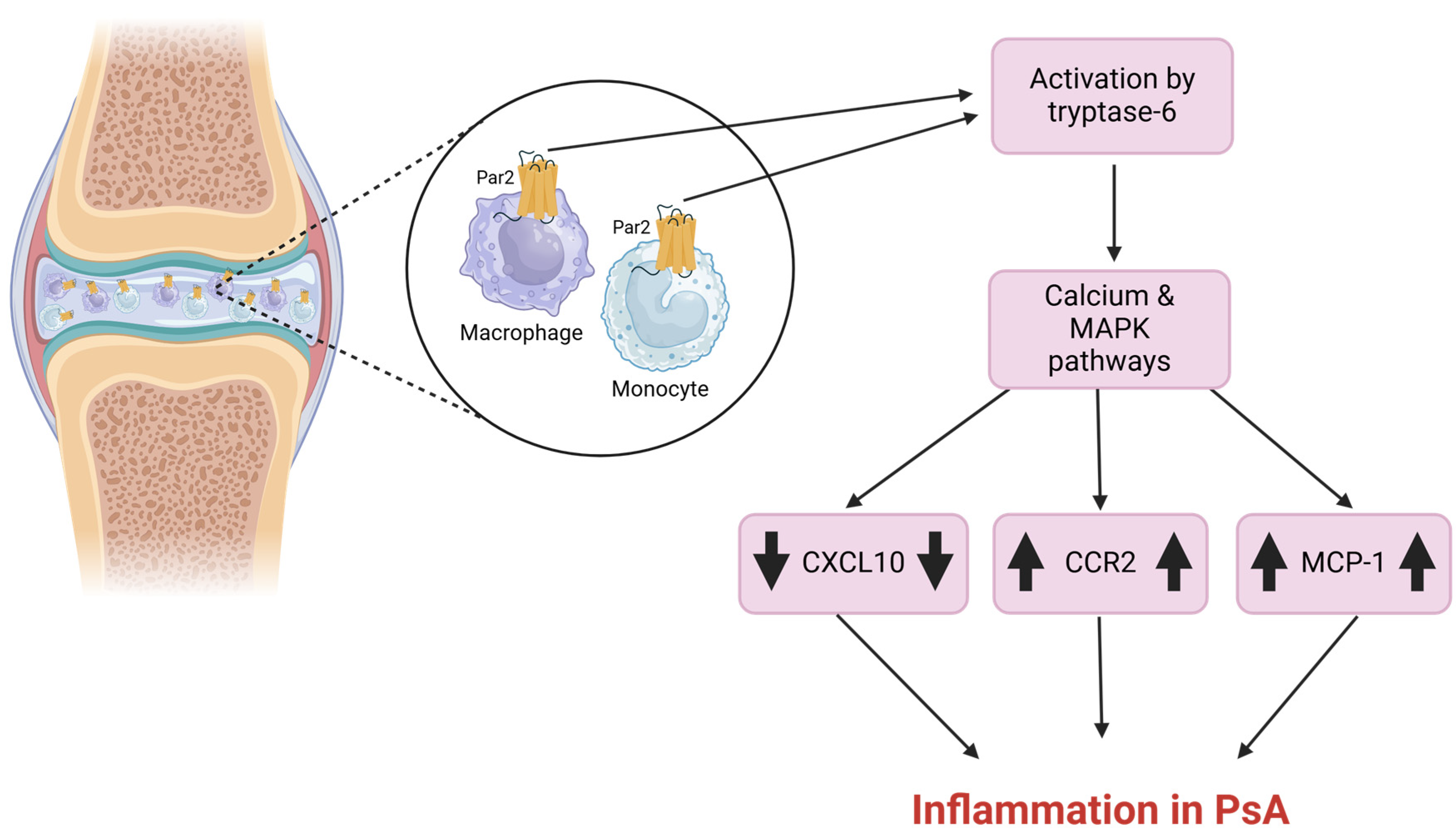
2.4. Psoriatic Arthritis
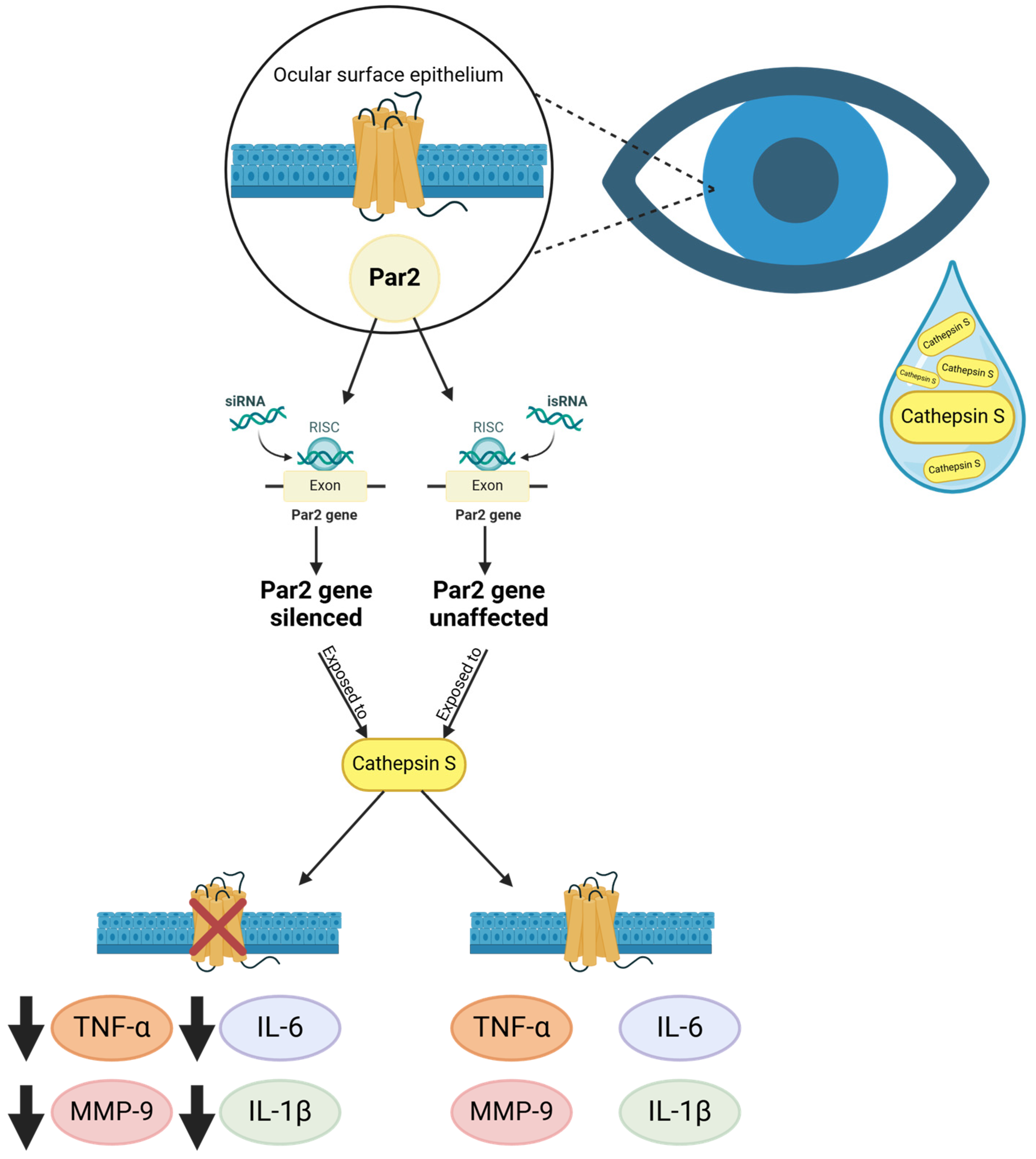
2.5. Sjögren’s Disease
2.6. Systemic Lupus Erythematosus
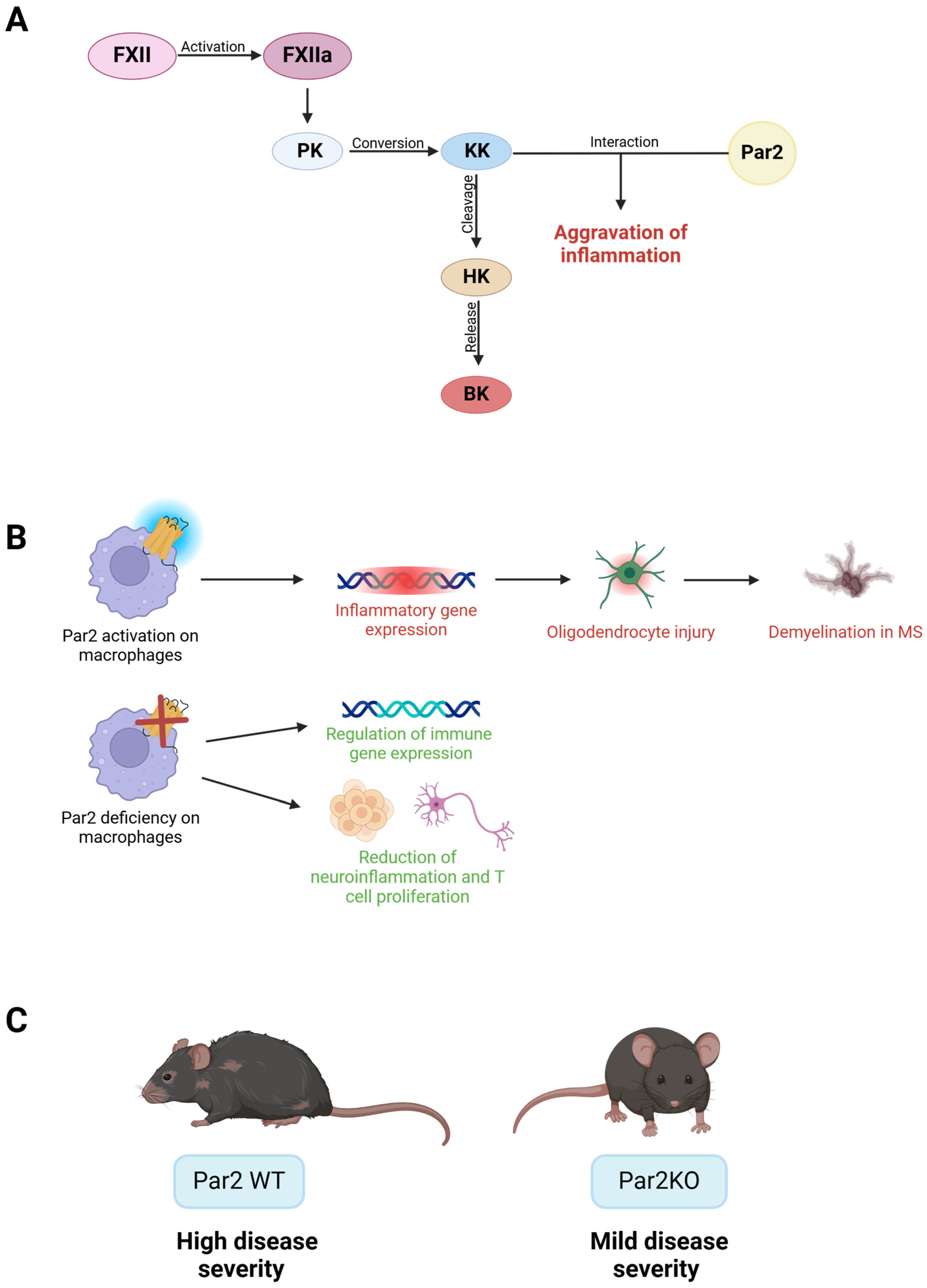
2.7. Multiple Sclerosis
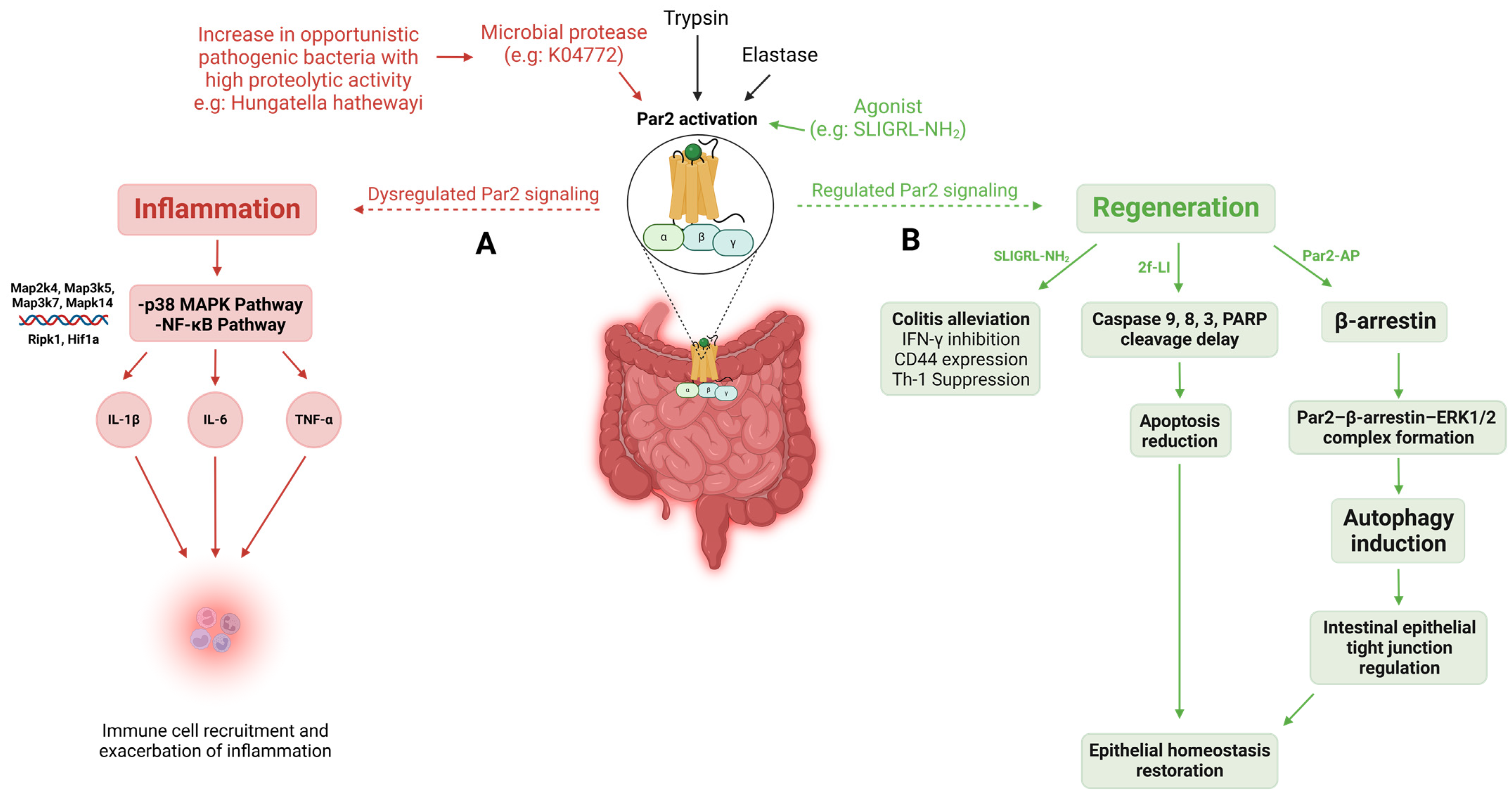
2.8. Inflammatory Bowel Disease
3. Discussion
4. Conclusions
Funding
Conflicts of Interest
References
- Miller, F.W. The increasing prevalence of autoimmunity and autoimmune diseases: An urgent call to action for improved understanding, diagnosis, treatment, and prevention. Curr. Opin. Immunol. 2023, 80, 102266. [Google Scholar] [CrossRef]
- Sun, L.; Wang, X.; Saredy, J.; Yuan, Z.; Yang, X.; Wang, H. Innate-adaptive immunity interplay and redox regulation in immune response. Redox Biol. 2020, 37, 101759. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Pepper, M.; Thomas, P.G. Principles and therapeutic applications of adaptive immunity. Cell 2024, 187, 2052–2078. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.; Jiang, W. Infectious diseases, autoantibodies, and autoimmunity. J. Autoimmun. 2023, 137, 102962. [Google Scholar] [CrossRef]
- Juarranz, Y. Molecular and cellular basis of autoimmune diseases. Cells 2021, 10, 474. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, K.; Cunningham, M.R.; Cadalbert, L.; Lockhart, J.; Boyd, G.; Ferrell, W.R.; Plevin, R. Proteinase-activated receptor-2 mediated inhibition of TNFalpha-stimulated JNK activation—A novel paradigm for G(q/11) linked GPCRs. Cell. Signal. 2010, 22, 265–273. [Google Scholar] [CrossRef]
- Scott, G.; Leopardi, S.; Parker, L.; Babiarz, L.; Seiberg, M.; Han, R. The proteinase-activated receptor-2 mediates phagocytosis in a Rho-dependent manner in human keratinocytes. J. Investig. Dermatol. 2003, 121, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Reches, G.; Piran, R. Par2-mediated responses in inflammation and regeneration: Choosing between repair and damage. Inflamm. Regen. 2024, 44, 26. [Google Scholar] [CrossRef] [PubMed]
- Chandrabalan, A.; Ramachandran, R. Molecular mechanisms regulating Proteinase-Activated Receptors (PARs). FEBS J. 2021, 288, 2697–2726. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Metcalf, M.; Bunnett, N.W. Biased signaling of protease-activated receptors. Front. Endocrinol. 2014, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Stalheim, L.; Ding, Y.; Gullapalli, A.; Paing, M.M.; Wolfe, B.L.; Morris, D.R.; Trejo, J. Multiple independent functions of arrestins in the regulation of protease-activated receptor-2 signaling and trafficking. Mol. Pharmacol. 2005, 67, 78–87. [Google Scholar] [CrossRef] [PubMed]
- de França, B.N.; Gasparoni, L.M.; Rovai, E.M.; Batitucci Ambrósio, L.M.; de Mendonça, N.F.; Hagy, M.H.; Mendoza, A.H.; Sipert, C.R.; Holzhausen, M. Protease-activated receptor type 2 activation downregulates osteogenesis in periodontal ligament stem cells. Braz. Oral Res. 2023, 37, e002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; He, Y.; Shi, Z.; Jiang, J.; He, B.; Xu, S.; Fang, Z. Small Intestine Protection of Mica Against Non-Steroidal Anti-Inflammatory Drugs-Injury Through ERK1/2 Signal Pathway in Rats. Front. Pharmacol. 2019, 10, 871. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, A.; Oono, Y.; Yonezawa, D.; Hiramatsu, K.; Inoi, N.; Sekiguchi, F.; Honjo, M.; Hirofuchi, M.; Kanke, T.; Ishiwata, H. 2-Furoyl-LIGRL-NH2, a potent agonist for proteinase-activated receptor-2, as a gastric mucosal cytoprotective agent in mice. Br. J. Pharmacol. 2005, 144, 212–219. [Google Scholar] [CrossRef]
- Boitano, S.; Flynn, A.N.; Schulz, S.M.; Hoffman, J.; Price, T.J.; Vagner, J. Potent agonists of the protease activated receptor 2 (PAR2). J. Med. Chem. 2011, 54, 1308–1313. [Google Scholar] [CrossRef] [PubMed]
- Ocak, U.; Ocak, P.E.; Huang, L.; Zhang, J.H. FSLLRY-NH2 Improves Neurological Outcome After Cardiac Arrest in Rats. Turk. Neurosurg. 2020, 30, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Ocasio-Rivera, M.; Marin-Maldonado, F.; Trossi-Torres, G.; Ortiz-Rosado, A.; Rodríguez-Irizarry, V.; Rodriguez-Lopez, E.; Martínez, S.; Almodóvar, S.; Suarez-Martínez, E. Targeting of protease activator receptor-2 (PAR-2) antagonist FSLLRY-NH2 as an asthma adjuvant therapy. Medicine 2020, 99, e22351. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Yu, S.-J.; Tsai, J.-J.; Yu, C.-H.; Liao, E.-C. Antagonism of Protease Activated Receptor-2 by GB88 Reduces Inflammation Triggered by Protease Allergen Tyr-p3. Front. Immunol. 2021, 12, 557433. [Google Scholar] [CrossRef] [PubMed]
- Soeda, M.; Ohka, S.; Nishizawa, D.; Iseki, M.; Yamaguchi, K.; Arita, H.; Hanaoka, K.; Kato, J.; Ogawa, S.; Hiranuma, A.; et al. Single-Nucleotide Polymorphisms of the PAR2 and IL-17A Genes Are Significantly Associated with Chronic Pain. Int. J. Mol. Sci. 2023, 24, 17627. [Google Scholar] [CrossRef]
- Kume, M.; Ahmad, A.; DeFea, K.A.; Vagner, J.; Dussor, G.; Boitano, S.; Price, T.J. Protease-Activated Receptor 2 (PAR2) Expressed in Sensory Neurons Contributes to Signs of Pain and Neuropathy in Paclitaxel Treated Mice. J. Pain 2023, 24, 1980–1993. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liu, N.; Li, M.; Liang, S. Blocking PAR2 alleviates bladder pain and hyperactivity via TRPA1 signal. Transl. Neurosci. 2016, 7, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Fan, X.; Lai, Y.; Chen, J.; Peng, Y.; Peng, Y.; Xiang, L.; Ma, Y. Protease-Activated Receptor 2 in inflammatory skin disease: Current evidence and future perspectives. Front. Immunol. 2024, 15, 1448952. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Munanairi, A.; Liu, X.Y.; Zhang, J.; Hu, L.; Hu, M.; Bu, D.; Liu, L.; Xie, Z.; Kim, B.S.; et al. PAR2 mediates itch via TRPV3 signaling in keratinocytes. J. Investig. Dermatol. 2020, 140, 1524–1532. [Google Scholar] [CrossRef]
- Kalogera, S.; He, Y.; Bay-Jensen, A.C.; Gantzel, T.; Sun, S.; Manon-Jensen, T.; Karsdal, M.A.; Thudium, C.S. The activation fragment of PAR2 is elevated in serum from patients with rheumatoid arthritis and reduced in response to anti-IL6R treatment. Sci. Rep. 2021, 11, 24285. [Google Scholar] [CrossRef]
- Ferrell, W.R.; Lockhart, J.C.; Kelso, E.B.; Dunning, L.; Plevin, R.; Meek, S.E.; Smith, A.J.H.; Hunter, G.D.; McLean, J.S.; McGary, F.; et al. Essential role for proteinase-activated receptor-2 in arthritis. J. Clin. Investig. 2003, 111, 35–41. [Google Scholar] [CrossRef]
- Cattaruzza, F.; Lyo, V.; Jones, E.; Pham, D.; Hawkins, J.; Kirkwood, K.; Valdez-Morales, E.; Ibeakanma, C.; Vanner, S.J.; Bogyo, M.; et al. Cathepsin S is activated during colitis and causes visceral hyperalgesia by a PAR2-dependent mechanism in mice. Gastroenterology 2011, 141, 1864–1874.e1. [Google Scholar] [CrossRef] [PubMed]
- Rondeau, L.E.; Barbosa Da Luz, B.; Santiago, A.; Bermudez-Brito, M.; Hann, A.; De Palma, G.; Jury, J.; Wang, X.; Verdu, E.F.; Galipeau, H.J.; et al. Proteolytic bacteria expansion during colitis amplifies inflammation through cleavage of the external domain of PAR2. Gut Microbes 2024, 16, 2387857. [Google Scholar] [CrossRef]
- Santiago, A.; Hann, A.; Constante, M.; Rahmani, S.; Libertucci, J.; Jackson, K.; Rueda, G.; Rossi, L.; Ramachandran, R.; Ruf, W.; et al. Crohn’s disease proteolytic microbiota enhances inflammation through PAR2 pathway in gnotobiotic mice. Gut Microbes 2023, 15, 2205425. [Google Scholar] [CrossRef]
- Reches, G.; Blondheim Shraga, N.R.; Carrette, F.; Malka, A.; Saleev, N.; Gubbay, Y.; Ertracht, O.; Haviv, I.; Bradley, L.M.; Levine, F.; et al. Resolving the conflicts around Par2 opposing roles in regeneration by comparing immune-mediated and toxic-induced injuries. Inflamm. Regen. 2022, 42, 52. [Google Scholar] [CrossRef]
- Reches, G.; Khoon, L.; Ghanayiem, N.; Malka, A.; Piran, R. Controlling autoimmune diabetes onset by targeting Protease-Activated Receptor 2. Biomed. Pharmacother. 2024, 175, 116622. [Google Scholar] [CrossRef]
- Yoon, H.; Radulovic, M.; Wu, J.; Blaber, S.I.; Blaber, M.; Fehlings, M.G.; Scarisbrick, I.A. Kallikrein 6 signals through PAR1 and PAR2 to promote neuron injury and exacerbate glutamate neurotoxicity. J. Neurochem. 2013, 127, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Bandara, M.; MacNaughton, W.K. Protease-activated receptor-2 activation enhances epithelial wound healing via epidermal growth factor receptor. Tissue Barriers 2022, 10, 1968763. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Radulovic, M.; Walters, G.; Paulsen, A.R.; Drucker, K.; Starski, P.; Wu, J.; Fairlie, D.P.; Scarisbrick, I.A. Protease activated receptor 2 controls myelin development, resiliency and repair. Glia 2017, 65, 2070–2086. [Google Scholar] [CrossRef] [PubMed]
- Piran, R.; Lee, S.H.; Kuss, P.; Hao, E.; Newlin, R.; Millán, J.L.; Levine, F. PAR2 regulates regeneration, transdifferentiation, and death. Cell Death Dis. 2016, 7, e2452. [Google Scholar] [CrossRef] [PubMed]
- Komori, A. Recent updates on the management of autoimmune hepatitis. Clin. Mol. Hepatol. 2021, 27, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Covelli, C.; Sacchi, D.; Sarcognato, S.; Cazzagon, N.; Grillo, F.; Baciorri, F.; Fanni, D.; Cacciatore, M.; Maffeis, V.; Guido, M. Pathology of autoimmune hepatitis. Pathologica 2021, 113, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.M. Type 1 diabetes: Pathogenesis and prevention. CMAJ 2006, 175, 165–170. [Google Scholar] [CrossRef]
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Mueller, A.-L.; Payandeh, Z.; Mohammadkhani, N.; Mubarak, S.M.H.; Zakeri, A.; Bahrami, A.A.; Brockmueller, A.; Shakibaei, M. Recent advances in understanding the pathogenesis of rheumatoid arthritis: New treatment strategies. Cells 2021, 10, 3017. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Luo, Y.; Li, T.; Zhao, X.; Lv, T.; Fang, G.; Ou, P.; Li, H.; Luo, X.; Huang, A.; et al. Systemic complications of rheumatoid arthritis: Focus on pathogenesis and treatment. Front. Immunol. 2022, 13, 1051082. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, K.; McGrath, S.; Huesa, C.; Dunning, L.; Litherland, G.; Crilly, A.; Hutlin, L.; Ferrell, W.R.; Lockhart, J.C.; Goodyear, C.S. Rheumatic Disease: Protease-Activated Receptor-2 in Synovial Joint Pathobiology. Front. Endocrinol. 2018, 9, 257. [Google Scholar] [CrossRef] [PubMed]
- Crilly, A.; Burns, E.; Nickdel, M.B.; Lockhart, J.C.; Perry, M.E.; Ferrell, P.W.; Baxter, D.; Dale, J.; Dunning, L.; Wilson, H.; et al. PAR(2) expression in peripheral blood monocytes of patients with rheumatoid arthritis. Ann. Rheum. Dis. 2012, 71, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Kelso, E.B.; Ferrell, W.R.; Lockhart, J.C.; Elias-Jones, I.; Hembrough, T.; Dunning, L.; Gracie, J.A.; McInnes, I.B. Expression and proinflammatory role of proteinase-activated receptor 2 in rheumatoid synovium: Ex vivo studies using a novel proteinase-activated receptor 2 antagonist. Arthritis Rheum. 2007, 56, 765–771. [Google Scholar] [CrossRef]
- Brand, D.D.; Latham, K.A.; Rosloniec, E.F. Collagen-induced arthritis. Nat. Protoc. 2007, 2, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Boissier, M.C.; Feng, X.Z.; Carlioz, A.; Roudier, R.; Fournier, C. Experimental autoimmune arthritis in mice. I. Homologous type II collagen is responsible for self-perpetuating chronic polyarthritis. Ann. Rheum. Dis. 1987, 46, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Holmdahl, R.; Jansson, L.; Larsson, E.; Rubin, K.; Klareskog, L. Homologous type II collagen induces chronic and progressive arthritis in mice. Arthritis Rheum. 1986, 29, 106–113. [Google Scholar] [CrossRef]
- Amiable, N.; Tat, S.K.; Lajeunesse, D.; Duval, N.; Pelletier, J.P.; Martel-Pelletier, J.; Boileau, C. Proteinase-activated receptor (PAR)-2 activation impacts bone resorptive properties of human osteoarthritic subchondral bone osteoblasts. Bone 2009, 44, 1143–1150. [Google Scholar] [CrossRef]
- Boileau, C.; Amiable, N.; Martel-Pelletier, J.; Fahmi, H.; Duval, N.; Pelletier, J.-P. Activation of proteinase-activated receptor 2 in human osteoarthritic cartilage upregulates catabolic and proinflammatory pathways capable of inducing cartilage degradation: A basic science study. Arthritis Res. Ther. 2007, 9, R121. [Google Scholar] [CrossRef]
- Sawamukai, N.; Yukawa, S.; Saito, K.; Nakayamada, S.; Kambayashi, T.; Tanaka, Y. Mast cell-derived tryptase inhibits apoptosis of human rheumatoid synovial fibroblasts via rho-mediated signaling. Arthritis Rheum. 2010, 62, 952–959. [Google Scholar] [CrossRef]
- Xue, M.; Lin, H.; Liang, H.P.H.; McKelvey, K.; Zhao, R.; March, L.; Jackson, C. Deficiency of protease-activated receptor (PAR) 1 and PAR2 exacerbates collagen-induced arthritis in mice via differing mechanisms. Rheumatology 2021, 60, 2990–3003. [Google Scholar] [CrossRef]
- Lohman, R.-J.; Cotterell, A.J.; Barry, G.D.; Liu, L.; Suen, J.Y.; Vesey, D.A.; Fairlie, D.P. An antagonist of human protease activated receptor-2 attenuates PAR2 signaling, macrophage activation, mast cell degranulation, and collagen-induced arthritis in rats. FASEB J. 2012, 26, 2877–2887. [Google Scholar] [CrossRef] [PubMed]
- Crilly, A.; Palmer, H.; Nickdel, M.B.; Dunning, L.; Lockhart, J.C.; Plevin, R.; McInnes, I.B.; Ferrell, W.R. Immunomodulatory role of proteinase-activated receptor-2. Ann. Rheum. Dis. 2012, 71, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, D.V.; Gladman, D. Psoriatic arthritis. [version 1; peer review: 2 approved]. F1000Research 2019, 8, 1665. [Google Scholar] [CrossRef]
- Azuaga, A.B.; Ramírez, J.; Cañete, J.D. Psoriatic arthritis: Pathogenesis and targeted therapies. Int. J. Mol. Sci. 2023, 24, 4901. [Google Scholar] [CrossRef]
- Gossec, L.; Baraliakos, X.; Kerschbaumer, A.; de Wit, M.; McInnes, I.; Dougados, M.; Primdahl, J.; McGonagle, D.G.; Aletaha, D.; Balanescu, A.; et al. EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2019 update. Ann. Rheum. Dis. 2020, 79, 700–712. [Google Scholar] [CrossRef]
- Abji, F.; Rasti, M.; Gómez-Aristizábal, A.; Muytjens, C.; Saifeddine, M.; Mihara, K.; Motahhari, M.; Gandhi, R.; Viswanathan, S.; Hollenberg, M.D.; et al. Proteinase-Mediated Macrophage Signaling in Psoriatic Arthritis. Front. Immunol. 2020, 11, 629726. [Google Scholar] [CrossRef]
- Chen, C.; Darrow, A.L.; Qi, J.-S.; D’Andrea, M.R.; Andrade-Gordon, P. A novel serine protease predominately expressed in macrophages. Biochem. J. 2003, 374 Pt 1, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, M.D.; Renaux, B.; Hyun, E.; Houle, S.; Vergnolle, N.; Saifeddine, M.; Ramachandran, R. Derivatized 2-furoyl-LIGRLO-amide, a versatile and selective probe for proteinase-activated receptor 2: Binding and visualization. J. Pharmacol. Exp. Ther. 2008, 326, 453–462. [Google Scholar] [CrossRef]
- Ramachandran, R.; Eissa, A.; Mihara, K.; Oikonomopoulou, K.; Saifeddine, M.; Renaux, B.; Diamandis, E.; Hollenberg, M.D. Proteinase-activated receptors (PARs): Differential signalling by kallikrein-related peptidases KLK8 and KLK14. Biol. Chem. 2012, 393, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.I. Sjögren’s syndrome. Lancet 2005, 366, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Bjordal, O.; Norheim, K.B.; Rødahl, E.; Jonsson, R.; Omdal, R. Primary Sjögren’s syndrome and the eye. Surv. Ophthalmol. 2020, 65, 119–132. [Google Scholar] [CrossRef]
- Klinngam, W.; Fu, R.; Janga, S.R.; Edman, M.C.; Hamm-Alvarez, S.F. Cathepsin S Alters the Expression of Pro-Inflammatory Cytokines and MMP-9, Partially through Protease-Activated Receptor-2, in Human Corneal Epithelial Cells. Int. J. Mol. Sci. 2018, 19, 3530. [Google Scholar] [CrossRef] [PubMed]
- Fava, A.; Petri, M. Systemic lupus erythematosus: Diagnosis and clinical management. J. Autoimmun. 2019, 96, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Arazi, A.; Rao, D.A.; Berthier, C.C.; Davidson, A.; Liu, Y.; Hoover, P.J.; Chicoine, A.; Eisenhaure, T.M.; Jonsson, A.H.; Li, S.; et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat. Immunol. 2019, 20, 902–914. [Google Scholar] [CrossRef]
- Seo, Y.; Hee Mun, C.; Park, S.H.; Jeon, D.; Jeong Kim, S.; Yoon, T.; Ko, E.; Jo, S.; Park, Y.B.; Namkung, W.; et al. Punicalagin ameliorates lupus nephritis via inhibition of PAR2. Int. J. Mol. Sci. 2020, 21, 4975. [Google Scholar] [CrossRef]
- Itto, R.; Oe, Y.; Imaruoka, K.; Sato, E.; Sekimoto, A.; Yamakage, S.; Kumakura, S.; Sato, H.; Ito, S.; Takahashi, N. Glomerular Injury Is Exacerbated in Lupus-Prone MRL/lpr Mice Treated with a Protease-Activated Receptor 2 Antagonist. Tohoku J. Exp. Med. 2019, 249, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Mey, G.M.; Mahajan, K.R.; DeSilva, T.M. Neurodegeneration in multiple sclerosis. WIREs Mech. Dis. 2023, 15, e1583. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Du, S.; Zhao, L.; Jain, S.; Sahay, K.; Rizvanov, A.; Lezhnyova, V.; Khaibullin, T.; Martynova, E.; Khaiboullina, S.; et al. Autoreactive lymphocytes in multiple sclerosis: Pathogenesis and treatment target. Front. Immunol. 2022, 13, 996469. [Google Scholar] [CrossRef]
- Hauser, S.L.; Cree, B.A.C. Treatment of multiple sclerosis: A review. Am. J. Med. 2020, 133, 1380–1390.e2. [Google Scholar] [CrossRef]
- Göbel, K.; Asaridou, C.M.; Merker, M.; Eichler, S.; Herrmann, A.M.; Geuß, E.; Ruck, T.; Schüngel, L.; Groeneweg, L.; Narayanan, V.; et al. Plasma kallikrein modulates immune cell trafficking during neuroinflammation via PAR2 and bradykinin release. Proc. Natl. Acad. Sci. USA 2019, 116, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Bernstein, C.N. Environmental risk factors for inflammatory bowel disease. United Eur. Gastroenterol. J. 2022, 10, 1047–1053. [Google Scholar] [CrossRef]
- Seyedian, S.S.; Nokhostin, F.; Malamir, M.D. A review of the diagnosis, prevention, and treatment methods of inflammatory bowel disease. J. Med. Life 2019, 12, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.L.; Zheng, L.-B.; Kanazawa, Y.; Shih, D.Q. Immunopathology of inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 6–21. [Google Scholar] [CrossRef]
- Petagna, L.; Antonelli, A.; Ganini, C.; Bellato, V.; Campanelli, M.; Divizia, A.; Efrati, C.; Franceschilli, M.; Guida, A.M.; Ingallinella, S.; et al. Pathophysiology of Crohn’s disease inflammation and recurrence. Biol. Direct 2020, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.-F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef] [PubMed]
- Ordás, I.; Eckmann, L.; Talamini, M.; Baumgart, D.C.; Sandborn, W.J. Ulcerative colitis. Lancet 2012, 380, 1606–1619. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.-N.; Wang, M.X.; Han, J.L.; Feng, C.Y.; Wang, M.; Wang, M.; Sun, J.Y.; Li, N.Y.; Simal-Gandara, J.; Liu, C. Improved colonic inflammation by nervonic acid via inhibition of NF-κB signaling pathway of DSS-induced colitis mice. Phytomedicine 2023, 112, 154702. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Zhao, Y.; Long, Y.; Ma, X.; Zeng, Y.; Wu, W.; Deng, C.; Li, M.; Peng, S.; Yang, H.; et al. TLR4 promoted endoplasmic reticulum stress induced inflammatory bowel disease via the activation of p38 MAPK pathway. Biosci. Rep. 2022, 42, BSR20220307. [Google Scholar] [CrossRef] [PubMed]
- Bohm, S.K.; Kong, W.; Bromme, D.; Smeekens, S.P.; Anderson, D.C.; Connolly, A.; Kahn, M.; Nelken, N.A.; Coughlin, S.R.; Payan, D.G.; et al. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem. J. 1996, 314 Pt 3, 1009–1016. [Google Scholar] [CrossRef]
- Berger, M.; Guiraud, L.; Dumas, A.; Sagnat, D.; Payros, G.; Rolland, C.; Vergnolle, N.; Deraison, C.; Cenac, N.; Racaud-Sultan, C. Prenatal stress induces changes in PAR2- and M3-dependent regulation of colon primitive cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 323, G609–G626. [Google Scholar] [CrossRef]
- D’Andrea, M.R.; Derian, C.L.; Leturcq, D.; Baker, S.M.; Brunmark, A.; Ling, P.; Darrow, A.L.; Santulli, R.J.; Brass, L.F.; Andrade-Gordon, P. Characterization of protease-activated receptor-2 immunoreactivity in normal human tissues. J. Histochem. Cytochem. 1998, 46, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Mencarelli, A.; Palazzetti, B.; Distrutti, E.; Vergnolle, N.; Hollenberg, M.D.; Wallace, J.L.; Morelli, A.; Cirino, G. Proteinase-activated receptor 2 is an anti-inflammatory signal for colonic lamina propria lymphocytes in a mouse model of colitis. Proc. Natl. Acad. Sci. USA 2001, 98, 13936–13941. [Google Scholar] [CrossRef] [PubMed]
- Saez, A.; Herrero-Fernandez, B.; Gomez-Bris, R.; Sánchez-Martinez, H.; Gonzalez-Granado, J.M. Pathophysiology of inflammatory bowel disease: Innate immune system. Int. J. Mol. Sci. 2023, 24, 1526. [Google Scholar] [CrossRef]
- Iablokov, V.; Hirota, C.L.; Peplowski, M.A.; Ramachandran, R.; Mihara, K.; Hollenberg, M.D.; MacNaughton, W.K. Proteinase-activated receptor 2 (PAR2) decreases apoptosis in colonic epithelial cells. J. Biol. Chem. 2014, 289, 34366–34377. [Google Scholar] [CrossRef] [PubMed]
- Michielan, A.; D’Incà, R. Intestinal permeability in inflammatory bowel disease: Pathogenesis, clinical evaluation, and therapy of leaky gut. Mediat. Inflamm. 2015, 2015, 628157. [Google Scholar] [CrossRef] [PubMed]
- Her, J.Y.; Lee, Y.; Kim, S.J.; Heo, G.; Choo, J.; Kim, Y.; Howe, C.; Rhee, S.H.; Yu, H.S.; Chung, H.Y.; et al. Blockage of protease-activated receptor 2 exacerbates inflammation in high-fat environment partly through autophagy inhibition. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G30–G42. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, Y.; Heo, G.; Jeong, S.; Park, S.; Yoo, J.W.; Jung, Y.; Im, E. Modulation of Intestinal Epithelial Permeability via Protease-Activated Receptor-2-Induced Autophagy. Cells 2022, 11, 878. [Google Scholar] [CrossRef]
- Lee, T.; Lee, E.; Irwin, R.; Lucas, P.C.; McCabe, L.R.; Parameswaran, N. β-Arrestin-1 deficiency protects mice from experimental colitis. Am. J. Pathol. 2013, 182, 1114–1123. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khoon, L.; Piran, R. A New Strategy in Modulating the Protease-Activated Receptor 2 (Par2) in Autoimmune Diseases. Int. J. Mol. Sci. 2025, 26, 410. https://doi.org/10.3390/ijms26010410
Khoon L, Piran R. A New Strategy in Modulating the Protease-Activated Receptor 2 (Par2) in Autoimmune Diseases. International Journal of Molecular Sciences. 2025; 26(1):410. https://doi.org/10.3390/ijms26010410
Chicago/Turabian StyleKhoon, Lynn, and Ron Piran. 2025. "A New Strategy in Modulating the Protease-Activated Receptor 2 (Par2) in Autoimmune Diseases" International Journal of Molecular Sciences 26, no. 1: 410. https://doi.org/10.3390/ijms26010410
APA StyleKhoon, L., & Piran, R. (2025). A New Strategy in Modulating the Protease-Activated Receptor 2 (Par2) in Autoimmune Diseases. International Journal of Molecular Sciences, 26(1), 410. https://doi.org/10.3390/ijms26010410