
Mixed-Lineage Leukaemia Gene Regulates Glucose-Sensitive Gene Expression and Insulin Secretion in Pancreatic Beta Cells

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Insulin Secretion Decreased in Response to Glucose Loading in βHC-9 Cells
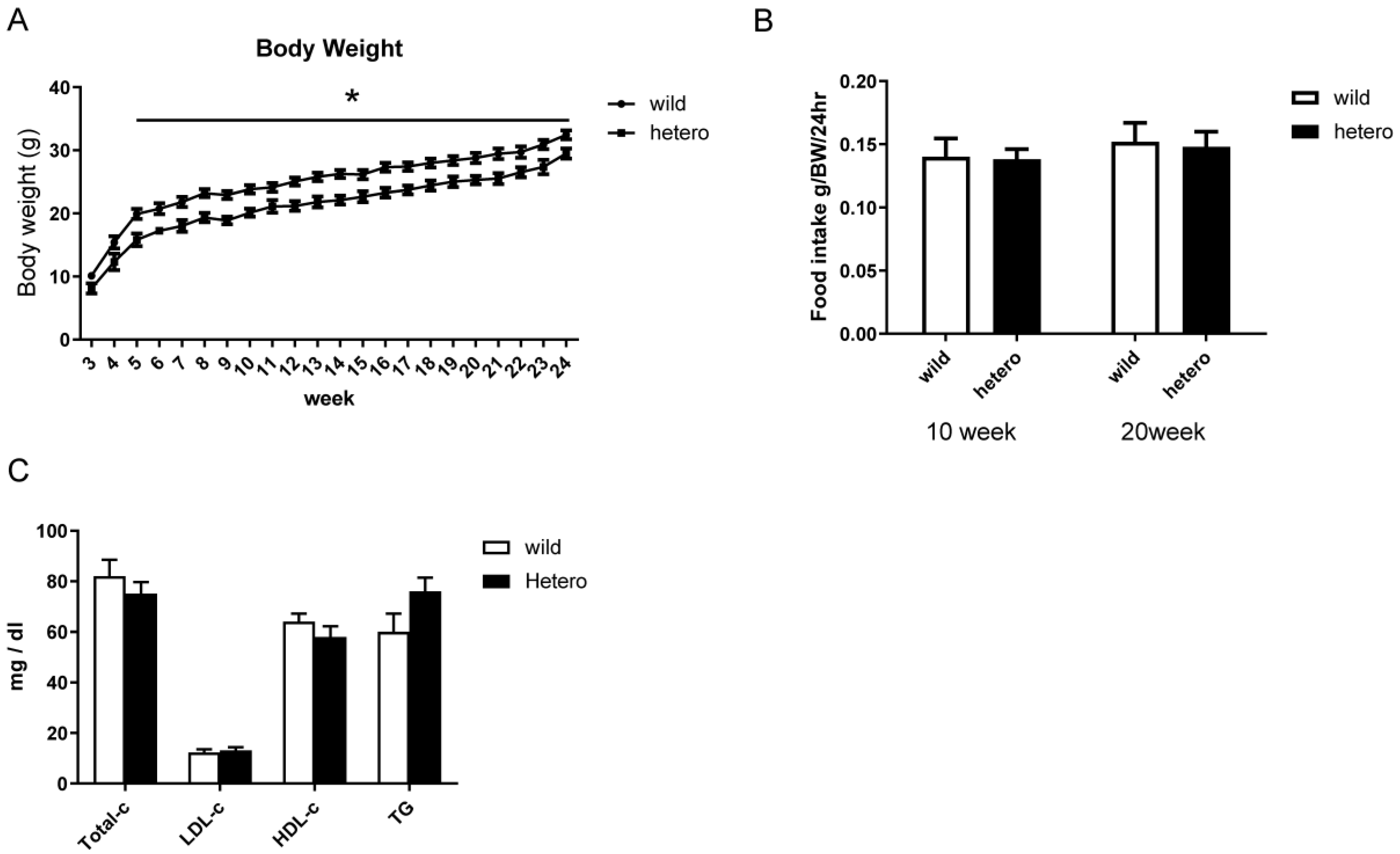
2.2. Metabolic Profile of MLL Heterozygous Knockout Mice
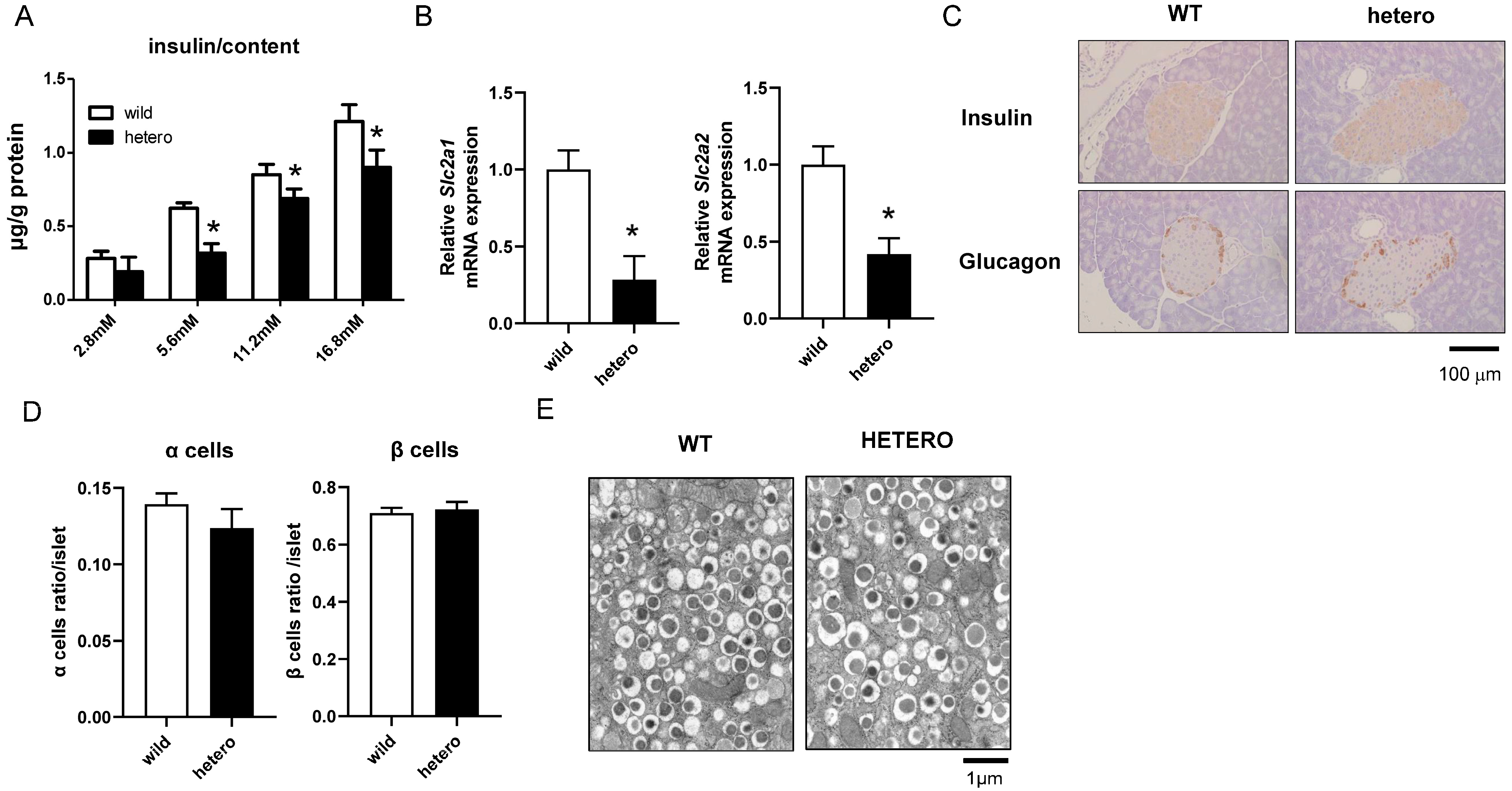
2.3. Insulin Secretion Decreased in MLL Heterozygous Knockout Mice
2.4. Impaired Insulin Secretion and Downregulation of Glucose-Sensitive Genes in MLL Heterozygous Knockout Mice
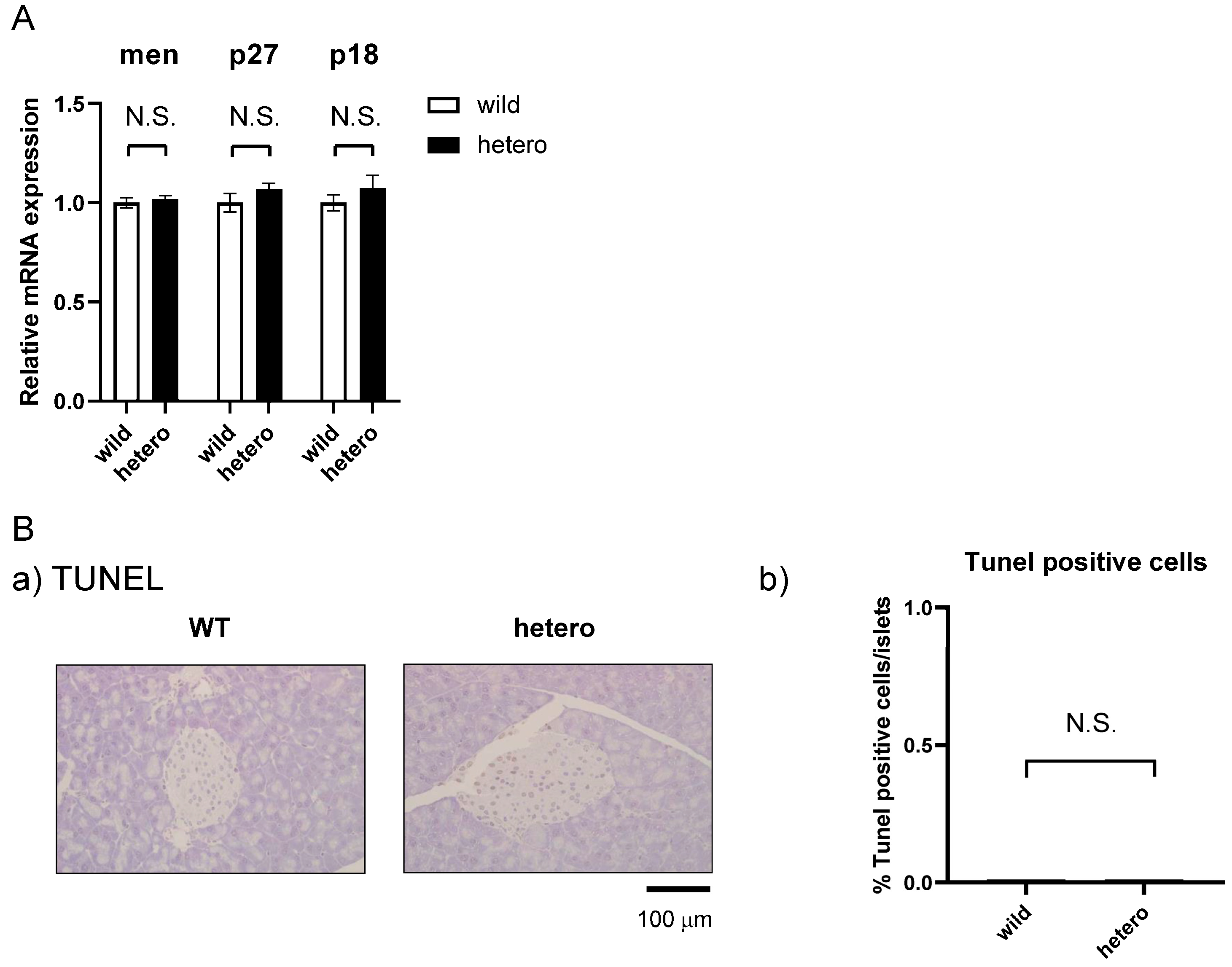
2.5. MLL Heterozygous Mouse Islets Did Not Exhibit Apoptosis
3. Discussion
4. Materials and Methods
4.1. Animals and Islet Extraction
4.2. Biochemical Parameters and Insulin and Glucose Tolerance Tests
4.3. Cell Culture
4.4. Knockdown of Kmt2A by siRNA
4.5. RNA Extraction and Real-Time Polymerase Chain Reaction (PCR)
4.6. Glucose-Stimulated Insulin Secretion (GSIS)
4.7. Histology and Electron Microscopy
4.8. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahrén, B.; Pacini, G. Glucose effectiveness: Lessons from studies on insulin-independent glucose clearance in mice. J. Diabetes Investig. 2021, 12, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Ziemin-van der Poel, S.; McCabe, N.R.; Gill, H.J.; Espinosa, R.; Patel, Y.; Harden, A.; Rubinelli, P.; Smith, S.D.; LeBeau, M.M.; Rowley, J.D. Identification of a gene, mll, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc. Natl. Acad. Sci. USA 1991, 88, 10735–10739. [Google Scholar] [CrossRef] [PubMed]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. Mll targets set domain methyltransferase activity to hox gene promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Milne, T.A.; Tackett, A.J.; Smith, E.R.; Fukuda, A.; Wysocka, J.; Allis, C.D.; Chait, B.T.; Hess, J.L.; Roeder, R.G. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 2005, 121, 873–885. [Google Scholar] [CrossRef]
- Dou, Y.; Milne, T.A.; Ruthenburg, A.J.; Lee, S.; Lee, J.W.; Verdine, G.L.; Allis, C.D.; Roeder, R.G. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 2006, 13, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Wysocka, J.; Swigut, T.; Milne, T.A.; Dou, Y.; Zhang, X.; Burlingame, A.L.; Roeder, R.G.; Brivanlou, A.H.; Allis, C.D. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 2005, 121, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Ansari, K.I.; Mishra, B.P.; Mandal, S.S. MLL histone methylases in gene expression, hormone signaling and cell cycle. Front. Biosci. 2009, 14, 3483–3495. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.M.; Rozenblatt-Rosen, O.; Milne, T.A.; Copeland, T.D.; Levine, S.S.; Lee, J.C.; Hayes, D.N.; Shanmugam, K.S.; Bhattacharjee, A.; Biondi, C.A.; et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol. Cell 2004, 13, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Wang, Z.; Wysocka, J.; Sanyal, M.; Aufiero, D.J.; Kitabayashi, I.; Herr, W.; Cleary, M.L. Leukemia proto-oncoprotein mll forms a set1-like histone methyltransferase complex with menin to regulate hox gene expression. Mol. Cell. Biol. 2004, 24, 5639–5649. [Google Scholar] [CrossRef] [PubMed]
- Karnik, S.K.; Hughes, C.M.; Gu, X.; Rozenblatt-Rosen, O.; McLean, G.W.; Xiong, Y.; Meyerson, M.; Kim, S.K. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18ink4c. Proc. Natl. Acad. Sci. USA 2005, 102, 14659–14664. [Google Scholar] [CrossRef]
- Milne, T.A.; Hughes, C.M.; Lloyd, R.; Yang, Z.; Rozenblatt-Rosen, O.; Dou, Y.; Schnepp, R.W.; Krankel, C.; Livolsi, V.A.; Gibbs, D.; et al. Menin and mll cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, R.; Yamada, M.; Horiguchi, K.; Tomaru, T.; Ozawa, A.; Shibusawa, N.; Hashimoto, K.; Okada, S.; Satoh, T.; Mori, M. Haploinsufficient and predominant expression of multiple endocrine neoplasia type 1 (MEN1)-related genes, MLL, p27Kip1 and p18Ink4C in endocrine organs. Biochem. Biophys. Res. Commun. 2011, 415, 378–383. [Google Scholar] [CrossRef]
- Katada, S.; Sassone-Corsi, P. The histone methyltransferase MLL1 permits the oscillation of circadian gene expression. Nat. Struct. Mol. Biol. 2010, 17, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.D.; Hess, J.L.; Horning, S.E.; Brown, G.A.J.; Korsmeyer, S.J. Altered Hox expression and segmental identity in Mll-mutant mice. Nature 1995, 378, 505–508. [Google Scholar] [CrossRef]
- Hess, J.L.; Yu, B.D.; Li, B.; Hanson, R.; Korsmeyer, S.J. Defects in yolk sac hematopoiesis in Mll-null embryos. Blood 1997, 90, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- Yagi, H.; Deguchi, K.; Aono, A.; Tani, Y.; Kishimoto, T.; Komori, T. Growth disturbance in fetal liver hematopoiesis of Mll-mutant mice. Blood 1998, 92, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Thakker, R.V.; Newey, P.J.; Walls, G.V.; Bilezikian, J.; Dralle, H.; Ebeling, P.R.; Melmed, S.; Sakurai, A.; Tonelli, F.; Brandi, M.L.; et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J. Clin. Endocrinol. Metab. 2012, 97, 2990–3011. [Google Scholar] [CrossRef]
- Mele, C.; Mencarelli, M.; Caputo, M.; Mai, S.; Pagano, L.; Aimaretti, G.; Scacchi, M.; Falchetti, A.; Marzullo, P. Phenotypes associated with MEN1 syndrome: A focus on genotype-phenotype correlations. Front. Endocrinol. 2020, 11, 591501. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Komatsu, M.; Sharp, G.W. The betaHC-9 pancreatic beta-cell line preserves the characteristics of progenitor mouse islets. Diabetes 1996, 45, 1766–1773. [Google Scholar] [CrossRef] [PubMed]
- Sugeedha, J.; Gautam, J.; Tyagi, S. SET1/MLL family of proteins: Functions beyond histone methylation. Epigenetics 2021, 16, 469–487. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Barve, A.; Vega, A.; Shah, P.P.; Ghare, S.; Casson, L.; Wunderlich, M.; Siskind, L.J.; Beverly, L.J. Perturbation of methionine/S-adenosylmethionine metabolism as a novel vulnerability in MLL rearranged leukemia. Cells 2019, 8, 1322. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Du, J.; Gale, R.P.; Wang, L.; Zhan, H.; Liu, F.; Huang, K.; Xu, H.; Zeng, H. Glucose partitioning in the bone marrow micro-environment in acute myeloid leukaemia. Leukemia 2023, 37, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Sayo, Y.; Murao, K.; Imachi, H.; Cao, W.M.; Sato, M.; Dobashi, H.; Wong, N.C.; Ishida, T. The multiple endocrine neoplasia type 1 gene product, menin, inhibits insulin production in rat insulinoma cells. Endocrinology 2002, 143, 2437–2440. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pahlavanneshan, S.; Behmanesh, M.; Oropeza, D.; Furuyama, K.; Tahamtani, Y.; Basiri, M.; Herrera, P.L.; Baharvand, H. Combined inhibition of menin-MLL interaction and TGF-β signaling induces replication of human pancreatic beta cells. Eur. J. Cell Biol. 2020, 99, 151094. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Karakose, E.; Liu, H.; Swartz, E.; Ackeifi, C.; Zlatanic, V.; Wilson, J.; González, B.J.; Bender, A.; Takane, K.K.; et al. Combined inhibition of DYRK1A, SMAD, and trithorax pathways synergizes to induce robust replication in adult human beta cells. Cell Metab. 2019, 29, 638–652.e5. [Google Scholar] [CrossRef]
- Barron, C.C.; Bilan, P.J.; Tsakiridis, T.; Tsiani, E. Facilitative glucose transporters: Implications for cancer detection, prognosis and treatment. Metabolism 2016, 65, 124–139. [Google Scholar] [CrossRef]
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, H.; Seino, S.; Imura, H.; Seino, Y.; Bell, G.I. Characterization and expression of human HepG2/erythrocyte glucose-transporter gene. Diabetes 1988, 37, 657–661. [Google Scholar] [CrossRef]
- Sun, B.; Chen, H.; Xue, J.; Li, P.; Fu, X. The role of GLUT2 in glucose metabolism in multiple organs and tissues. Mol. Biol. Rep. 2023, 50, 6963–6974. [Google Scholar] [CrossRef]
- Berger, C.; Zdzieblo, D. Glucose transporters in pancreatic islets. Pflug. Arch. 2020, 472, 1249–1272. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, K.T.; Wiberg, A.; Amirian, N.; Kay, T.W.; von Herrath, M.G. Persistent glucose transporter expression on pancreatic beta cells from longstanding type 1 diabetic individuals. Diabetes Metab. Res. Rev. 2011, 27, 746–754. [Google Scholar] [CrossRef]
- Ohtsubo, K.; Chen, M.Z.; Olefsky, J.M.; Marth, J.D. Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport. Nat. Med. 2011, 17, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B.; Sarkar, H.K.; Kaback, H.R.; Lodish, H.F. Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and beta-pancreatic islet cells. Cell 1988, 55, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.H.; Newgard, C.B.; Milburn, J.L.; Lodish, H.F.; Thorens, B. The high Km glucose transporter of islets of Langerhans is functionally similar to the low affinity transporter of liver and has an identical primary sequence. J. Biol. Chem. 1990, 265, 6548–6551. [Google Scholar] [CrossRef] [PubMed]
- Guillam, M.T.; Hümmler, E.; Schaerer, E.; Yeh, J.I.; Birnbaum, M.J.; Beermann, F.; Schmidt, A.; Dériaz, N.; Thorens, B. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat. Genet. 1997, 17, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Valera, A.; Solanes, G.; Fernández-Alvarez, J.; Pujol, A.; Ferrer, J.; Asins, G.; Gomis, R.; Bosch, F. Expression of GLUT-2 antisense RNA in beta cells of transgenic mice leads to diabetes. J. Biol. Chem. 1994, 269, 28543–28546. [Google Scholar] [CrossRef] [PubMed]
- Del Guerra, S.; Lupi, R.; Marselli, L.; Masini, M.; Bugliani, M.; Sbrana, S.; Torri, S.; Pollera, M.; Boggi, U.; Mosca, F.; et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes 2005, 54, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Marcheva, B.; Ramsey, K.M.; Buhr, E.D.; Kobayashi, Y.; Su, H.; Ko, C.H.; Ivanova, G.; Omura, C.; Mo, S.; Vitaterna, M.H.; et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 2010, 466, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, M.S.; Li, R.; Liu, V.Y.; Fu, L.; Moore, D.D.; Ma, K.; Yechoor, V.K. Loss of Bmal1 leads to uncoupling and impaired glucose-stimulated insulin secretion in β-cells. Islets 2011, 3, 381–388. [Google Scholar] [CrossRef]
- Chen, L.; Alam, T.; Johnson, J.H.; Hughes, S.; Newgard, C.B.; Unger, R.H. Regulation of beta-cell glucose transporter gene expression. Proc. Natl. Acad. Sci. USA 1990, 87, 4088–4092. [Google Scholar] [CrossRef] [PubMed]
- Frese, T.; Bazwinsky, I.; Mühlbauer, E.; Peschke, E. Circadian and age-dependent expression patterns of GLUT2 and glucokinase in the pancreatic beta-cell of diabetic and nondiabetic rats. Horm. Metab. Res. 2007, 39, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Novosadová, Z.; Polidarová, L.; Sládek, M.; Sumová, A. Alteration in glucose homeostasis and persistence of the pancreatic clock in aged mPer2Luc mice. Sci. Rep. 2018, 8, 11668. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, G.; Hager, J.H.; Volik, S.; Hariono, S.; Wernick, M.; Moore, D.; Nowak, N.; Albertson, D.G.; Pinkel, D.; Collins, C.; et al. Genome scanning with array cgh delineates regional alterations in mouse islet carcinomas. Nat. Genet. 2001, 29, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, S.; Satoh, T.; Yamada, M.; Hashimoto, K.; Tomaru, T.; Katano-Toki, A.; Kakizaki, S.; Okada, S.; Shimizu, H.; Ozawa, A.; et al. Protection against high-fat diet-induced obesity in Helz2-deficient male mice due to enhanced expression of hepatic leptin receptor. Endocrinology 2014, 155, 3459–3472. [Google Scholar] [CrossRef] [PubMed]
- Sait, T.; Okada, S.; Yamada, E.; Ohshima, K.; Shimizu, H.; Shimomura, K.; Sato, M.; Pessin, J.E.; Mori, M. Syntaxin 4 and Synip (syntaxin 4 interacting protein) regulate insulin secretion in the pancreatic βHC-9 cell. J. Biochem. Chem. 2003, 278, 36718–36725. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshino, S.; Ishida, E.; Horiguchi, K.; Matsumoto, S.; Nakajima, Y.; Ozawa, A.; Yamada, M.; Yamada, E. Mixed-Lineage Leukaemia Gene Regulates Glucose-Sensitive Gene Expression and Insulin Secretion in Pancreatic Beta Cells. Int. J. Mol. Sci. 2024, 25, 4704. https://doi.org/10.3390/ijms25094704
Yoshino S, Ishida E, Horiguchi K, Matsumoto S, Nakajima Y, Ozawa A, Yamada M, Yamada E. Mixed-Lineage Leukaemia Gene Regulates Glucose-Sensitive Gene Expression and Insulin Secretion in Pancreatic Beta Cells. International Journal of Molecular Sciences. 2024; 25(9):4704. https://doi.org/10.3390/ijms25094704
Chicago/Turabian StyleYoshino, Satoshi, Emi Ishida, Kazuhiko Horiguchi, Shunichi Matsumoto, Yasuyo Nakajima, Atsushi Ozawa, Masanobu Yamada, and Eijiro Yamada. 2024. "Mixed-Lineage Leukaemia Gene Regulates Glucose-Sensitive Gene Expression and Insulin Secretion in Pancreatic Beta Cells" International Journal of Molecular Sciences 25, no. 9: 4704. https://doi.org/10.3390/ijms25094704
APA StyleYoshino, S., Ishida, E., Horiguchi, K., Matsumoto, S., Nakajima, Y., Ozawa, A., Yamada, M., & Yamada, E. (2024). Mixed-Lineage Leukaemia Gene Regulates Glucose-Sensitive Gene Expression and Insulin Secretion in Pancreatic Beta Cells. International Journal of Molecular Sciences, 25(9), 4704. https://doi.org/10.3390/ijms25094704