
IQGAP3 Is an Important Mediator of Skin Inflammatory Diseases

Abstract
1. Introduction
2. Results
2.1. IQGAP3 Knockdown in Keratinocytes
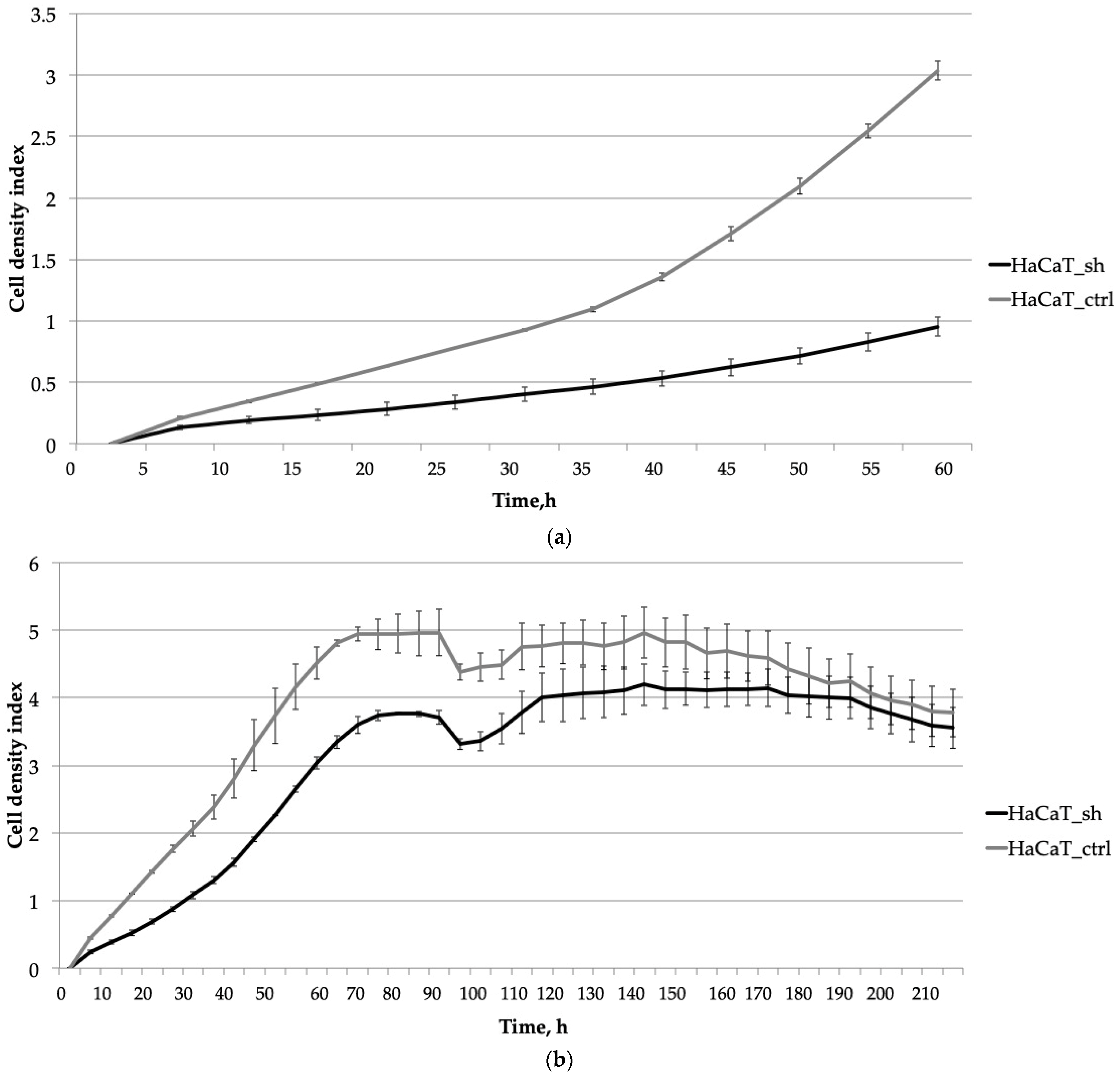
2.2. The Knockdown of IQGAP3 Significantly Alters the Growth Dynamics of Cells
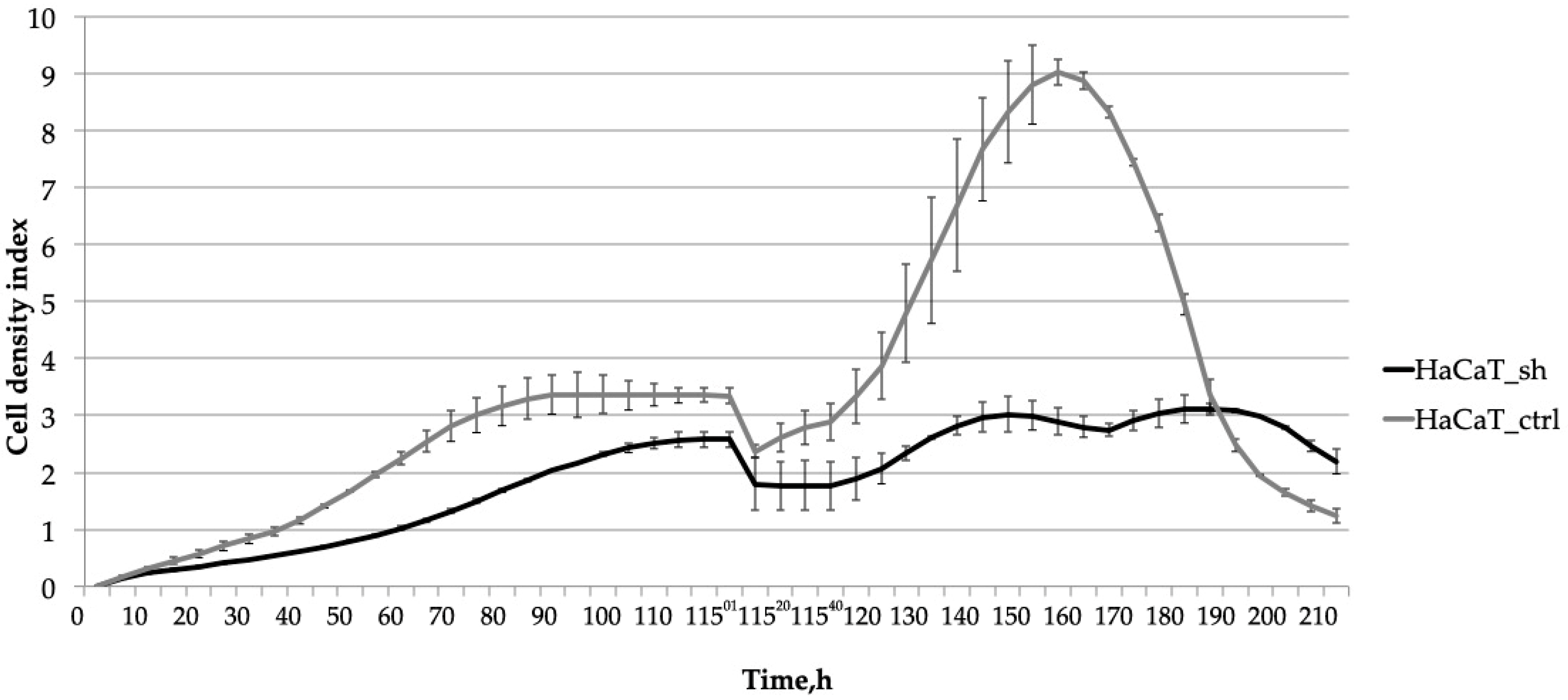
2.3. Knocking down IQGAP3 Reduces the Effect of Pro-Inflammatory Cytokine Stimulation
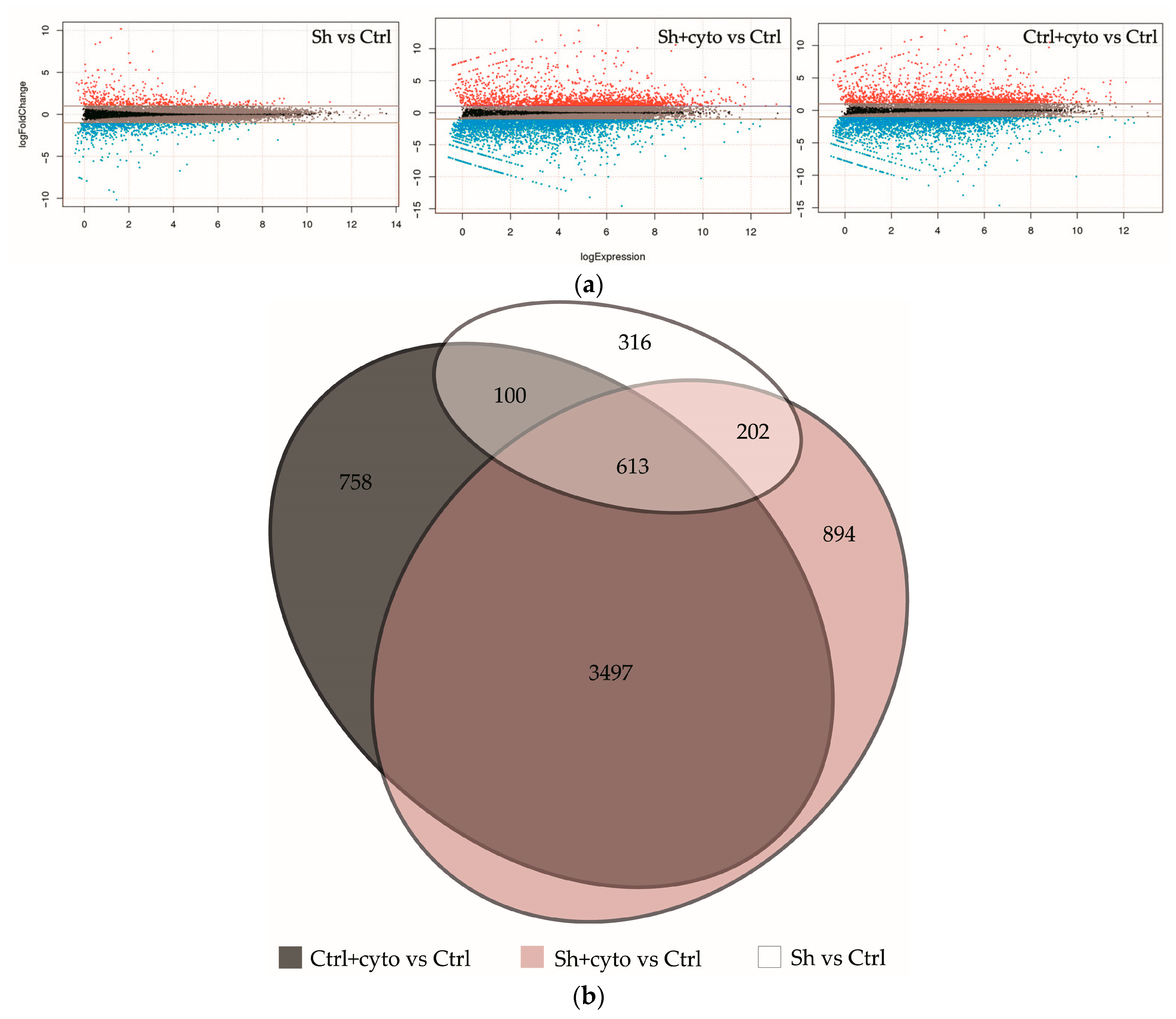
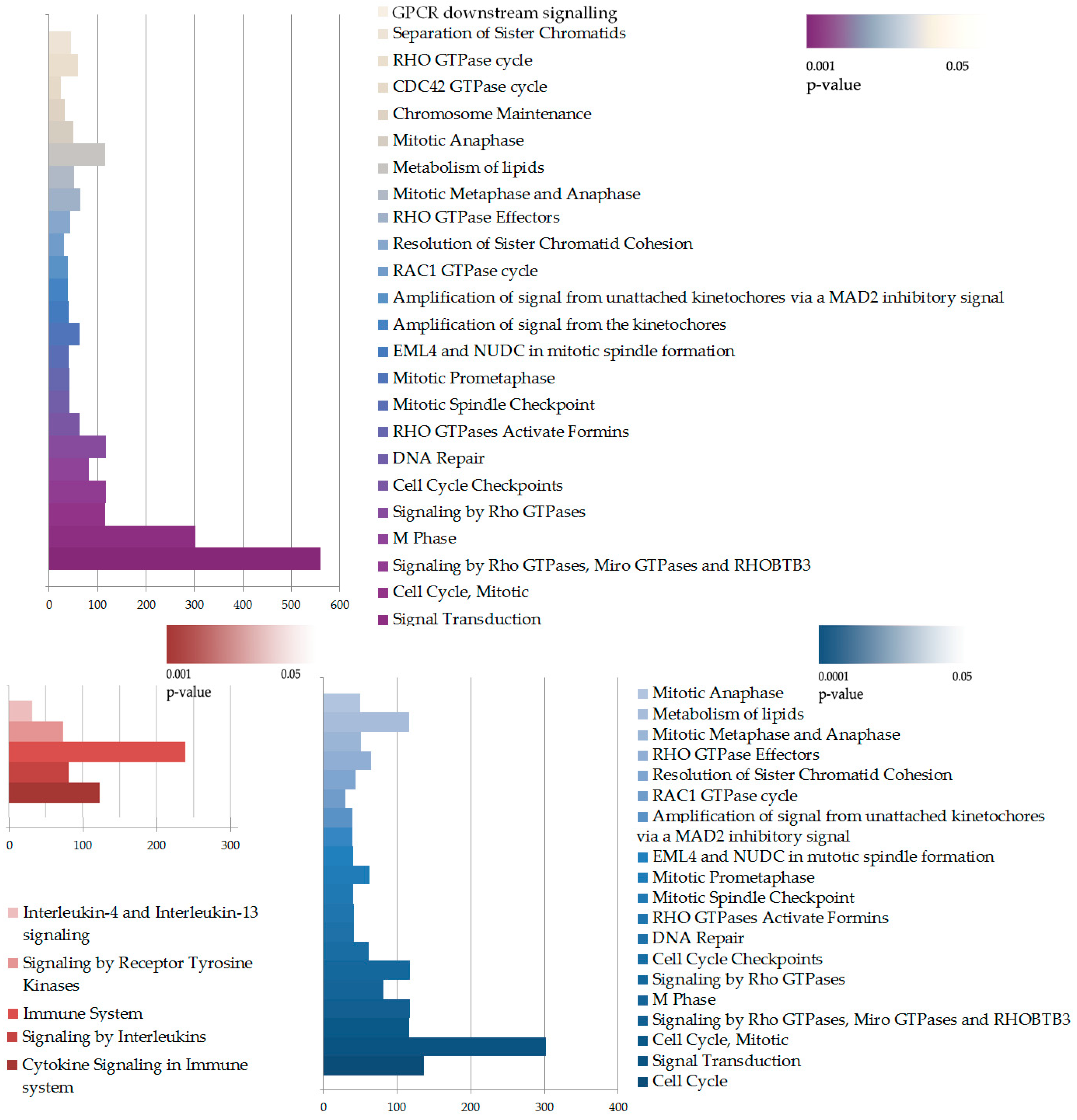
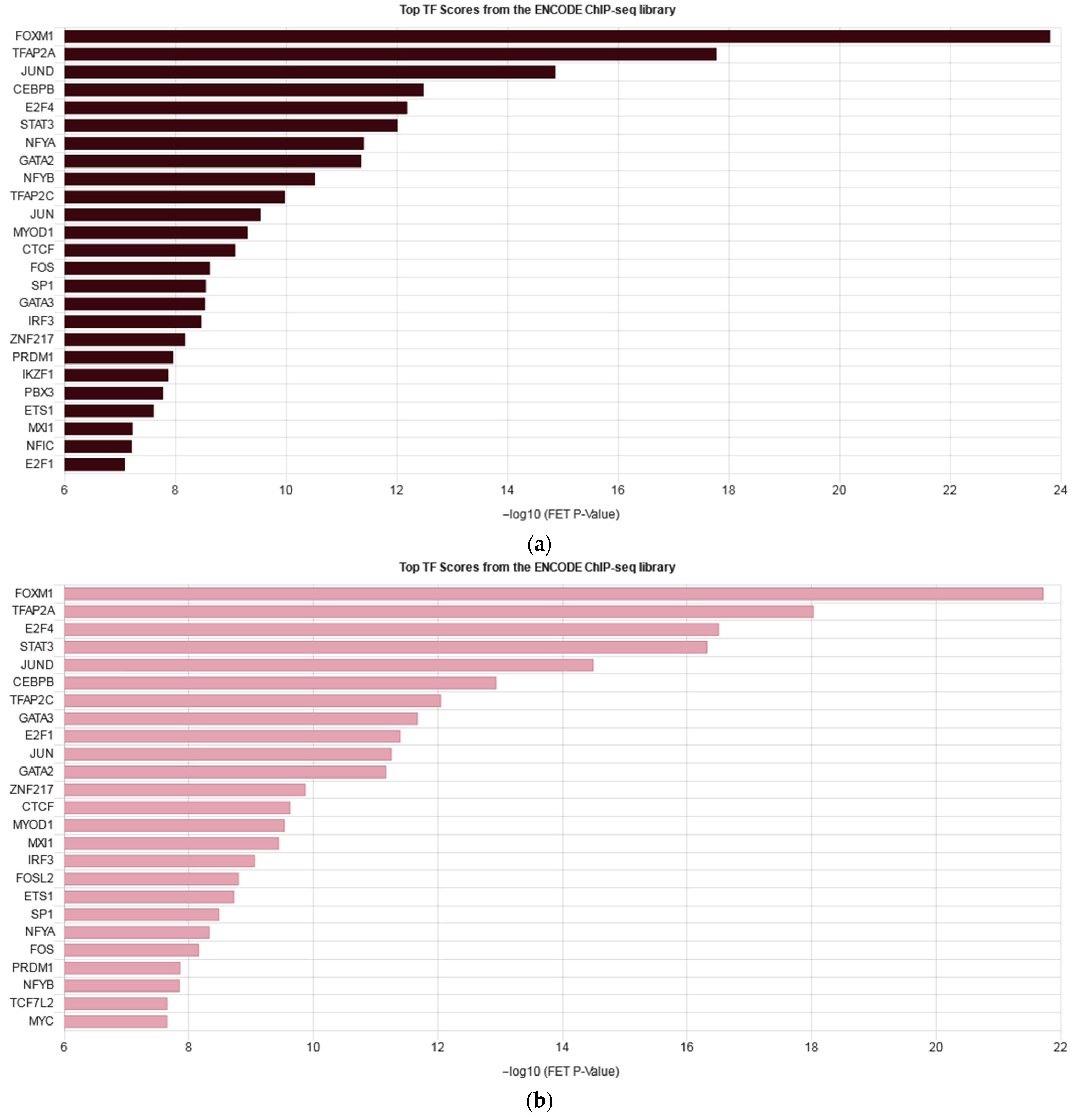
2.4. RNA Sequencing Analysis Unveils the Pathways Affected by IQGAP3 Knockdown
2.5. IQGAP3-Mediated Pathways under Inflammatory Stimulation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Oligos
4.3. Lentiviral Transduction
4.4. RNA Isolation, Reverse Transcription and Quantitative PCR
4.5. Western Blotting and Antibodies
4.6. RNAseq Library Preparation
4.7. RNA Sequencing and Data Analysis
4.8. Real-Time Cell Proliferation Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lowes, M.A.; Suárez-Fariñas, M.; Krueger, J.G. Immunology of psoriasis. Annu. Rev. Immunol. 2014, 32, 227–255. [Google Scholar] [CrossRef]
- Ran, D.; Cai, M.; Zhang, X. Genetics of psoriasis: A basis for precision medicine. Precis. Clin. Med. 2019, 2, 120–130. [Google Scholar] [CrossRef]
- Conrad, C.; Gilliet, M. Psoriasis: From Pathogenesis to Targeted Therapies. Clin. Rev. Allerg. Immunol. 2018, 54, 102–113. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- McKay, M.M.; Ritt, D.A.; Morrison, D.K. Signaling dynamics of the KSR1 scaffold complex. Proc. Natl. Acad. Sci. USA 2009, 106, 11022–11027. [Google Scholar] [CrossRef]
- Thines, L.; Roushar, F.J.; Hedman, A.C.; Sacks, D.B. The IQGAP scaffolds: Critical nodes bridging receptor activation to cellular signaling. J. Cell Biol. 2023, 222, e202205062. [Google Scholar] [CrossRef]
- Briggs, M.W.; Sacks, D.B. IQGAP proteins are integral components of cytoskeletal regulation. EMBO Rep. 2003, 4, 571–574. [Google Scholar] [CrossRef]
- Monteleon, C.L.; McNeal, A.; Duperret, E.K.; Oh, S.J.; Schapira, E.; Ridky, T.W. IQGAP1 and IQGAP3 Serve Individually Essential Roles in Normal Epidermal Homeostasis and Tumor Progression. J. Investig. Dermatol. 2015, 135, 2258–2265. [Google Scholar] [CrossRef]
- Zoheir, K.M.; Abd-Rabou, A.A.; Harisa, G.I.; Ashour, A.E.; Ahmad, S.F.; Attia, S.M.; Bakheet, S.A.; Abdel-Hamied, H.E.; Abd-Allah, A.R.; Kumar, A. Gene expression of IQGAPs and Ras families in an experimental mouse model for hepatocellular carcinoma: A mechanistic study of cancer progression. Int. J. Clin. Exp. Pathol. 2015, 8, 8821–8831. [Google Scholar]
- Hua, X.; Long, Z.Q.; Guo, L.; Wen, W.; Huang, X.; Zhang, W.W. IQGAP3 Overexpression Correlates With Poor Prognosis and Radiation Therapy Resistance in Breast Cancer. Front. Pharmacol. 2021, 11, 584450. [Google Scholar] [CrossRef]
- Wang, S.; Watanabe, T.; Noritake, J.; Fukata, M.; Yoshimura, T.; Itoh, N.; Harada, T.; Nakagawa, M.; Matsuura, Y.; Arimura, N.; et al. IQGAP3, a novel effector of Rac1 and Cdc42, regulates neurite outgrowth. J. Cell Sci. 2007, 120, 567–577. [Google Scholar] [CrossRef]
- Chekalin, E.V.; Zolotarenko, A.D.; Bruskin, S.A. IQGAP Genes in Psoriasis. Russ. J. Genet. 2020, 56, 345–353. [Google Scholar] [CrossRef]
- Tanos, B.E.; Yeaman, C.; Rodriguez-Boulan, E. An emerging role for IQGAP1 in tight junction control. Small GTPases 2018, 9, 375–383. [Google Scholar] [CrossRef]
- Shi, Y.; Qin, N.; Zhou, Q.; Chen, Y.; Huang, S.; Chen, B.; Shen, G.; Jia, H. Role of IQGAP3 in metastasis and epithelial–mesenchymal transition in human hepatocellular carcinoma. J. Transl. Med. 2017, 15, 176. [Google Scholar] [CrossRef]
- Kumar, D.; Hassan, M.K.; Pattnaik, N.; Mohapatra, N.; Dixit, M. Reduced expression of IQGAP2 and higher expression of IQGAP3 correlates with poor prognosis in cancers. PLoS ONE 2017, 12, e0186977. [Google Scholar] [CrossRef]
- Tae Kim, W.; Hwan Kim, Y.; Jeong, P.; Seo, S.; Kang, H.; Kim, Y.; Joong Yun, S.; Lee, S.; Moon, S.; Choi, Y.; et al. Urinary cell-free nucleic acid IQGAP3: A new non-invasive diagnostic marker for bladder cancer. Oncotarget 2018, 9, 14354–14365. [Google Scholar]
- Qian, E.N.; Han, S.Y.; Ding, S.Z.; Lv, X. Expression and diagnostic value of CCT3 and IQGAP3 in hepatocellular carcinoma. Cancer Cell Int. 2016, 16, 55. [Google Scholar] [CrossRef]
- Wu, K.; Zhang, X.; Li, F.; Xiao, D.; Hou, Y.; Zhu, S.; Liu, D.; Ye, X.; Ye, M.; Yang, J.; et al. Frequent alterations in cytoskeleton remodelling genes in primary and metastatic lung adenocarcinomas. Nat. Commun. 2015, 6, 10131. [Google Scholar] [CrossRef]
- Hu, G.; Xu, Y.; Chen, W.; Wang, J.; Zhao, C.; Wang, M. RNA Interference of IQ Motif Containing GTPase-Activating Protein 3 (IQGAP3) Inhibits Cell Proliferation and Invasion in Breast Carcinoma Cells. Oncol. Res. 2016, 24, 455–461. [Google Scholar] [CrossRef]
- Lin, M.; Liu, Y.; Ding, X.; Ke, Q.; Shi, J.; Ma, Z.; Gu, H.; Wang, H.; Zhang, C.; Yang, C.; et al. E2F1 transactivates IQGAP3 and promotes proliferation of hepatocellular carcinoma cells through IQGAP3-mediated PKC-alpha activation. Am. J. Cancer Res. 2019, 9, 285–299. [Google Scholar]
- Li, W.; Wang, Z.; Wang, H.; Zhang, J.; Wang, X.; Xing, S.; Chen, S. IQGAP3 in clear cell renal cell carcinoma contributes to drug resistance and genome stability. PeerJ 2022, 10, e14201. [Google Scholar] [CrossRef]
- Matsuo, J.; Chuang, L.S.H.; Tong, J.J.L.; Douchi, D.; Ito, Y. Identifying Adult Stomach Tissue Stem/Progenitor Cells Using the Iqgap3-2A-CreERT2 Mouse. Methods Mol. Biol. 2023, 2691, 3–17. [Google Scholar] [CrossRef]
- Man, X.Y.; Chen, X.B.; Li, W.; Landeck, L.; Dou, T.T.; Chen, J.Q.; Zhou, J.; Cai, S.Q.; Zheng, M. Analysis of epithelial-mesenchymal transition markers in psoriatic epidermal keratinocytes. Open Biol. 2015, 5, 150032. [Google Scholar] [CrossRef]
- Zolotarenko, A.; Chekalin, E.; Piruzian, E.; Bruskin, S. FRA1 mediates the activation of keratinocytes: Implications for the development of psoriatic plaques. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3726–3734. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Wang, S.; Kaibuchi, K. IQGAPs as Key Regulators of Actin-cytoskeleton Dynamics. Cell Struct. Funct. 2015, 40, 69–77. [Google Scholar] [CrossRef]
- Laurin, M.; Gomez, N.C.; Levorse, J.; Sendoel, A.; Sribour, M.; Fuchs, E. An RNAi screen unravels the complexities of Rho GTPase networks in skin morphogenesis. Elife 2019, 8, e50226. [Google Scholar] [CrossRef]
- Reeb, T.; Rhea, L.; Adelizzi, E.; Garnica, B.; Dunnwald, E.; Dunnwald, M. ARHGAP29 is required for keratinocyte proliferation and migration. bioRxiv 2023. [Google Scholar] [CrossRef]
- Matsuo, J.; Douchi, D.; Myint, K.; Mon, N.N.; Yamamura, A.; Kohu, K.; Heng, D.L.; Chen, S.; Mawan, N.A.; Nuttonmanit, N.; et al. Iqgap3-Ras axis drives stem cell proliferation in the stomach corpus during homoeostasis and repair. Gut 2021, 70, 1833–1846. [Google Scholar] [CrossRef]
- Leone, M.; Cazorla-Vázquez, S.; Ferrazzi, F.; Wiederstein, J.L.; Gründl, M.; Weinstock, G.; Vergarajauregui, S.; Eckstein, M.; Krüger, M.; Gaubatz, S.; et al. IQGAP3, a YAP Target, Is Required for Proper Cell-Cycle Progression and Genome Stability. Mol. Cancer Res. 2021, 19, 1712–1726. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Z.; Peng, H.; Zeng, K. Recent advances on the roles of epidermal growth factor receptor in psoriasis. Am. J. Transl. Res. 2019, 11, 520–528. [Google Scholar]
- Yang, Y.; Zhao, W.; Xu, Q.W.; Wang, X.S.; Zhang, Y.; Zhang, J. IQGAP3 promotes EGFR-ERK signaling and the growth and metastasis of lung cancer cells. PLoS ONE 2014, 9, e97578. [Google Scholar] [CrossRef]
- Mavropoulos, A.; Rigopoulou, E.I.; Liaskos, C.; Bogdanos, D.P.; Sakkas, L.I. The role of p38 MAPK in the aetiopathogenesis of psoriasis and psoriatic arthritis. Clin. Dev. Immunol. 2013, 2013, 569751. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, Y.; Cui, L.; Shi, Y.; Guo, C. Advances in the pathogenesis of psoriasis: From keratinocyte perspective. Cell Death Dis. 2022, 13, 81. [Google Scholar] [CrossRef]
- Zhang, J.; Shu, J.; Sun, H.; Zhai, T.; Li, H.; Li, H.; Sun, Y.; Huo, R.; Shen, B.; Sheng, H. CCN1 upregulates IL-36 via AKT/NF-κB and ERK/CEBP β-mediated signaling pathways in psoriasis-like models. J Dermatol. 2023, 50, 337–348. [Google Scholar] [CrossRef]
- Fréchette, L.; Degrandmaison, J.; Binda, C.; Boisvert, M.; Côté, L.; Michaud, T.; Lalumière, M.P.; Gendron, L.; Parent, J.L. Identification of the interactome of the DP1 receptor for Prostaglandin D2: Regulation of DP1 receptor signaling and trafficking by IQGAP1. Biochim. Et Biophys. Acta (BBA) Gen. Subj. 2021, 1865, 129969. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.B.; Schulz, V.V.; Miranti, C.K. A streamlined method for the design and cloning of shRNAs into an optimized Dox-inducible lentiviral vector. BMC Biotechnol. 2017, 17, 24. [Google Scholar] [CrossRef]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Keenan, A.B.; Torre, D.; Lachmann, A.; Leong, A.K.; Wojciechowicz, M.L.; Utti, V.; Jagodnik, K.M.; Kropiwnicki, E.; Wang, Z.; Ma’ayan, A. ChEA3: Transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019, 47, W212–W224. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO Enriched Only in HaCaT_Ctrl + Cyto | GO Enriched Only in HaCaT_sh + Cyto | ||||||
|---|---|---|---|---|---|---|---|
| GO ID | Term | % of DEGs | FDR | GO ID | Term | % of DEGs | FDR |
| GO:0070373 | negative regulation of ERK1 and ERK2 cascade | 0.78 | 0.000178 | GO:0050679 | positive regulation of epithelial cell proliferation | 1.45 | 0.010 |
| GO:0051781 | positive regulation of cell division | 0.74 | 0.012 | GO:0031099 | regeneration | 1.26 | 0.024 |
| GO:0008625 | extrinsic apoptotic signaling pathway via death domain receptors | 0.67 | 0.006 | GO:0010634 | positive regulation of epithelial cell migration | 1.22 | 0.006 |
| GO:0006692 | prostanoid metabolic process | 0.47 | 0.013 | GO:0051092 | positive regulation of NF-kappa B transcription factor activity | 1.15 | 0.005 |
| GO:1900744 | regulation of p38MAPK cascade | 0.43 | 0.022 | GO:1904019 | epithelial cell apoptotic process | 0.98 | 0.018 |
| GO:1900745 | positive regulation of p38MAPK cascade | 0.34 | 0.020 | GO:0002821 | positive regulation of adaptive immune response | 0.94 | 0.013 |
| GO:0038134 | ERBB2-EGFR signaling pathway | 0.16 | 0.016 | GO:0032649 | regulation of interferon-gamma production | 0.85 | 0.022 |
| GO:0007229 | integrin-mediated signaling pathway | 0.85 | 0.036 | ||||
| GO:0002224 | toll-like receptor signaling pathway | 0.83 | 0.040 | ||||
| GO:0032755 | positive regulation of interleukin-6 production | 0.72 | 0.038 | ||||
| GO:0031424 | keratinization | 0.70 | 0.043 | ||||
| GO:0032088 | negative regulation of NF-kappa B transcription factor activity | 0.66 | 0.048 | ||||
| GO:0032689 | negative regulation of interferon-gamma production | 0.41 | 0.024 | ||||
| GO:0010837 | regulation of keratinocyte proliferation | 0.41 | 0.038 | ||||
| GO:0033561 | regulation of water loss via skin | 0.38 | 0.384 | ||||
| GO:0061436 | establishment of skin barrier | 0.36 | 0.001 | ||||
| GO:0043276 | anoikis | 0.34 | 0.025 | ||||
| GO:0070268 | cornification | 0.28 | 0.001 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zolotarenko, A.; Bruskin, S. IQGAP3 Is an Important Mediator of Skin Inflammatory Diseases. Int. J. Mol. Sci. 2024, 25, 4545. https://doi.org/10.3390/ijms25084545
Zolotarenko A, Bruskin S. IQGAP3 Is an Important Mediator of Skin Inflammatory Diseases. International Journal of Molecular Sciences. 2024; 25(8):4545. https://doi.org/10.3390/ijms25084545
Chicago/Turabian StyleZolotarenko, Alena, and Sergey Bruskin. 2024. "IQGAP3 Is an Important Mediator of Skin Inflammatory Diseases" International Journal of Molecular Sciences 25, no. 8: 4545. https://doi.org/10.3390/ijms25084545
APA StyleZolotarenko, A., & Bruskin, S. (2024). IQGAP3 Is an Important Mediator of Skin Inflammatory Diseases. International Journal of Molecular Sciences, 25(8), 4545. https://doi.org/10.3390/ijms25084545