4. Materials and Methods
1H NMR and
13C NMR spectra were recorded with a Bruker Avance DRX 400 [400 and 100 MHz, respectively,
δ = 0 ppm (tetramethylsilane)] and Bruker Avance DRX 500 [500 and 125 MHZ, respectively,
δ = 0 ppm (tetramethylsilane)] (both Bruker Biospin, Karlsruhe, Baden Württemberg, Germany). Chemical shifts (
δ) were expressed in ppm relative to tetramethylsilane as an internal reference.
J values were given in Hz.. GC measurements were made with a PerkinElmer Autiosystem KL GC consisting of a flame ionisation detector and a Turbochrom Workstation data system (PerkinElmer Corporation, Norwalk, CT, USA). Separation of the enantiomers of the
O-acetyl derivatives of 1-phenyl-1-propanol was performed on a CHIRASIL-DEX CB column (2500 × 0.265 mm inner diameter, Agilent Technologies, Inc., Santa Clara, CA, USA; see
Supporting Information, Figures S147–S156). Chiral HPLC analysis was performed on a Chiralcel OD-H column (250 × 4.6 mm, see
Supporting Information, Figures S157–S162). UV detection was monitored at 210 nm or at 254 nm.
Optical rotations were performed with a PerkinElmer 341 polarimeter (PerkinElmer Inc., Shelton, CT, USA). Melting points were determined with a Kofler apparatus (Nagema, Dresden, Germany). Chromatographic separations were performed with Merck Kieselgel 60 (230–400 mesh ASTM, Merck Co., Darmstadt, Germany). Reactions were monitored with Merck Kieselgel 60 F254-precoated TLC plates (0.25 mm thickness, Merck Co., Darmstadt, Germany). All
1H-,
13C- NMR, HMQC, HMBC, and NOESY spectra are found in the
Supporting Information (see Figures S1–S144).
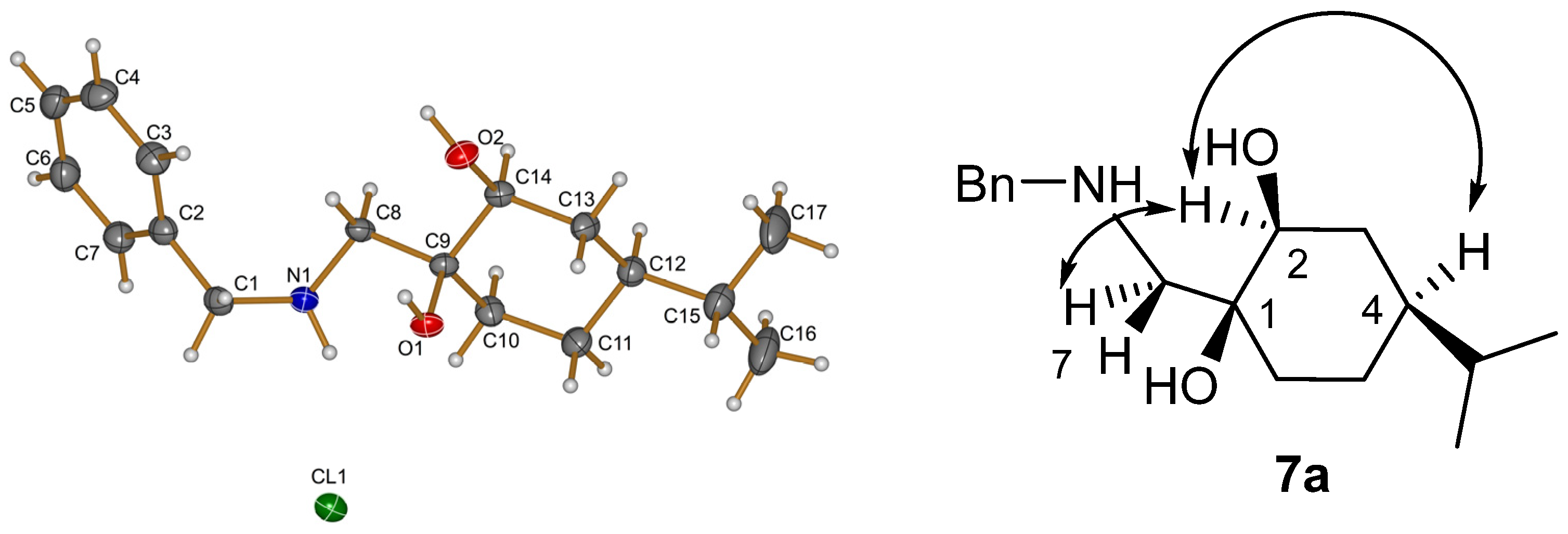
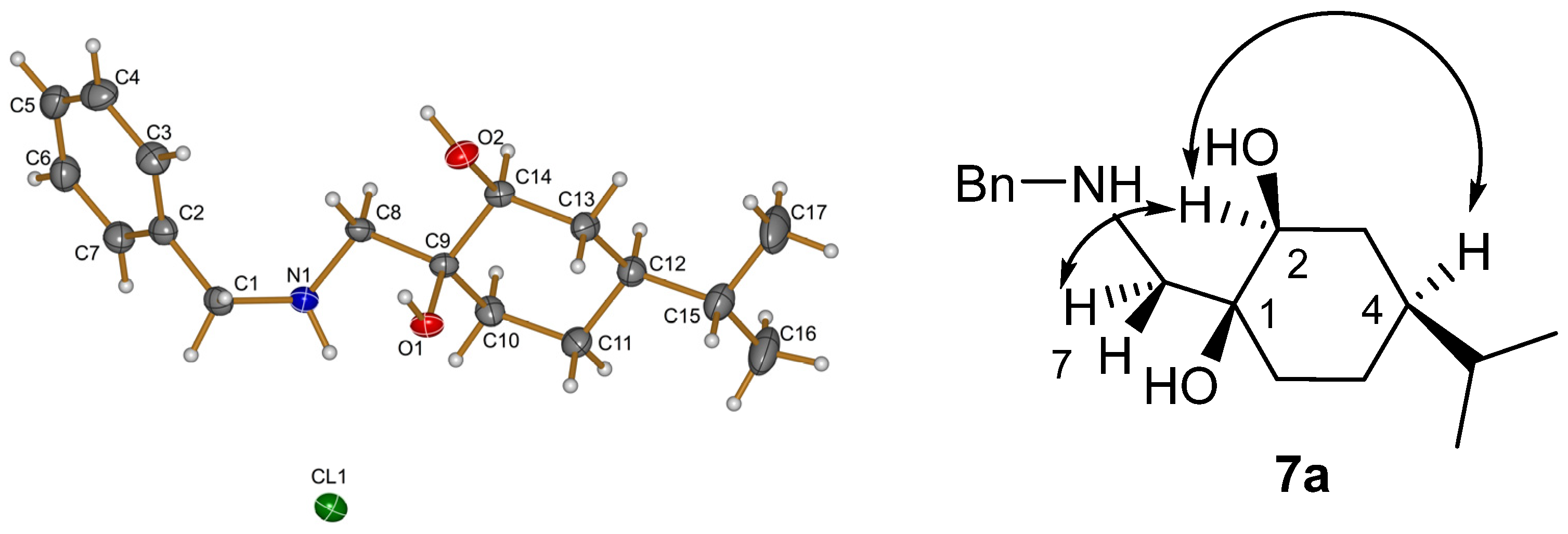
X-Ray structural determination of compound
7a was determined by a Rigaku Oxford Diffraction Supernova diffractometer ((Rigaku Oxford Diffraction, Yarnton, UK) using Cu Kα radiation. The
CrysAlisPro software package (version: 1.171.37.35) was used for cell refinement and data reduction (see
Supporting Information part, Table S1).
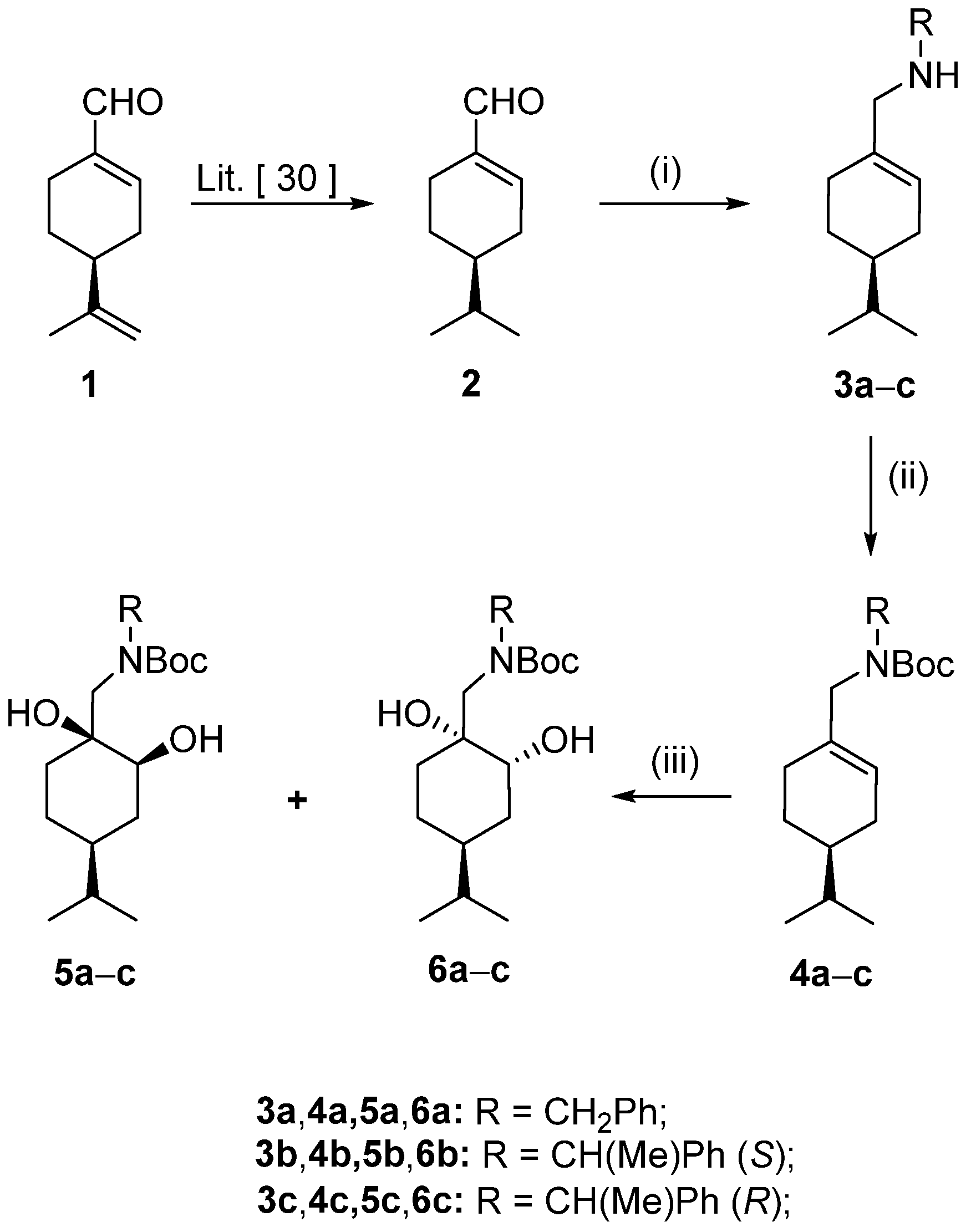
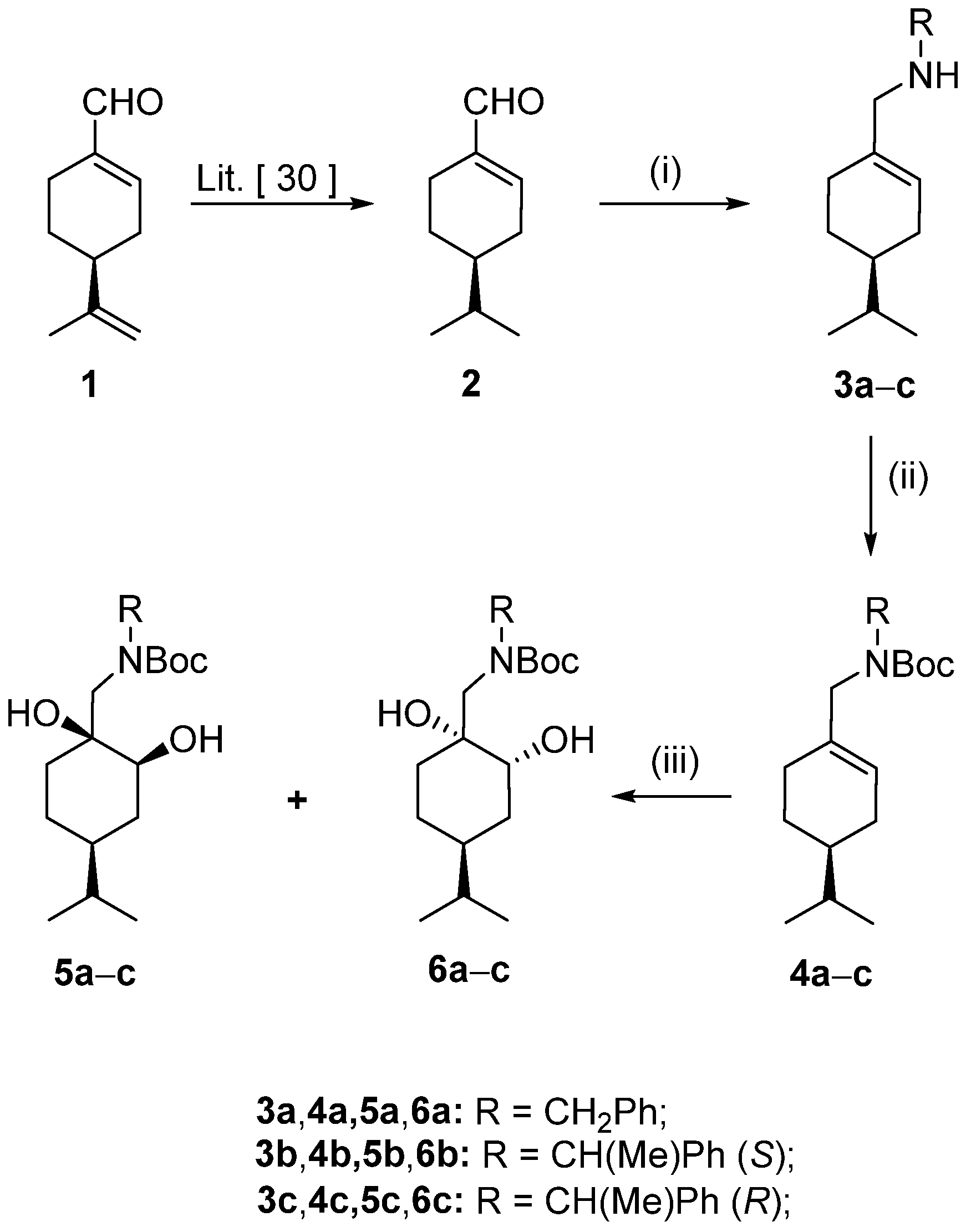
Starting materials: (−)-(
S)-peryllaldehyde (
1) was sourced commercially from Merck Co (Merck Co., Darmstadt, Germany). All chemicals and solvents were used as supplied. THF and toluene were dried over Na wire. (
S)-4-Isopropylcyclohex-1-ene carbaldehyde (
2) and (
S)-4-isopropylcyclohex-1-ene-1-ylmethanol (
15) were prepared according to literature procedures, and all spectroscopic data were similar to those reported therein [
30].
General method for the reductive amination of 2 with amines
The appropriate benzylamine (1.05 equiv, 27.6 mmol) was added to a stirred solution of compound 2 (4.00 g, 26.28 mmol) in dry EtOH (200 mL). The mixture was stirred at room temperature for 1 h. Afterwards, the solvent was evaporated, and the residue was redissolved in dry EtOH (200 mL) and then stirred for an additional hour. NaBH4 (2.98 g, 78.84 mmol) was added in small portions to the reaction mixture, which was stirred at room temperature (for compound 3a) or under reflux (for compounds 3b and 3c) for 2 h. Next, the solvent was removed under vacuum, and the crude product was dissolved in ice-cold H2O (70 mL) and extracted with dichloromethane (DCM) (3 × 100 mL). The combined organic phase was dried (Na2SO4), filtered, and evaporated. The crude product was purified via column chromatography on silica gel by applying toluene/EtOH 9:1, and then hydrochloride salts of the compounds were formed to yield 3a–c.
Prepared with benzylamine according to the general method. Yield: 6.32 g (86%); white crystals; m.p.: 178–185 °C; [α = −92 (c 0.25, MeOH). 1H NMR (DMSO-d6, 400.1 MHz): δ = 0.86 (3H, d, J = 6.8 Hz), 0.87 (3H, d, J = 6.8 Hz), 1.10–1.27 (2H, m), 1.41–1.51 (1H, m), 1.67–1.79 (2H, m), 1.99–2.17 (3H, m), 3.39 (2H, br s), 4.04 (2H, br s), 5.80–5.85 (1H, m), 7.38–7.45 (3H, m), 7.55–7.61 (2H, m), 9.50 (2H, br s); 13C NMR (100.6 MHz, DMSO-d6): δ = 20.3, 20.6, 26.2, 28.0, 29.2, 31.2, 32.4, 39.8, 50.1, 52.3, 129.4, 129.7, 129.9, 130.4, 131.1, 132.8.; HR-MS (ESI): m/z calcd for C17H26N [M + H]+: 244.20598; found: 244.20532.
Prepared with (S)-1-phenylethylamine according to the general method. Yield: 5.64 g (73%), white crystals, m.p.: 208–212 °C; [α = −43 (c 0.25, MeOH); 1H NMR (DMSO-d6, 400.1 MHz): δ = 0.85 (3H, d, J = 6.8 Hz), 0.86 (3H, d, J = 6.8 Hz), 1.08–1.24 (2H, m), 1.41–1.50 (1H, m), 1.56 (3H, d, J = 6.6 Hz), 1.64–1.77 (2H, m), 1.88–2.14 (3H, m), 3.08 (1H, d, J = 13.3 Hz), 3.24 (1H, d, J = 13.4 Hz), 4.13–4.26 (1H, m), 5.65–5.73 (1H, m), 7.34–7.46 (3H, m), 7.50–7.61 (2H, m), 9.24 (2H, br s); 13C NMR (100.6 MHz, DMSO-d6): δ = 20.3, 20.5, 20.6, 26.3, 28.0, 29.1, 32.4, 39.8, 51.3, 57.7, 128.6, 129.0, 129.4, 129.7, 138.4; HR-MS (ESI): m/z calcd for C18H28N [M + H]+: 258.22163; found: 258.22098.
Prepared with (R)-1-phenylethylamine according to the general method. Yield: 5.10 g (66%), white crystals, m.p.: 204–206 °C; [α = −76 (c 0.25 MeOH); 1H NMR (DMSO-d6, 400.1 MHz): δ = 0.86 (3H, d, J = 6.7 Hz), 0.87 (3H, d, J = 6.7 Hz), 1.02–1.14 (1H, m), 1.16–1.26 (1H, m), 1.40–1.51 (1H, m), 1.61 (3H, d, J = 6.8 Hz), 1.63–1.75 (2H, m), 1.97–2.07 (3H, m), 3.11 (1H, d, J = 13.5 Hz), 3.27 (1H, d, J = 13.5 Hz), 4.26 (1H, dd, J = 6.8, 13.8 Hz), 5.66–5.73 (1H, m), 7.35–7.47 (3H, m), 7.56–7.65 (2H, m), 9.30 (1H, br s), 9.73 (1H, br s); 13C NMR (100.6 MHz, DMSO-d6): δ = 20.3, 20.6, 26.2, 28.0, 29.2, 31.2, 32.4, 39.8, 50.1, 52.3, 129.4, 129.7, 129.9, 130.4, 131.1, 132.8; HR-MS (ESI): m/z calcd for C18H28N [M + H]+: 258.22163; found: 258.22099.
General procedure for tert-butyloxycarbonyl (BOC) protection of compounds 3a–c
To a stirred solution of the liberated bases of allylamines 3a–c (12 mmol) in dry THF (30 mL), di-tert-butyl dicarbonate (2.88 g, 13.2 mmol for 4a; 5.76 g, 26.4 mmol for 4b, and 3.56 g, 16.32 mmol for 4c), TEA (3.64 g 36 mmol), and a catalytic amount of DMAP (0.15 g, 1.2 mmol) were added. The mixture was stirred overnight at rt. After completion of the reaction, indicated by means of TLC, the solvent was evaporated. The crude product was purified by column chromatography on silica gel by using n-hexane/EtOAc 9:1 for 4a, n-hexane/EtOAc 19:1 for 4b, or n-hexane/Et2O 19:1 for 4c.
Prepared from 3a according to the general method. Yield: product: 3.92 g (95%) (mixture of two rotamers in CDCl3); colourless oil, [α = −55 (c 0.25, MeOH). 1H NMR (CDCl3, 400.1 MHz): δ = 0.88 (3H, d, J = 6.8 Hz), 0.89 (3H, d, J = 6.8 Hz), 1.08–1.31 (2H, m), 1.40–1.51 (1H, m, overlapped with s), 1.46 (9H, s), 1.68–1.79 (2H, m), 1.82–2.08 (3H, m), 3.57–3.85 (2H, m), 4.26–4.46 (2H, m), 5.41–5.49 (1H, m), 7.16–7.34 (5H, m); 13C NMR (100.6 MHz, CDCl3): δ = 19.8, 19.9, 20.1, 24.1, 26.2, 26.9, 28.6, 28.9, 29.5, 31.3, 32.3, 33.0, 40.2, 44.3, 48.6, 49.1, 51.6, 51.9, 79.7, 124.3, 126.7, 127.1, 127.6, 128.2, 128.5, 133.7, 138.7, 156.1; HR-MS (ESI): m/z calcd for C18H26NO2 [M − CH(CH3)3 + H + H]+: 288.19581; found: 288.19506.
Prepared from 3b according to the general method. Yield: 3.99 g (93%), colourless oil, [α = −120 (c 0.25, MeOH) 1H NMR (DMSO-d6, 400.1 MHz): δ = 0.83 (3H, d, J = 6.1 Hz), 0.84 (3H, d, J = 6.1 Hz), 0.93–0.95 (1H, m), 1.09–1.19 (1H, m), 1.33 (9H, s), 1.35–1.53 (2H, m), 1.46 (3H, d, J = 7.0 Hz), 1.56–1.70 (2H, m), 1.74–1.96 (3H, m), 3.60 (2H, br s), 5.35 (1H, s), 7.19–7.36 (5H, m); 13C NMR (100.6 MHz, DMSO-d6): δ = 17.9, 20.0, 20.2, 26.0, 26.8, 28.5, 28.6, 32.1, 32.8, 40.1, 44.0, 53.4, 79.0, 127.2, 127.3, 128.5, 135.2, 142.2, 155.4. HR-MS (ESI): m/z calcd for C19H28NO2 [M − CH(CH3)3 + H + H]+: 302.21146; found: 302.21089.
Prepared from 3c according to the general method. Yield: 3.86 g (90%) colourless oil, [α = +4 (c 0.25, MeOH) 1H NMR (DMSO-d6, 400.1 MHz): δ = 0.83 (3H, d, J = 6.1 Hz), 0.84 (3H, d, J = 6.1 Hz), 0.93–0.95 (1H, m), 1.09–1.19 (1H, m), 1.33 (9H, s), 1.35–1.53 (2H, m), 1.46 (3H, d, J = 7.0 Hz), 1.56–1.70 (2H, m), 1.74–1.96 (3H, m), 3.60 (2H, br s), 5.35 (1H, s), 7.19–7.36 (5H, m); 13C NMR (100.6 MHz, DMSO-d6): δ = 17.9, 20.0, 20.2, 26.0, 26.8, 28.5, 28.6, 32.1, 32.8, 40.1, 44.0, 53.4, 79.0, 127.2, 127.3, 128.5, 135.2, 142.2, 155.4. HR-MS (ESI): m/z calcd for C19H28NO2 [M -CH(CH3)3 + H + H]+: 302.21146; found: 302.21089.
General method for dihydroxylation of 4a–c
To a solution of 4a–c (10 mmol) in acetone (50 mL), an aqueous solution of 4-methylmorpholine-4-oxide (NMO) (8.5 mL, 50% aqueous sol.) and a solution of OsO4 in tert-BuOH (4.5 mL, 2% tert-BuOH solution) were added in one portion. The mixture was stirred at room temperature overnight, and then quenched by the addition of a saturated aqueous solution of Na2SO3 (80 mL) and extracted with EtOAc (3 × 60 mL). The combined organic phase was dried, filtered, and evaporated. The products were purified by means of column chromatography on silica gel in n-hexane/EtOAc 4:1 mixture.
Prepared from 4a according to the general method. Yield: 0.95 g (25%) light yellow oil [α = −15 (c 0.25, MeOH) 1H NMR (DMSO-d6, 400.1 MHz): δ = 0.85 (3H, d, J = 6.0 Hz), 0.86 (3H, d, J = 6.0 Hz), 1.09–1.20 (1H, m), 1.34–1.71 (6H, m), 1.44 (9H, s), 1.80–1.87 (1H, m), 3.31 (3H, dd, overlapped with br s, J = 15.5, 35.6 Hz), 3.59–3.65 (1H, m), 4.33 (1H, br s), 4.50 (2H, br s), 7.15–7.37 (5H, m); 13C NMR (100.6 MHz, DMSO-d6): δ = 20.4, 20.5, 25.2, 28.4, 29.8, 30.1, 30.4, 32.2, 37.5, 53.4, 53. 6, 70.0, 74.8, 81.5, 127.2, 127.5, 128.8, 138.0, 154.6. HR-MS (ESI): m/z calcd for C22H36NO4 [M + H]+: 378.26389; found: 378.26325, calcd for C22H35NO4Na [M + Na]+: 400.24583; found: 400.24510.
Prepared from 4a according to the general method. Yield: 1.72 g (46%) white crystals, m.p. 103–106 °C; [α = −36 (c 0.25, MeOH); 1H NMR (CDCl3, 400.1 MHz): δ = 0.88 (6H, d, J = 6.8 Hz), 1.06–1.16 (1H, m), 1.24–1.55 (5H, m), 1.41 (9H, s), 1.70–1.79 (2H, m), 2.95 (1H, br s), 2.99 (1H, d, J = 15.1 Hz), 3.42 (1H, dt, J = 5.0, 11.5 Hz), 3.54 (1H, d, J = 14.7 Hz), 4.28 (2H, d, overlapped with br s, J = 15.1 Hz), 4.65 (1H, d, J = 15.1 Hz), 7.12–7.36 (5H, m); 13C NMR (100.6 MHz, CDCl3): δ = 20.2, 20.3, 24.3, 28.7, 32.6, 32.9, 34.2, 42.9, 53.8, 55.5, 71.8, 74.2, 81.6, 127.4, 127.7, 129.0, 138.3, 158.5. HR-MS (ESI): m/z calcd for C22H36NO4 [M + H]+: 378.26389; found: 378.26332, calcd for C22H35NO4Na [M + Na]+: 400.24583; found: 400.24518.
Prepared from 4b according to the general method. Yield: 1.68 g (43%), oil, [α = +2 (c 0.26, MeOH); 1H NMR (CDCl3, 400.1 MHz): δ = 0.88 (3H, d, J = 6.8 Hz), 0.87 (3H, d, J = 6.2 Hz), 0.91–1.01 (1H, m), 1.23–1.46 (5H, m), 1.32 (9H, s), 1.68 (3H, d, J = 7.1 Hz), 1.69–1.77 (2H, m), 3.19 (1H, d, J = 14.8 Hz), 3.38 (1H, dd, J = 4.6, 11.3), 3.57 (1H, d, J = 14.8 Hz), 4.73 (1H, dd, J = 6.9, 14.1 Hz), 7.19–7.35 (5H, m); 13C NMR (100.6 MHz, CDCl3): δ = 19.6, 20.2, 20.3, 24.4, 28.6, 32.9, 33.0, 34.2, 42.9, 57.7, 58.9, 72.4, 73.5, 81.7, 126.5, 127.4, 128.7, 142.7, 158.8. HR-MS (ESI): m/z calcd for C23H38NO4 [M + H]+: 392.27954; found: 392.27903, calcd for C23H37NO4Na [M + Na]+: 414.26148; found: 414.26077.
Prepared from 4b according to the general method. Yield: 1.58 g (38%), oil, [α = −5 (c 0.26, MeOH); 1H NMR (CDCl3, 400.1 MHz): δ = 0.79 (3H, d, J = 6.2 Hz), 1.02–1.14 (1H, m), 1.24–1.53 (5H, m), 1.30 (9H, s), 1.48–1.60 (2H, m), 1.65 (3H, d, J = 7.0 Hz), 1.74–1.82 (1H, m), 3.29 (1H, d, J = 14.8 Hz), 3.44 (1H, d, J = 14.8), 3.57–3.62 (1H, m), 4.81 (1H, dd, J = 6.9, 14.1 Hz), 7.21–7.36 (5H, m); 13C NMR (100.6 MHz, CDCl3): δ = 18.5, 20.8, 21.0, 25.0, 28.6, 30.1, 30.5, 32.6, 38.1, 54.9, 59.1, 70.8, 74.1, 81.8, 127.0, 127.5, 128.7, 142.4, 159.0. HR-MS (ESI): m/z calcd for C23H38NO4 [M + H]+: 392.27954; found: 392.27901, calcd for C23H37NO4Na [M + Na]+: 414.26148; found: 414.26077.
tert-Butyl (((1S,2S,4S)-1,2-dihydroxy-4-isopropylcyclohexyl)methyl)((R)-1-phenylethyl)carbamate (5c)
Prepared from 4c according to the general method. Yield: 1.92 g (48%) oil, [α = −18 (c 0.255, MeOH), 1H NMR (400.1 MHz, CDCl3): δ = 0.92 (6H, d, J = 6.8 Hz), 1.06–1.17 (1H, m), 1.34 (9H, s), 1.32–1.56 (7H, m), 1.72 (3H, d, J = 7.0 Hz), 1.73–1.81 (2H, m), 3.22 (1H, d, J = 14.6 Hz), 3.37–3.49 (2H, m), 3.60 (3H, d, J = 14.6 Hz), 4.77 (1H, q, J = 7.0, 13.8 Hz), 7.22–7.39 (5H, m). 13C NMR (100.6 MHz, CDCl3): δ = 19.8, 20.3, 20.5, 24.5, 28.8, 33.0, 33.1, 43.3, 43.1, 57.9, 59.1, 72.6, 73.7, 81.9, 126.7, 127.5, 128.9, 142.8, 158.9; HR-MS (ESI): m/z calcd for C23H38NO4 [M + H]+: 392.27954; found: 392.27901, calcd for C23H37NO4Na [M + Na]+: 414.26148; found: 414.26072.
Prepared from 4c according to the general method. Yield: 1.88 g (48%), oil, [α = −15 (c 0.275, MeOH), 1H NMR (DMSO-d6, 400.1 MHz): mixture of two rotamers, δ = 0.88 (3H, d, J = 6.8 Hz), 0.87 (3H, d, J = 6.2 Hz), 0.91–1.01 (1H, m), 1.23–1.46 (5H, m), 1.32 (9H, s), 1.68 (3H, d, J = 7.1 Hz), 1.69–1.77 (2H, m), 3.19 (1H, d, J = 14.8 Hz), 3.38 (1H, dd, J = 4.6, 11.3), 3.57 (1H, d, J = 14.8 Hz), 4.73 (1H, dd, J = 6.9, 14.1 Hz), 7.19–7.35 (5H, m); 13C NMR (100.6 MHz, DMSO-d6): δ = 20.2, 20,3, 20.7, 20.8, 24.0, 25.3, 28.3, 30.7, 32.5, 33.2, 33.7, 42.6, 54.6, 69.7, 72.6, 74.4, 79.0, 79.2, 126.4, 126.7, 128.2, 128.3, 144.3, 156.1. HR-MS (ESI): m/z calcd for C23H38NO4 [M + H]+: 392.27954; found: 392.27882, calcd for C23H37NO4Na [M + Na]+: 414.26148; found: 414.26057.
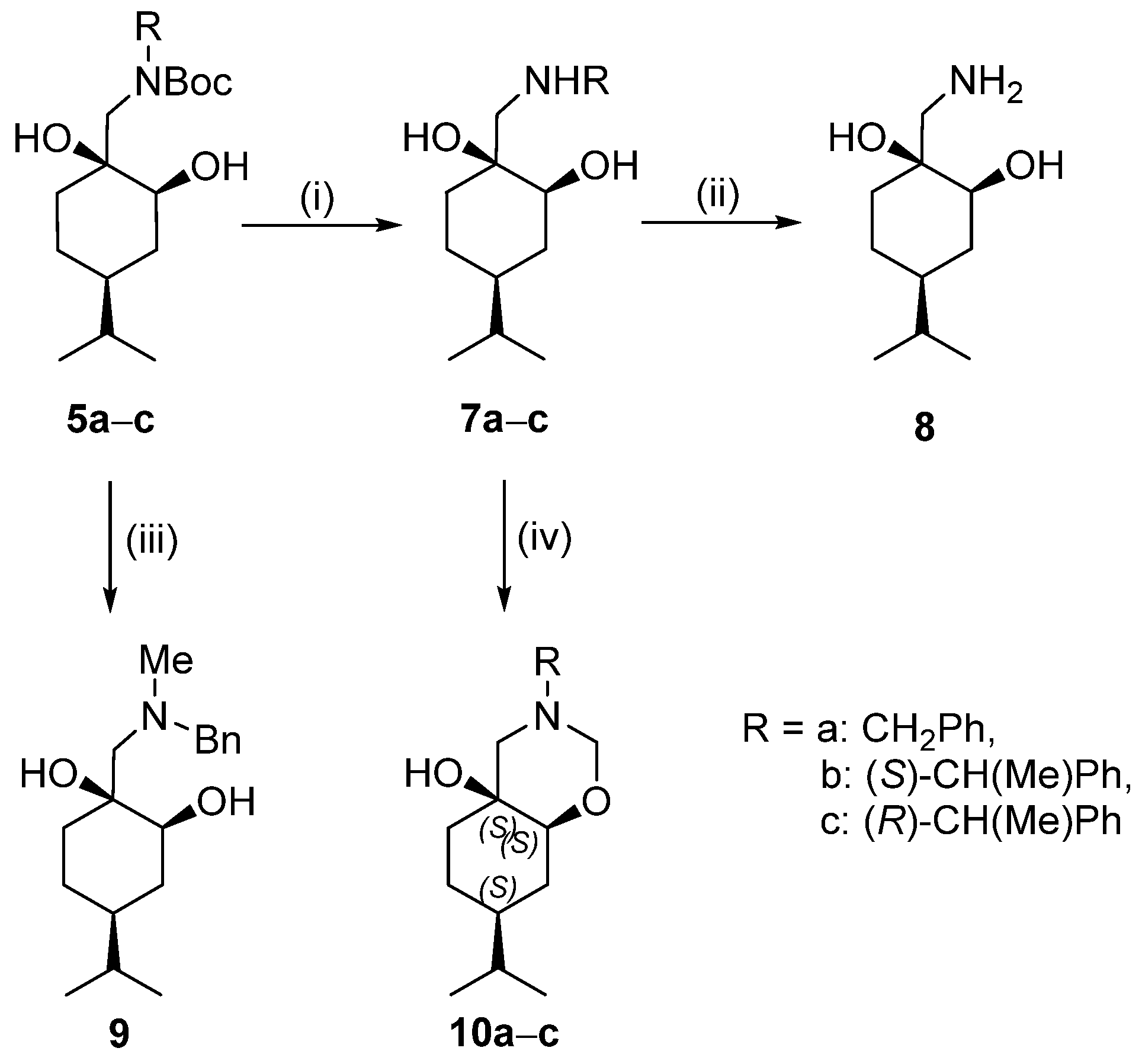
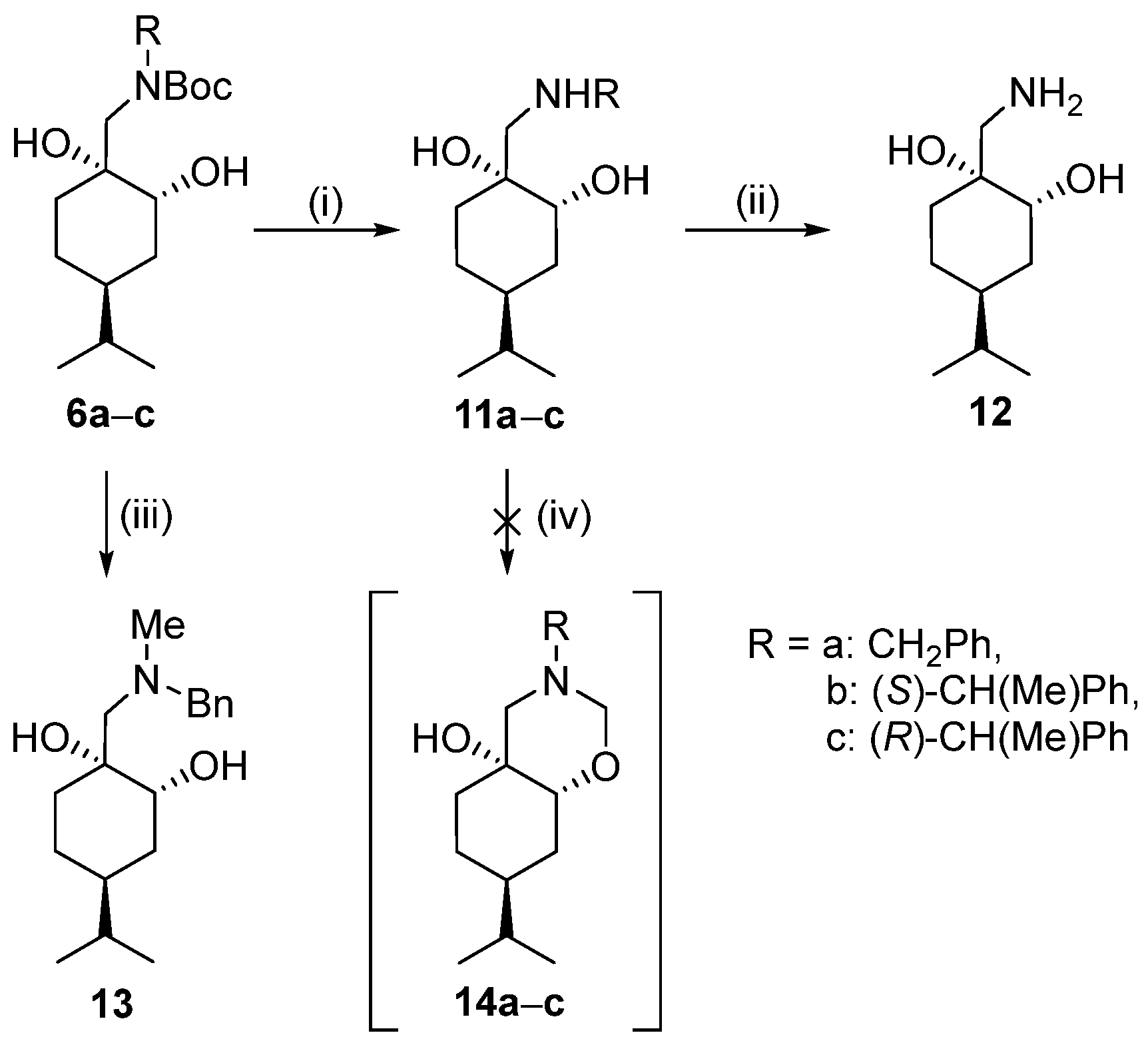
General procedure for the Boc deprotection of 5a–c and 6a–c
To a stirred solution of compounds 5a–c or 6a–c (3 mmol) in Et2O (25 mL), an 18% aqueous solution of HCl (70 mL) was added and the mixture was stirred overnight. After the reaction was complete (indicated by TLC), the solvent was removed by vacuum evaporation. The resulting hydrochloride salt was filtered off and washed with Et2O.
Prepared from 5a according to the general method. Yield: 0.83 g (88%); white crystals, m.p. 215–218 °C; [α = −5 (c 0.225, MeOH). 1H NMR (DMSO-d6, 400.1 MHz): δ = 0.84 (6H, d, J = 6.7 Hz, CHMe2), 1.01–1.12 (1H, m, H4β), 1.15–1.46 (5H, m, CH2(5), CHMe2, H4α, H6β), 1.48–1.56 (1H, m, 6Hα), 1.67–1.78 (1H, m, 3Hα), 2.73–2.83 (1H, m, CH2NH), 3.01–3.11 (1H, m, CH2NH), 3.37 (1H, dd, J = 4.4, 11.3 Hz, CHOH), 4.10–4.24 (2H, m, CH2Ph), 7.37–7.48 (3H, m, 3 × CHAr), 7.55–7.66 (2H, m, 2 × CHAr), 8.77 (1H, br s, NH2+), 9.15 (1H, br s, NH2+); 13C NMR (100.6 MHz, DMSO-d6): δ = 20.6 and 20.7 (CHMe2), 23.9 (C5), 32.8 (CHMe2), 33.9 (C3), 34.0 (C6), 42.5 (C4), 51.8 (CH2NH), 55.6 (CH2Ph), 71.1 (C1), 73.8 (C2), 129.5 (2 × CHAr), 129.8 (2 × CHAr), 131.1, 132.7 (CqAr). HR-MS (ESI): m/z calcd for C17H28NO2 [M + H]+: 278.21146; found: 278.21069.
Prepared from 5b according to the general method. Yield: 0.81 g (82%); white crystals, m.p. 205–207 °C; [α = +12 (c 0.255, MeOH). 1H NMR (DMSO-d6, 400.1 MHz): δ = 0.82 (6H, d, J = 6.7 Hz), 0.97–1.54 (7H, m), 1.64 (3H, d, J = 6.6 Hz), 1.77 (1H, d, J = 12.3 Hz), 2.43–2.51 (1H, m), 2.99–3.09 (1H, m), 3.32 (1H, d, J = 9.4 Hz), 3.37 (1H, br s,), 4.33–4.44 (1H, m), 4.67 (1H, s), 5.11 (1H, br s), 7.36–7.47 (3H, m), 7.60–7.67 (2H, m), 9.03 (1H, br s), 9.12 (1H, br s); 13C NMR (100.6 MHz, DMSO-d6): δ = 20.4, 20.5, 20.6, 23.8, 32.8, 33.8, 33.9, 42.5, 54.8, 59.4, 71.3, 73.3, 128.8, 129.6, 129.7, 138.1. HR-MS (ESI): m/z calcd for C18H30NO2 [M + H]+: 292.22711; found: 292.22641.
Prepared from 5c according to the general method. Yield: 0.73 g (84%), viscous oil; [α = −18 (c 0.26, MeOH). 1H NMR (DMSO-d6, 400.1 MHz): δ = 0.82 (6H, d, J = 6.7 Hz), 0.93–1.03 (1H, m), 1.30–1.54 (1H, m), 1.11 (1H, dt, J = 4.4, 12.9 Hz), 1.16–1.48 (7H, m), 1.26 (3H, d, J = 6.8 Hz), 2.29 (1H, d, J = 12.2 Hz), 2.48 (1H, d, J = 12.2 Hz), 3.30 (1H, dd, J = 4.3, 11.6 Hz), 3.67 (1H, q, J = 6.5, 12.9 Hz), 7.18–7.35 (5H, m); 13C NMR (100.6 MHz, DMSO-d6): δ = 20.2, 20.3, 23.9, 24.8, 32.5, 33.4, 34.3, 42.1, 57.4, 58.5, 71.4, 74.6, 126.9, 127.2, 128.8, 146.0. HR-MS (ESI): m/z calcd for C18H30NO2 [M + H]+: 292.22711; found: 292.22652.
Prepared from 6a according to the general method. Yield: 0.91 g (97%); white crystals, m.p. 172–174 °C; [α = −13 (c 0.265, MeOH). 1H NMR (DMSO-d6, 400.1 MHz): δ = 0.78 (3H, d, J = 9.4 Hz, CHMe2), 0.80 (3H, d, J = 9.4 Hz, CHMe2), 0.96–1.08 (1H, m, H5α), 1.18–1.28 (1H, m, H6α), 1.30–1.54 (4H m, H5β, CHMe2, CH2(3)), 1,56–1.67 (2H, m, H4, H6β), 2.85 (2H, br s, CH2NH), 3.53 (1H, d, J = 3.3 Hz, H2), 4.15 (2H, s, CH2Ph), 4.80 (1H, br s, NH2+), 4.89 (1H, s, NH2+), 7.37–7.46 (3H, m, 3 × CHAr), 7.55–7.64 (2H, m, 2 × CHAr); 13C NMR (100.6 MHz, DMSO-d6): δ = 21.1 (CHMe2), 24.9 (C5), 30.2 (CHMe2), 30.6 (C3), 33.3 (C6), 37.8 (C4), 51.5 (CH2NH), 52.3 (CH2Ph), 70.0 (C2), 71.6 (C1), 129.5 (2 × CHAr), 129.8 (CHAr), 131.2 (2 × CHAr), 132.5 (CqAr). HR-MS (ESI): m/z calcd for C17H28NO2 [M + H]+: 278.21146; found: 278.21068.
Prepared from 6b according to the general method. Yield: 0.78 g (79%), white crystals. m.p. 172–174 °C, [α = −15 (c 0.26, MeOH) 1H NMR (DMSO-d6, 400.1 MHz): δ = 0.83 (3H, d, J = 6.6 Hz), 1.00–1.53 (7H, m), 1.63 (3H, d, J = 6.8 Hz), 1.66–1.76 (1H, m), 2.72–2.89 (1H, m), 3.28–3.38 (1H, m), 4.30–4.43 (1H, m), 4.61 (1H, s), 5.09 (1H, br s), 7.35–7.48 (3H, m), 7.55–7.65 (2H, m), 8.69 (1H, br s), 9.27 (1H, br s); 13C NMR (100.6 MHz, DMSO-d6): 20.2, 20.5, 20.7, 32.8, 33.8, 42.5, 54.7, 59.2, 71.1, 73.7, 128.8, 129.7, 129.8, 138.1. HR-MS (ESI): m/z calcd for C18H30NO2 [M + H]+: 292.22711; found: 292.22634.
Prepared from 6c according to the general method. Yield: 0.77 g (78%); white crystals m.p. 198–200 °C; [α = +6 (c 0.26, MeOH). 1H NMR (DMSO-d6, 400.1 MHz): δ = 0.76 (3H, d, J = 8.3 Hz), 0.78 (3H, d, J = 8.3 Hz), 0.98–1.14 (1H, m), 1.26–1.63 (6H, m), 1.65 (3H, d, J = 6.9 Hz), 2.51–2.60 (1H, m), 2.84–2.95 (1H, m), 3.37 (1H, br s), 3.45 (1H, br s), 4.30–4.41 (1H, m), 4.78 (1H, br s), 4.93 (1H, s), 7.35–7.47 (3H, m), 7.57–7.65 (2H, m), 8.67 (1H, br s), 9.35 (1H, br s); 13C NMR (100.6 MHz, DMSO-d6): δ = 20.2, 21.1, 24.9, 30.2, 30.6, 33.2, 37.7, 51.8, 59.0, 69.7, 71.6, 128.9, 129.7, 129.8, 138.0. HR-MS (ESI): m/z calcd for C18H30NO2 [M + H]+: 292.22711; found: 292.22647.
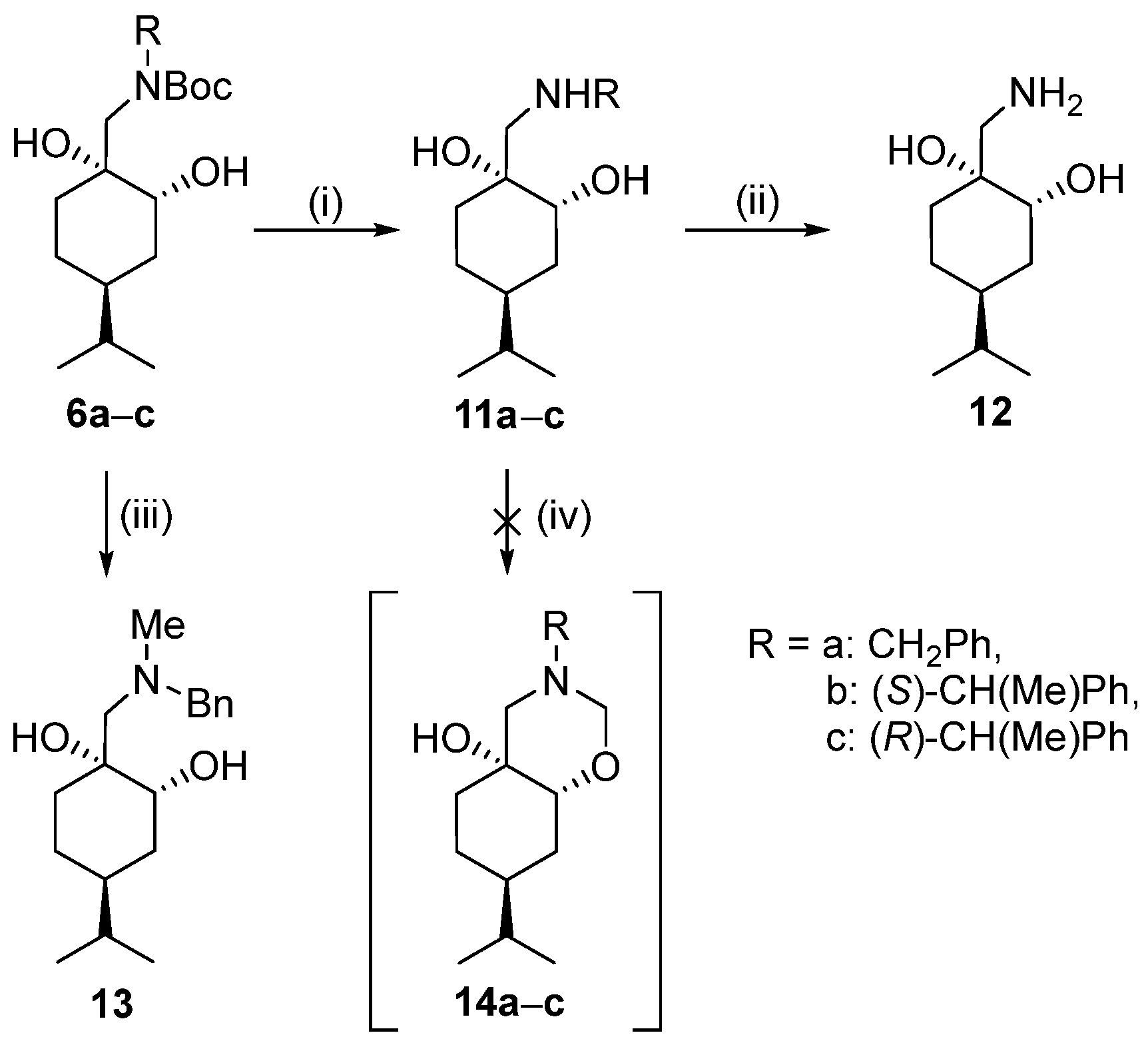
General procedure for debenzylation of 7a and 11a
To a stirred suspension of palladium-on-carbon (10% Pd/C, 0.10 g) in a mixture of n-hexane/EtOAc (24 mL) (1:2 mixture for 7a, 1:1 mixture for 11a), 7a or 11a (1.8 mmol) was added and the reaction mixture was stirred under a H2 atmosphere at room temperature and normal pressure. After the reaction was completed (monitored by TLC), the mixture was filtered through a short Celite pad, the solvent was concentrated, and the hydrochloride salts of compounds were formed.
Prepared from 7a according to the general method. Yield: 0.14 g (35%); white crystals m.p. 136–137 °C; [α = −4 (c 0.265, MeOH). 1H NMR (DMSO-d6, 400.1 MHz): δ = 0.81 (6H, d, J = 6.8 Hz, CHMe2), 0.98–1.44 (6H, m, H3α, H(4), CH2(5), CHMe2, H6β), 1.46–1.54 (1H, m, H6α), 1.60–1.69 (1H, m, H3α), 2.57–2.69 (1H, m, CH2NH), 2.91–3.02 (1H, m, CH2NH), 3.28–3.38 (1H, m, overlapped with H2O, CHOH), 4.46 (1H, br s, CHOH), 4.93 (1H, br s, CqOH), 7.78 (3H, br s, NH3+); 13C NMR (100.6 MHz, DMSO-d6): δ = 20.6 and 20.7 (CHMe2), 24.0 (C3), 32.8 (CHMe2), 33.5 (C5), 34.0 (C6), 42.6 (C4), 47.9 (CH2NH), 70.9 (C1), 73.6 (C2). HR-MS (ESI): m/z calcd for C10H22NO2 [M + H]+: 188.16451; found: 188.16423.
Prepared from 11a according to the general method. Yield: 0.18 g (45%); white crystals m.p. 176–178 °C; [α = −14 (c 0.265, MeOH). 1H NMR (DMSO-d6, 400.1 MHz,): δ = 0.82 (3H, d, J = 8.4 Hz, CHMe2), 0.84 (3H, d, J = 8.4 Hz, CHMe2), 1.04–1.18 (1H, m, CHMe2), 1.27–1.69 (7H, m, CH(4), CH2(3), CH2(5), CH2(6)), 2.80 (2H, br s, (CH2NH)), 3.54 (1H, br d, J = 3.7 Hz, CHOH), 4.69 (1H, br s, CHOH), 4.72 (1H, br s, CqOH), 7.83 (3H, br s, NH3+); 13C NMR (100.6 MHz, DMSO-d6): δ = 21.19 and 21.20 (CHMe2), 25.1 (C5), 30.2 (C3), 30.5 (CHMe2), 33.5 (C6), 37.8 (C4), 45.1 (CH2NH), 69.8 (C2), 71.4 (C1). HR-MS (ESI): m/z calcd for C10H22NO2 [M + H]+: 188.16451; found: 188.16419.
General procedure used to form the N-methyl derivatives of compounds 9 and 13
A solution of appropriate compound (5a or 6a) (1.00 g, 2.65 mmol) in dry THF (12 mL) was added to a stirred suspension of LiAlH4 (0.30 g, 7.95 mmol) in dry THF (15 mL) carefully at 0 °C. The mixture was stirred at reflux for 6 h. When the TLC indicated, a mixture of H2O (1 mL) and THF (16 mL) was added dropwise with cooling. The precipitated material was filtered off and washed with THF. The filtrate was dried (Na2SO4), filtered, and concentrated in a vacuum. The crude products were purified via column chromatography on silica gel by applying DCM/MeOH 19:1 (for compound 9) n-hexane/EtOAc 2:1 (for compound 13).
Prepared from 5a according to the general method. Yield: 0.46 g (60%), colourless oil, [α = −44 (c 0.25, MeOH). 1H NMR (DMSO-d6, 400.1 MHz): δ = 0.84 (6H, d, J = 6.6 Hz), 0.77–0.87 (1H, m), 0.81–0.86 (1H, m), 0.98–1.09 (1H, m), 1.21–1.34 (5H, m), 1.37–1.57 (3H, m), 2.20 (3H, s), 2.41 (1H, d, J = 13.5 Hz), 2.53 (1H, d, J = 13.5 Hz), 3.39 (1H, dd, J = 4.4, 11.4 Hz), 3.50 (1H, d, J = 13.3 Hz), 3.67 (1H, d, J = 13.3 Hz), 3.72 (1H, br s), 5.06 (1H, br s), 7.21–7.26 (1H, m), 7.28–7.35 (4H, m); 13C NMR (100.6 MHz, DMSO-d6): δ = 20.2, 20.3, 23.9, 28.7, 32.5, 33.5, 34.2, 42.4, 44.6, 64.0, 66.3, 72.9, 73.5, 127.3, 128.6, 129.1, 139.9. HR-MS (ESI): m/z calcd for C18H30NO2 [M + H]+: 292.22711; found: 292.22635.
Prepared from 6a according to the general method. Yield: 0.45 g (58%), colourless oil, [α = −8 (c 0.25, MeOH). 1H NMR (DMSO-d6, 400.1 MHz): δ = 0.73 (3H, d, J = 9.0 Hz), 0.74 (3H, d, J = 9.0 Hz), 0.77–0.87 (1H, m), 0.99–1.07 (1H, m), 1.25–1.44 (4H, m), 1.49–1.56 (1H, m), 2.25 (3H, s), 2.37 (2H, dd, J = 1.0, 14.1 Hz), 3.48 (1H, d, J = 12.8 Hz), 3.56 (1H, d, J = 12.8 Hz), 3.59–3.63 (1H, m), 3.79 (1H, br s), 4.39 (1H, br s), 7.19–7.25 (1H, m), 7.27–7.33 (4H, m); 13C NMR (100.6 MHz, DMSO-d6): δ = 20.4, 20.5, 25.5, 30.9, 31.8, 33.0, 36.7, 45.1, 61.9, 64.0, 69.6, 73.9, 127.4, 128.5, 140.1. HR-MS (ESI): m/z calcd for C18H30NO2 [M + H]+: 292.22711; found: 292.22635.
General method for the preparation of 1,3 oxazines 10a–c
To a stirred solution of aminodiol (7a–c) (0.15 g) in Et2O (6 mL), a 35% aqueous solution of formaldehyde (4.5 mL) was added. The reaction mixture was stirred at room temperature for 3 h, then extracted with a 10% aqueous solution of KOH (10 mL). The aqueous layer was extracted with Et2O (3 × 25 mL), and then the combined organic phase was washed with brine (3 × 25 mL). The organic layer was dried (NaSO4), filtered, and evaporated. The crude products were purified by column chromatography on silica gel by using an n-hexane/EtOAc 9:1 mixture for compound 10a and a 4:1 mixture for compounds 10b and 10c.
Prepared from 7a according to the general method. Yield: 0.11 g (70%); white crystals; m.p. 155–157 °C; [α = +38 (c 0.25, MeOH). 1H NMR (DMSO-d6, 400.1 MHz,): δ = 0.84 (3H, d, J = 6.8 Hz, CHMe2), 0.85 (3H, d, J = 6.8 Hz, CHMe2), 1.04–1.21 (2H, m, CH2(6)), 1.27–1.48 (6H, m, CH2(8), CHMe2, H7, CH2(5)), 2.18 (1H, d, J = 11.3 Hz, H4α), 2.63 (1H, d, J = 11.5 Hz, H4β), 3.14 (1H, dd, J = 4.8, 11.9 Hz, H8a), 3.58 (1H, d, J = 13.6 Hz, CH2Ph), 3.69 (1H, s, OH), 3.70 (1H, d, J = 13.8 Hz, CH2Ph), 3.80 (1H, d, J = 8.1 Hz, H2α), 4.36 (1H, dd, J = 1.2, 8.2 Hz, H2β), 7.20–7.27 (1H, m, CHAr), 7.29–7.37 (4H, m, 4 × CHAr); 13C NMR (100.6 MHz, DMSO-d6): δ = 20.5 and 20.7 (CHMe2), 24.1 (C6), 30.0 (C8), 33.0 (CHMe2), 33.5 (C5), 42.8 (C7), 57.3 (C4), 62.3 (CH2Ph), 67.2 (C4a), 82.1 (C8a), 85.5 (C2), 127.8 (CHAr), 129.0 (2 × CHAr), 129.5 (2 × CHAr), 139.0 (CqAr). HR-MS (ESI): m/z calcd for C18H28NO2 [M + H]+: 290.21146 found: 290.21090.
Prepared from 7b according to the general method. Yield: 0.15 g (96%), [α = −19 (c 0.05, MeOH), 1H NMR (DMSO-d6, 400.1 MHz): δ = 0.83 (6H, dd, J = 1.2, 6.9 Hz), 1.00–1.18 (2H, m), 1.24 (1H, s), 1.27–1.35 (5H, m), 1.36–1.47 (3H, m), 2.00 (1H, d, J = 11.32 Hz), 2.68 (1H, dd, J = 1.6, 11.3 Hz), 3.04 (1H, dd, J = 4.7, 11.0 Hz), 3.57 (1H, d, J = 1.35 Hz), 3.70 (1H, d, J = 8.1 Hz), 3.81 (1H, q, J = 6.8, 13.5 Hz,), 7.19–7.26 (1H, m), 7.26–7.35 (4H, m); 13C NMR (100.6 MHz, DMSO-d6): δ = 20.4, 20.5, 20.7, 24.1, 30.0,32.9, 33.3, 42.8, 59.1, 60.0, 67.1, 82.1, 84.2, 86.4, 127.8, 128.2, 129.1, 143.8 HR-MS (ESI): m/z calcd for C19H30NO2 [M + H]+: 304.22711; found: 304.22631.
Prepared from 7c according to the general method. Yield: 0.13 g (83%) [α = −28 (c 0.07, MeOH) 1H NMR (CDCl3, 400.1 MHz): δ = 0.88 (6H, dd, J = 2.4, 6.8 Hz), 1.09–1.18 (2H, m), 1.26 (1H, s), 1.36 (3H, d, J = 6.9 Hz), 1.42–152 (4H, m), 1.57 (dt, J = 3.7, 11.9 Hz), 1.65 (dt, J = 3.3, 13.5 Hz), 2.07 (1H, d, J = 10.6 Hz), 2.86 (1H, dd, J = 2.2, 10.6 Hz), 3.08 (1H, dd, J = 4.4, 11.4 Hz), 3.54 (1H, dd, J = 6.8, 13.5 Hz), 3.66 (1H, d, J = 7.8 Hz), 4.45 (1H, dd, J = 2.1, 7.8 Hz), 7.22–7.26 (1H, m), 7.27–7.35 (4H, m); 13C NMR (100.6 MHz, CDCl3): δ = 19.1, 19.7, 20.0, 23.8, 29.5, 32.5, 32.7, 59.6, 59.8, 66.7, 82.0, 84.4, 127.2, 127.3, 128.4, 142.8. HR-MS (ESI): m/z calcd for C19H30NO2 [M + H]+: 304.22711; found: 304.22646.
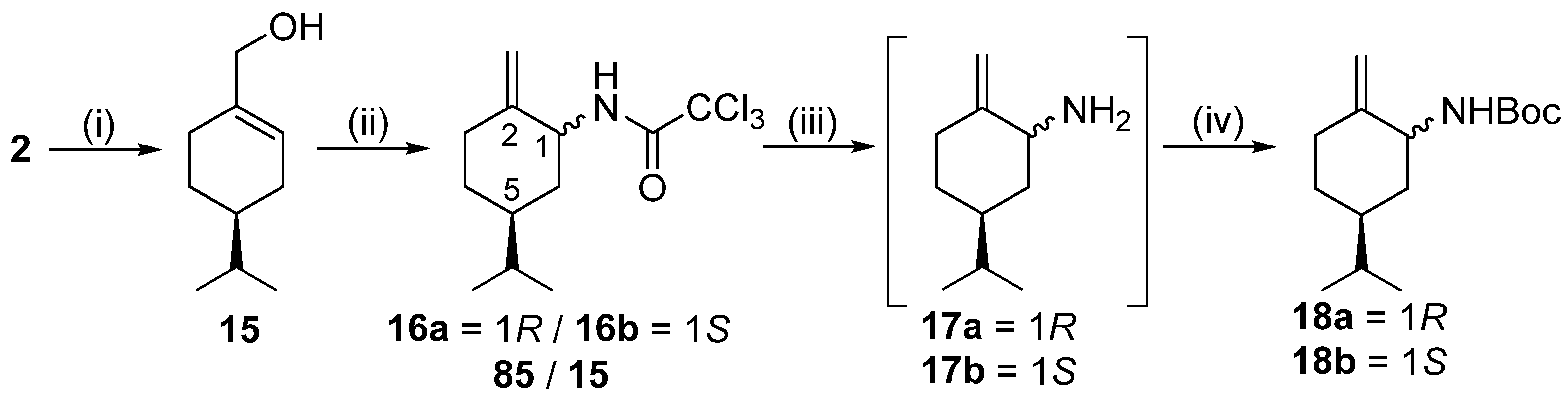
To a solution of
2 (5.36 g, 35.2 mmol) in MeOH (110 mL), NaBH
4 (3.99 g, 105.6 mmol) was added in small portions at 0 °C. The reaction mixture was stirred at room temperature for 1 h. When the reaction was completed (indicated by TLC), the solvent was evaporated. The residue was redissolved in water (100 mL) and extracted with DCM (3 × 100 mL). The combined organic phase was dried (Na
2SO
4), filtered, and concentrated to dryness. The crude product was used for the next step without further purification. Isolated product: 5.16 g (95%), light green transparent oil. All its spectroscopic data and physical properties agreed with those reported in the literature [
30].
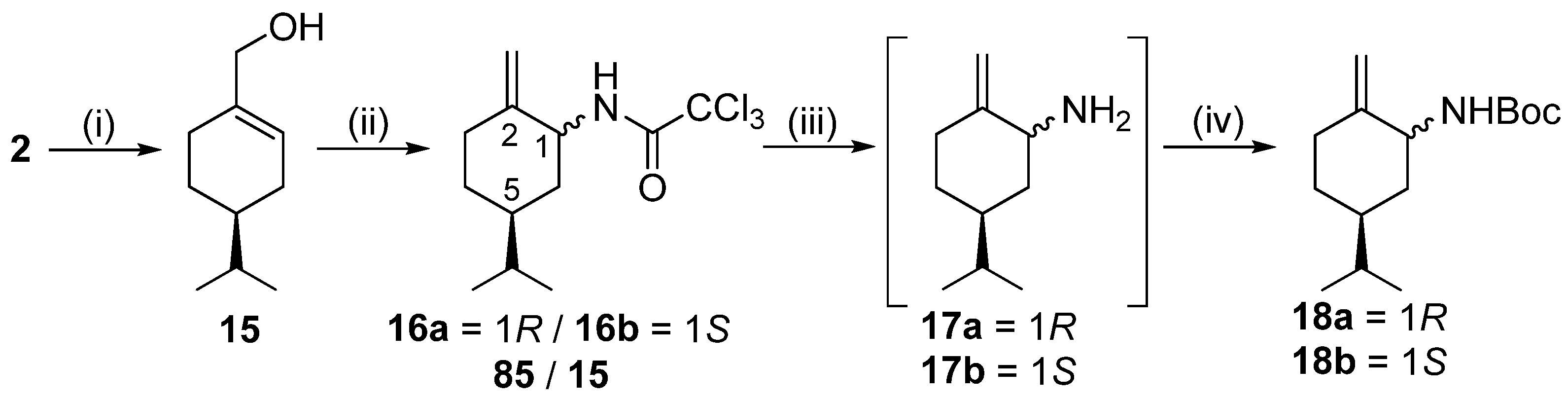
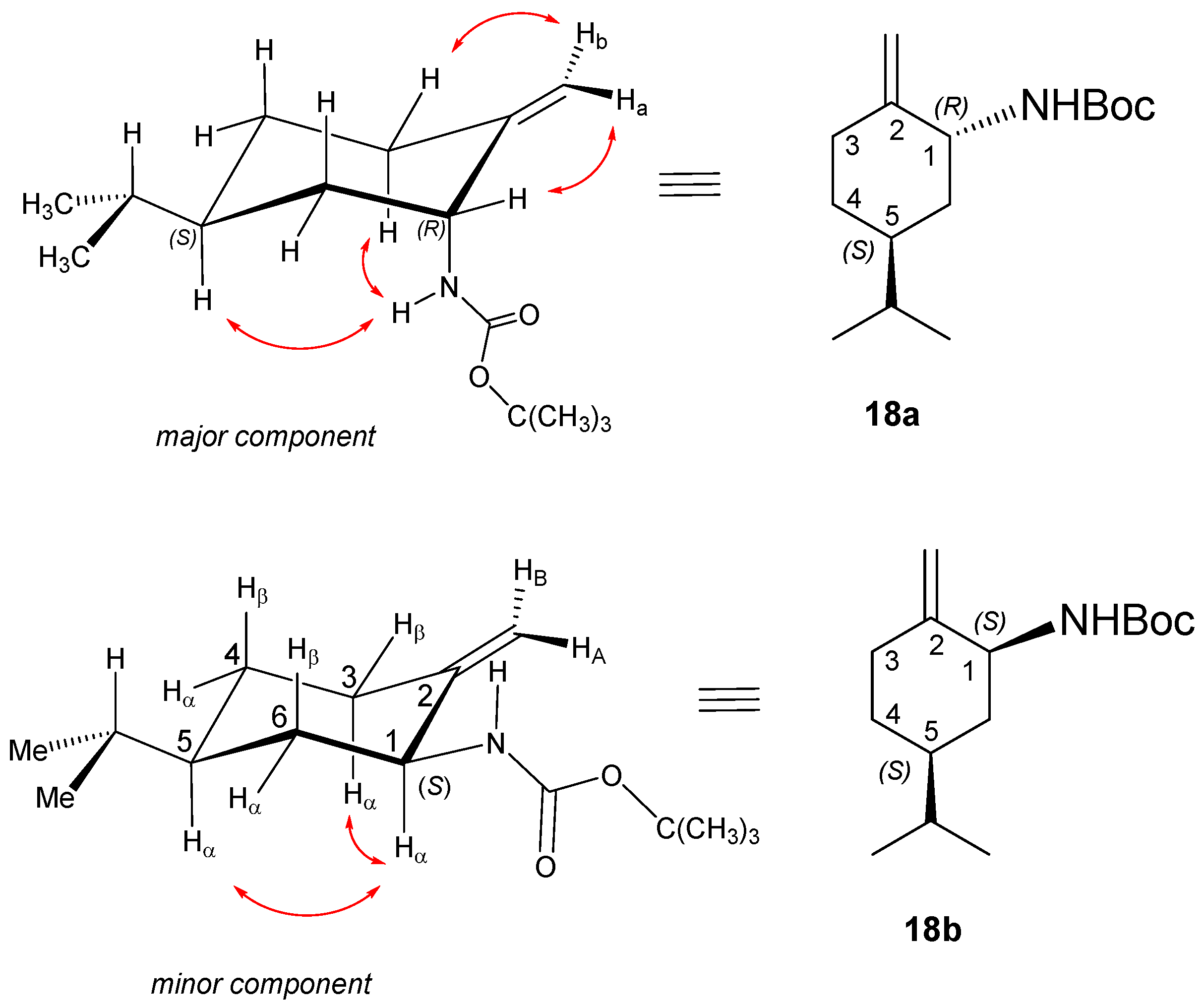
2,2,2-Trichloro-N-((1R,5S)-5-isopropyl-2-metilenecyclohexyl)acetamide (intermediate) 16a and 2,2,2-trichloro-N-((1S,5S)-5-isopropyl-2-methylenecyclohexyl)acetamide 16b
To a solution of
15 (10.48 g, 67.9 mmol) in dry DCM (320 mL), 1,8-diazabicyclo [5.4.0]undec-7-ene (12.41 g, 81.5 mmol, 12.2 mL) was added applying an Ar atmosphere. Trichloroacetonitrile (10.3 mL, 97.4 mmol) was added in small portions to the reaction mixture, and it was stirred for 2 h at room temperature. Upon completion of the reaction (indicated by TLC), the solvent was evaporated. The crude product was purified via chromatography on silica gel by using DCM. The top one-third of the column was dry Na
2SO
4, and the bottom two-thirds of the column were silica gel. The fractions that contained the imidate intermediate were collected, and the solvent was evaporated. The slightly yellow oil product was dissolved in dry toluene (300 mL) and reacted in an autoclave at 130 °C in an Ar atmosphere for 24 h. When the reaction was completed, the mixture was extracted with a cold aqueous solution of HCl (5%, 3 × 100 mL), and the organic layer was dried, filtered, and evaporated. Based on
1H NMR measurements and GC determination (Chirasil-DEX CB column, 2 mL flow rate, 110 °C,
Figure S145), the diastereomers formed a mixture with a ratio of 85:15. Although various column chromatography methods were carried out, the diastereomers were inseparable.
Yield: 14.52 g (72%) (two diastereomers); orange oil; [α = −33 (c 0.505, MeOH); 1H NMR (CDCl3, 500.2 MHz): δ = 0.87 (3H, d, J = 1.6 Hz, minor), 0.88 (3H, d, J = 1.6 Hz, minor), 0.90 (3H, d, J = 6.7 Hz, major), 0.92 (3H, d, J = 6.8 Hz, major), 0.97–1.08 (1H, m), 1.23–1.41 (2H, m), 1.49–1.63 (4H, m), 1.73–1.86 (2H, m), 1.90–1.98 (1H, m), 2.05–2.21 (2H, m), 2.35 (1H, dt, J = 4.3; 14.4 Hz, major), 2.50 (1H, dt, J = 3.3; 13.8 Hz, minor), 4.34–4.43 (1H, m, minor), 4.49–4.57 (1H, m, major), 4.71 (1H, s, minor), 4.81 (1H, s, minor), 4.88 (1H, s, major), 4.97 (1H, s, major), 6.66 (1H, br s, minor), 6.79 (1H, br s, major); 13C NMR (125.8 MHz, CDCl3): δ = 19.7, 19.8, 19.9, 20.0, 28.8, 29.4, 30.0, 30.7, 31.0, 32.2, 34.9, 37.6, 38.8, 43.1, 52.8, 53.5, 71.7, 74.3, 104.9, 111.4, 145.1, 147.4, 160.7, 161.0; HR-MS (ESI): m/z calcd for C12H19Cl3NO [M + H]+: 298.05322; found: 298.05359.
To a solution of
16a and
16b (11.11 g, 37.2 mmol) in EtOH/DCM 2:1 (66 mL), an aqueous solution of NaOH (5M solution, 525.6 mL) was added. The reaction was stirred at 50 °C for 15 h. As the TLC indicated, the reaction was cooled to room temperature, then extracted with DCM (3 × 150 mL). The combined organic phase was washed with brine (100 mL), dried, filtered, and evaporated. The residue was dissolved in dry THF (100 mL), and then triethylamine (11.29 g, 111.6 mmol), DMAP (0.45 g, 3.72 mmol), and di-
tert-butyl dicarbonate (8.93 g, 40.9 mmol) were added to the reaction mixture. The mixture was stirred at room temperature for 12 h. After that, the solvent was evaporated, and the crude product was purified via column chromatography on silica gel by applying
n-hexane/Et
2O 9:1. The diastereomers were still inseparable in this step. The ratio of the formed diastereomers remained 85:15 based on the determination by GC (Chirasil-DEX CB column, 2 mL flow rate, 160 °C,
Figure S146).
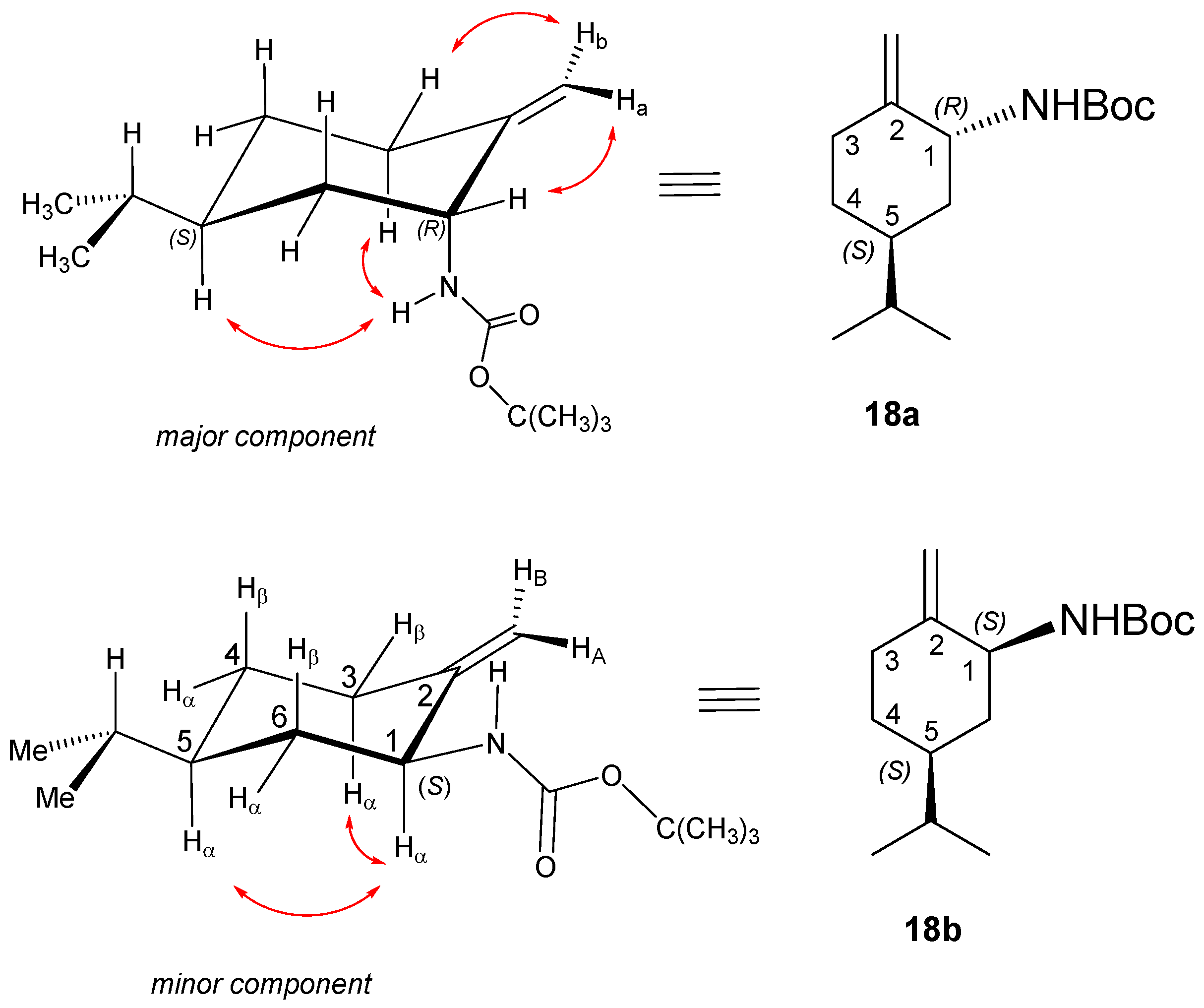
Yield (for the mixture of isomers): 5.56 g (59%); colourless viscous oil; [α = −48 (c 0.250, MeOH); 1H NMR (DMSO-d6, 500.2 MHz) of 18a (major): δ = 0.79 and 0.78 (6H, overlapping d’s, J = 6.7 Hz, CHMe2), 1.06 (1H, qa, J = 12.3 Hz, 4Hβ), 1.25 (1H, m, 6Hβ), 1.32 (9H, s, CMe3), 1.41 (1H, oct, J = 6.7 Hz, CHMe2), 1.48 (1H, m, 5Hα), 1.56 (1H, m, 4Hα), 4.67 (1H, brs, HA); 1.61 (1H, br d, J = 12.2 Hz, 6Hα), 2.13 (1H, td, J = 4.3, 13.9 Hz, 3Hβ), 2.22 (m, 1H, 3Hα), 4.60 (1H, br s, HB); 4.03 (1H, br s, 1Hβ), 6.85 (1H, br s, NH). 13C-NMR (DMSO-d6, 125 MHz): δ = 20.3 and 20.4 (CHMe2), 28.7 (CMe3), 30.4 (4C), 30.5 (3C), 30.9 (CHMe2), 36.2 (6C), 37.7 (4C), 52.2 (1C), 78.0 (CMe3), 108.9 (=CH2), 149.0 (C2),155.3 (C=O).
18b/minor: 1H-NMR (DMSO-d6) separated diagnostic signals: δ = 0.94 (1H, qa, J = 10.6 Hz, 6Hβ), 1.92 (1H, ddd, J = 13.5 Hz, 3.9, 9.5 Hz, 3Hα), 2.32 (dt, J = 13.5 Hz and 4.2 Hz, 1H, 3Hβ), 3.78 (1H, br t, J = 8.5 Hz, 1Hα). 13C-NMR (DMSO-d6, 125 MHz) diagnostic signals separated in the 1D spectrum or identified on the basis of 2D-HSQC: δ = 34.3 (3C), 37.7 (6C, coalesced with 4C line of 18a/major), 53.1 (1C), 104.9 (=CH2), 149.7 (C2), 155.5 (C=O). HR-MS (ESI): m/z calcd for C11H20NO2 [M − CH(CH3)3 + H + H]+: 198.14886; found: 198.14866.
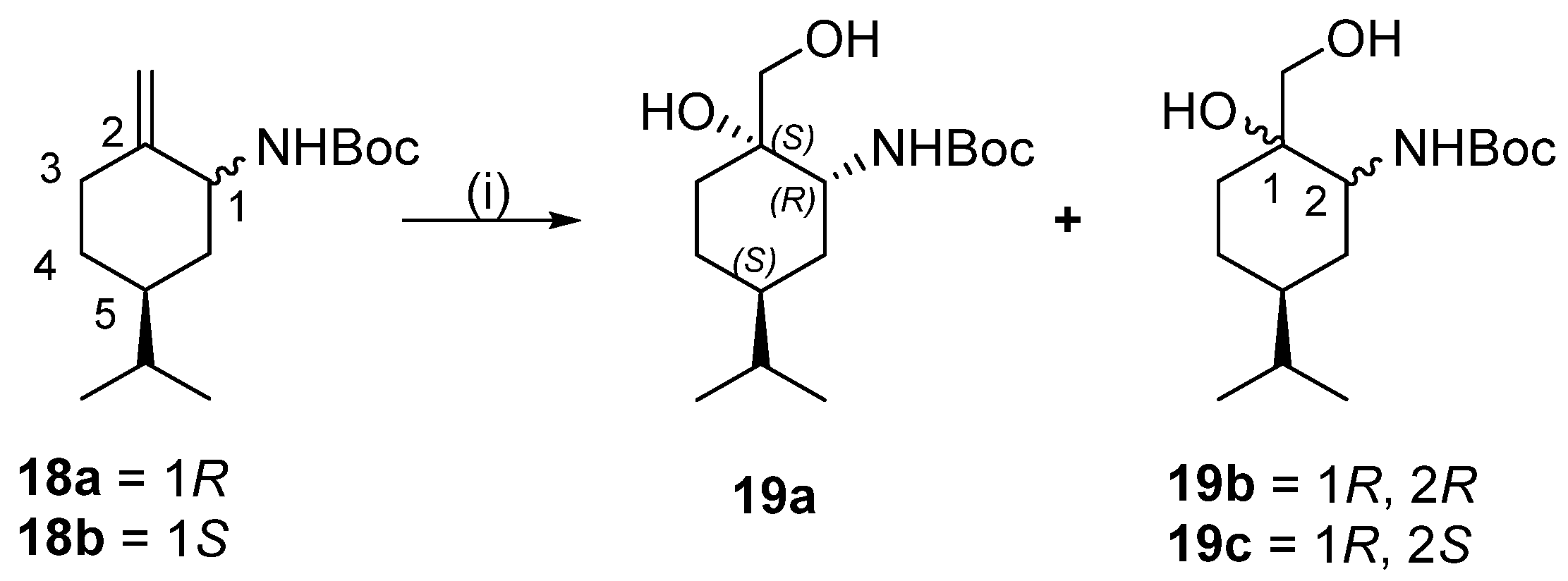
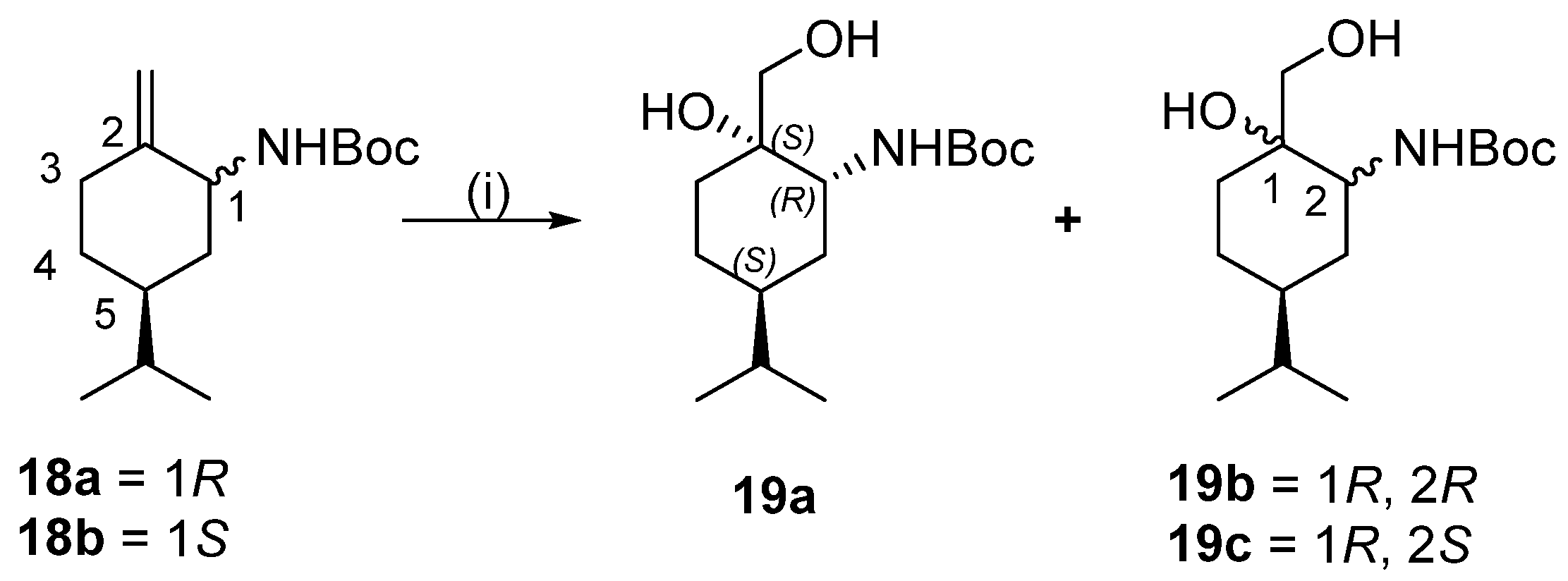
Dihydroxylation of compound mixture of 18a and 18b
To a solution of 18a and 18b (5.40 g, 21.3 mmol) in acetone (100 mL), 4-methylmorpholine-4-oxide (22.5 mL, 96 mmol, 50% aq. solution) and OsO4 (1.5 mL, 2.0% tert-BuOH solution) were added. The reaction mixture was stirred for 72 h at room temperature, then was quenched with a saturated aqueous solution of Na2SO3 (20 mL), and extracted with ethyl acetate (3 × 100 mL). The combined organic phase was dried, filtered, and concentrated to dryness. Based on the 1H NMR spectra, 19a, 19b, and 19c were formed in the ratio of 71:16:13. The crude product was purified by column chromatography on silica gel by using n-hexane/EtOAc 2:1). We found that 19a was successfully isolated, while 19a and 19b remained as a mixture. Total yield: 4.96 g (81%).
Yield: 2.94 g (48%); white crystals; m.p.: 111–112 °C; [α = −41 (c 0.275, MeOH); 1H NMR (CDCl3, 500.2 MHz): δ = 0.89 (3H, d, J = 6.7 Hz), 0.90 (3H, d, J = 6.7 Hz), 0.97–1.08 (1H, m), 1.19–1.28 (1H, m), 1.39–1.60 (5H, m), 1.46 (9H, s), 1.88 (1H, dt, J = 4.1; 13.2 Hz), 3.08 (1H, d, J = 12.8 Hz), 3.47 (1H, d, J = 12.1 Hz), 3.60–3.66 (1H, m), 4.82 (1H, d, J = 8.1 Hz); 13C NMR (125.8 MHz, CDCl3): δ = 19.6, 20.0, 23.2, 23.4, 28.3, 28.5, 29.7, 32.5, 38.5, 50.1, 67.2, 72.5, 80.7, 157.4; HR-MS (ESI): m/z calcd for C17H26N [M + H]+: 436.2410; found: 258.22099.
(1R,2R,5S)- and (1S,2R,5S)-tert-Butyl 2-hydroxy-2-hydroxymethyl-5-isopropylcyclohexyl)carbamate mixture 19a and 19b
Compounds 19b and 19c (2.02 g (33%); white crystals; were used in acetal preparation without further separation. HR-MS (ESI): m/z calcd for C15H30NO4 [M + H]+: 288.21693; found: 288.21649, calcd for C15H29NO4Na [M + Na]+: 310.19888; found: 310.19816.
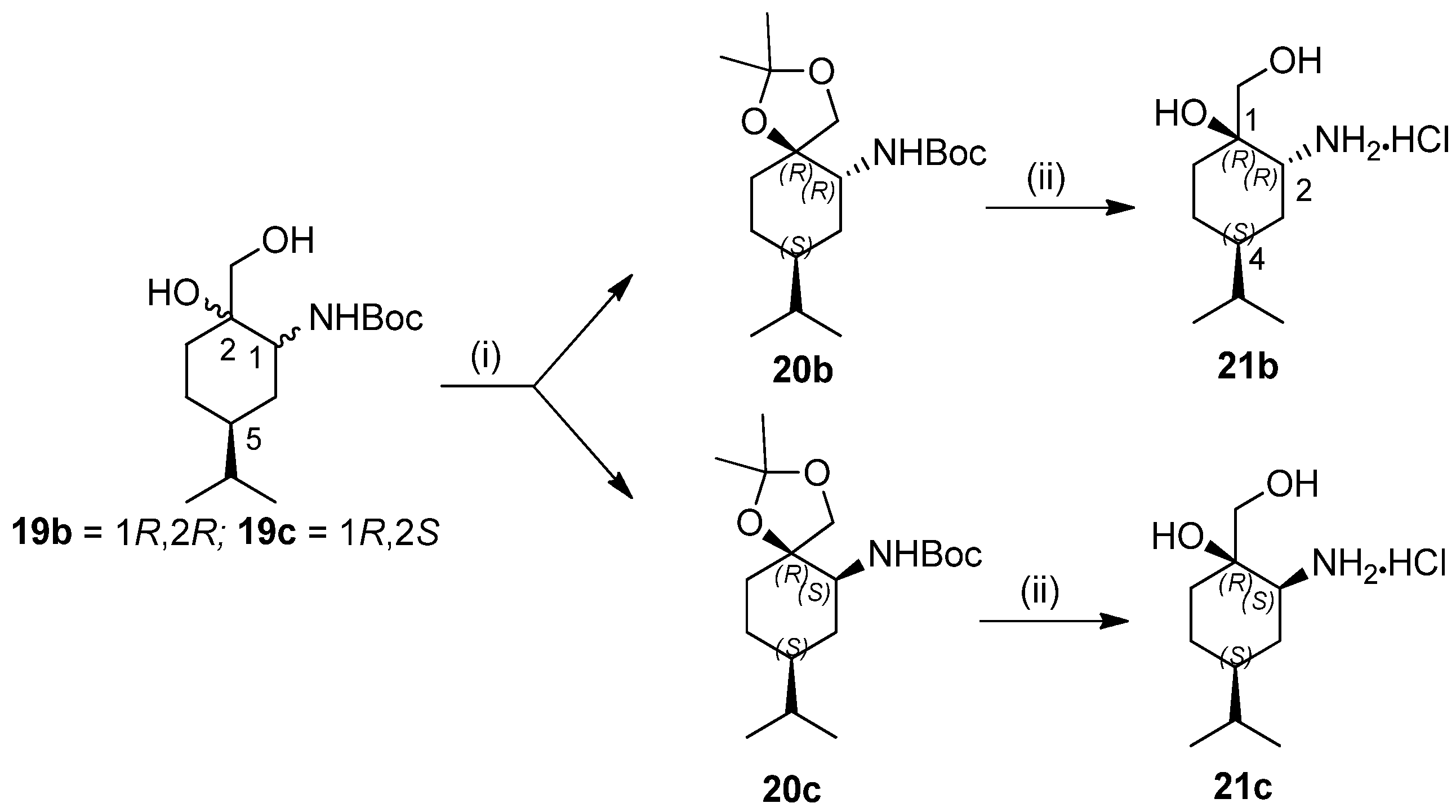
Acetal synthesis starting from 19b and 19c
To a solution of Boc-protected aminodiol mixture 19a and 19b (1.20 g, 4.2 mmol) in dry acetone (100 mL), 4-methylbenzene-1-sulfonic acid (0.10 g 0.6 mmol) was added and stirred at room temperature. When the TLC indicated, the solvent was concentrated in a vacuum. The crude product was purified by column chromatography on silica gel, applying n-hexane/EtOAc 1:1 and then n-hexane/EtOAc 9:1 to yield compounds 20b and 20c.
tert-Butyl ((5R,6R,8S)-8-isopropyl-2,2-dimethyl-1,3-dioxaspiro [4,5]decane-6-yl)carbamate 20b
Yield: 0.77 g (56%); white crystals; m.p.: 75–78 °C; [α = −9 (c 0.490, MeOH); 1H NMR (CDCl3, 500.2 MHz): δ = 0.87 (3H, d, J = 6.8 Hz), 0.90 (3H, d, J = 6.8 Hz), 1.11–1.31 (2H, m), 1.18–1.29 (1H, m), 1.34–1.43 (1H, m), 1.39 (3H, s), 1.41 (3H, s), 1.46 (9H, s), 1.49–1.76 (5H, m, 1.83–1.93 (1H, m), 3.66–3.73 (1H, m), 3.73 (1H, d, J = 8.4 Hz), 3.93 (1H, d, J = 8.8 Hz), 4.72 (1H, br d, J = 5.8 Hz); 13C NMR (125.8 MHz, DMSO-d6): δ = 20.4, 20.6, 25.5, 27.0, 27.2, 28.4, 29.5, 32.1, 32.6, 38.0, 51.2 72.2, 82.0, 109.3, 155.9; HR-MS (ESI): m/z calcd for C18H34NO4 [M + H]+: 328.24823; found: 328.24784.
tert-Butyl ((5R,6S,8S)-8-isopropyl-2,2-dimethyl-1,3-dioxaspiro [4,5]decane-6-yl)carbamate 20c
Yield: 0.24 g (18%); white crystals; m.p.: 167–168 °C; [α = +11 (c 0.255, MeOH); 1H NMR (DMSO-d6, 500.2 MHz), δ = 0.79–0.94 (2H, m), 0.81 (3H, d, J = 5.8 Hz), 0.83 (3H, d, J = 5.8 Hz), 1.09–1.19 (1H, m), 1.23 (3H, s), 1.26 (3H, s), 1.32–1.41 (1H, m), 1.37 (9H, s), 1.46 (1H, dt, J = 2.5, 12.9 Hz), 1.57–1.66 (2H, m), 1.75–1.80 (1H, m), 3.43–3.51 (1H, m), 3.62 (1H, d, J = 9.0 Hz), 4.07 (1H, d, J = 9.0 Hz), 6.56 (1H, br d, J = 9.5 Hz); 13C NMR (125.8 MHz, DMSO-d6): δ = 20.2, 20.4, 26.7, 27.3, 27.7, 28.8, 32.2, 35.2, 37.8, 42.8, 54.2, 67.3, 77.7, 84.0, 108.3, 155.5; HR-MS (ESI): m/z calcd for C18H34NO4 [M + H]+: 328.24823; found: 328.24781.
General procedure for the Boc and acetonide deprotection of compounds 19a, 20b, and 20c
To a stirred solution of compounds 19a, 20b, and 20c (0.70 mmol) in Et2O (6 mL), an aqueous solution of HCl (10%, 10 mL) was added. The reaction mixture was stirred vigorously for 24 h at room temperature. When the reaction was completed (indicated by TLC), it was extracted with Et2O (2 × 10 mL). The compounds were further purified as hydrochloride salts.
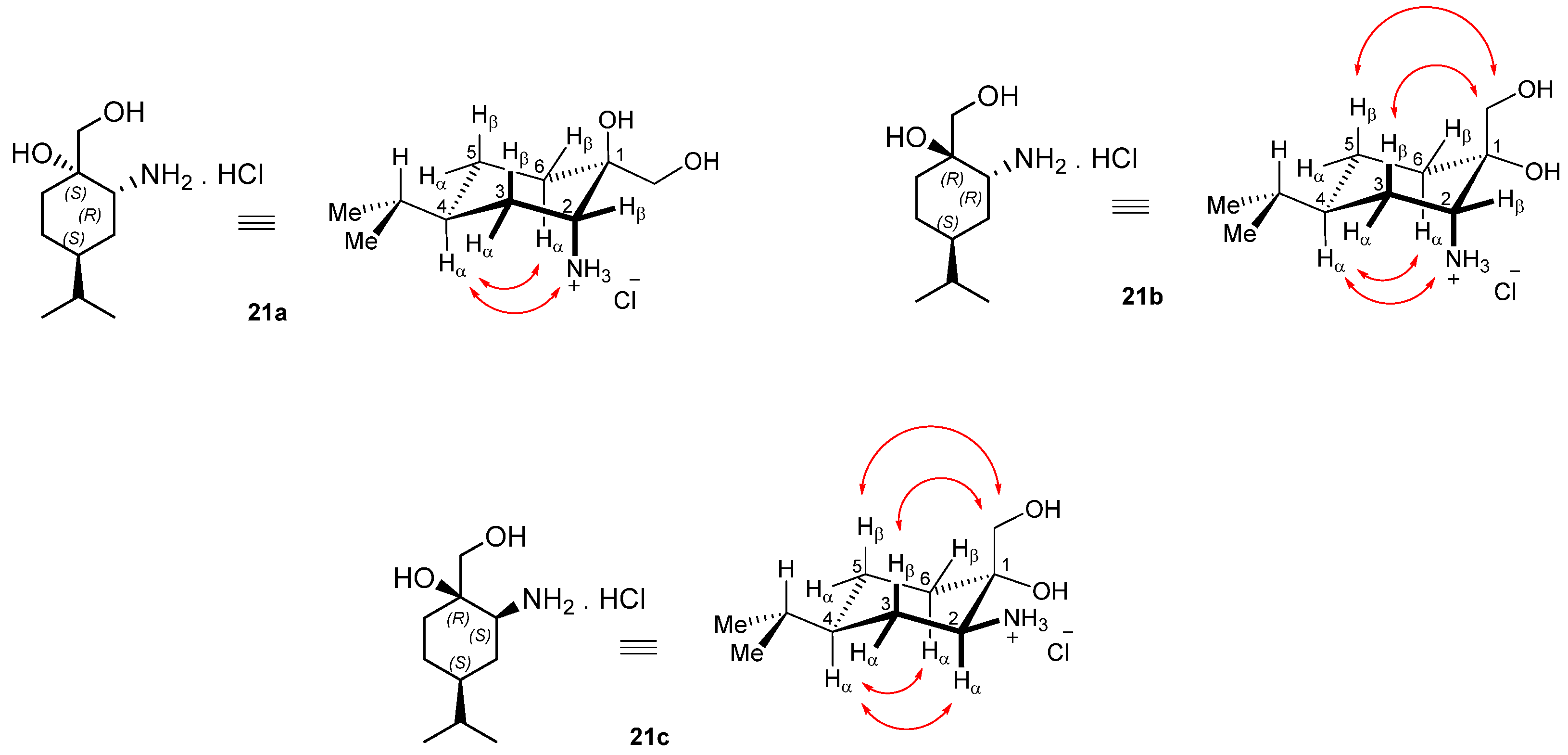
Prepared from 19a according to the general method. Yield: 0.133 g (86%); white crystals, m.p.: 247–250 °C; [α = −16 (c 0.700, MeOH); 1H-NMR (DMSO-d6): δ = 0.78 and 0.79 (6H, overlapping d’s, J = 6.7 Hz, CHMe2), 1.27–1.30 (4H, m, 3Hα, 4Hβ and 5Hα and 6Hβ), 1.32–1.36 (2H, m, CHMe2 and 4Hα), 1.55–1.67 (2H, m, 3Hβ and 6Hα), 3.12 (m 1H, 1Hβ), 3.23 and 3.34 (2H, 2xd, J = 11.9 Hz, OCH2), 7.96 (3H, s, NH3+). The signals of the rapidly exchanging OH protons are merged in the broadened HDO signal of the solvent centred at c.a. 3.3 ppm. 13C-NMR (DMSO-d6): δ = 20.0 and 20.1 (CHMe2), 23.3 (C4), 28.5 (two coalesced lines (C3 and C6), 31.9 (CHMe2), 36.9 (C5), 52.0 (C1), 67.0 (OCH2), 70.5 (C2); HR-MS (ESI): m/z calcd for C10H22NO2 [M + H]+: 188.16451; found: 188.16451.
Prepared from 20b according to the general method. Yield: 0.131 g (86%); white crystals m.p.: 142–143 °C; [α = −7 (c 0.260, MeOH); 1H-NMR (DMSO-d6): δ = 0.78 and 0.80 (overlapping d’s, J = 6.7 Hz, 6H, CHMe2), 1.17 (m, H4β), 1.29 (m, 1H, 5Hα), 1.38 (m, 1H, 3Hα), 1.44 (oct, J = 6.7 Hz, 1H, CHMe2), 1.52–1.55 (m, 3H, 3Hβ, 4Hα and 6Hβ), 1.72 (br d, J = 11.3 Hz, 1H, 6Hα), 3.19 (m 1H, 1Hβ), 3.29 and 3.36 (2 × d, J = 11.9 Hz, 2 × 1H, OCH2), 7.73 (s, 3H, NH3+). The signals of the rapidly exchanging OH protons are merged in the broadened HDO signal of the solvent centred at c.a. 3.3 ppm. 13C-NMR (DMSO-d6): δ = 20.78 and 20.80 (CHMe2), 24.0 (C4), 28.7 (C6), 28.8 (CHMe2), 29.4 (C3), 37.5 (C5), 50.1 (C1), 65.8 (OCH2), 71.0 (C2); HR-MS (ESI): m/z calcd for C10H22NO2 [M + H]+: 188.16451; found:188.16434.
Prepared from 20c according to the general method. Yield: 0.124 g (80%); white crystals m.p.: 247–250 °C; [α = +8 (c 0.320, MeOH); 1H-NMR (DMSO-d6): δ = 0.79 (6H, d, J = 6.7 Hz, CHMe2), 0.99 (1H, qad, J = 12.8 Hz and 3.6 Hz, H4β), 1.22 (1H, m, 5Hα); 1.19 (1H, m, 3Hα), 1.26 (1H, qa, J = 11.9 Hz; 6Hβ), 1.41 (1H, oct, J = 6.7 Hz, CHMe2), 1.46 (1H, br d, J = 13.0 Hz, 4Hα), 1.79–1.83 (2H, m, 3Hβ and 6Hα), 2.93 (1H, m 1Hα), 3.39 and 3.60 (2H, 2xd, J = 11.7 Hz, OCH2), 4.96 (1H, br s, CH2OH), 5.00 (1H, s, OH), 7.85 (3H, s, NH3+); 13C-NMR (DMSO-d6): δ = 20.6 and 20.8 (CHMe2), 26.0 (C4), 31.0 (C6), 32.1 (CHMe2), 35.3 (C3), 42.4 (C5), 58.6 (C1), 62.6 (OCH2), 71.8 (C2); HR-MS (ESI): m/z calcd for C10H22NO2 [M + H]+: 188.16451; found: 188.16430.
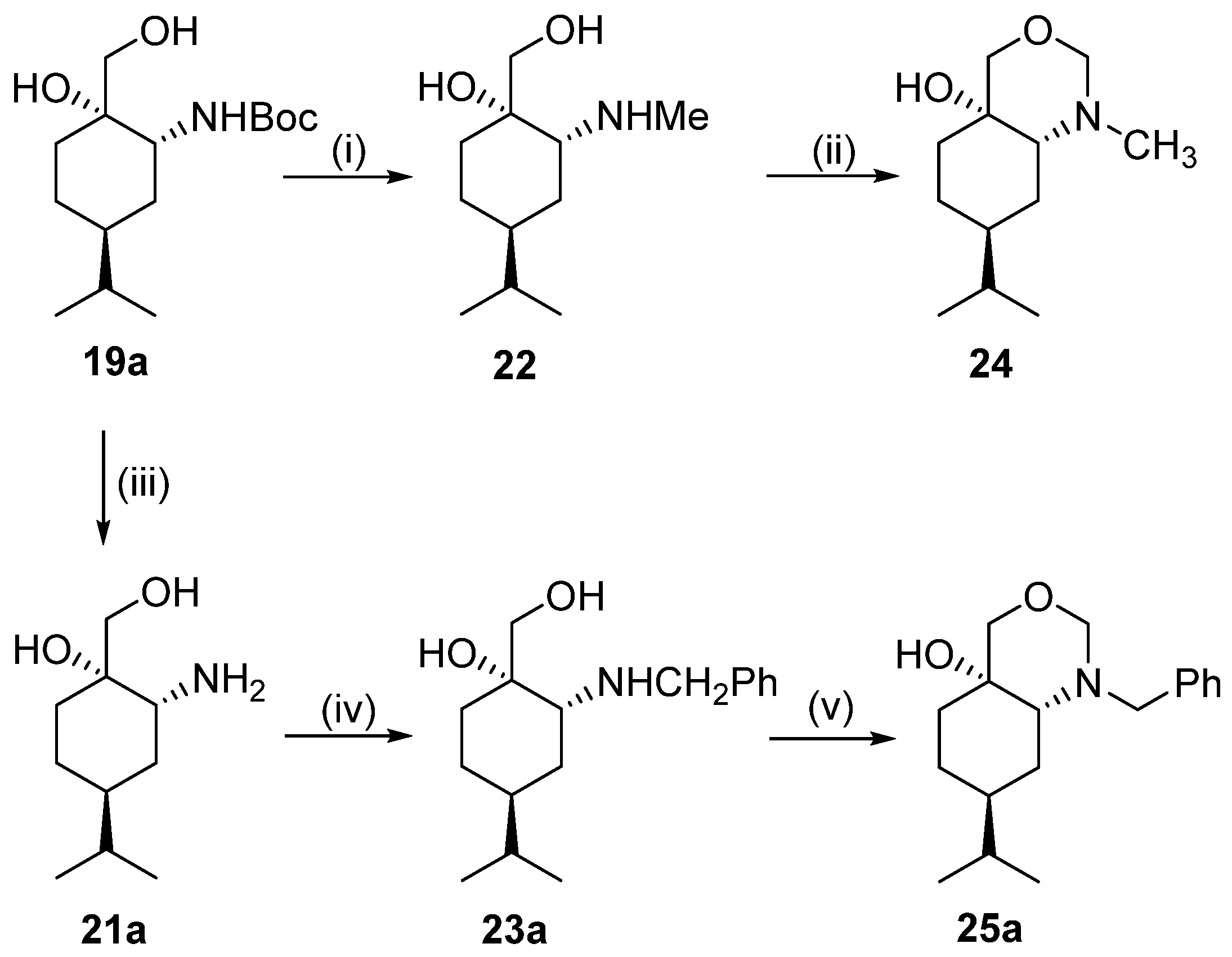
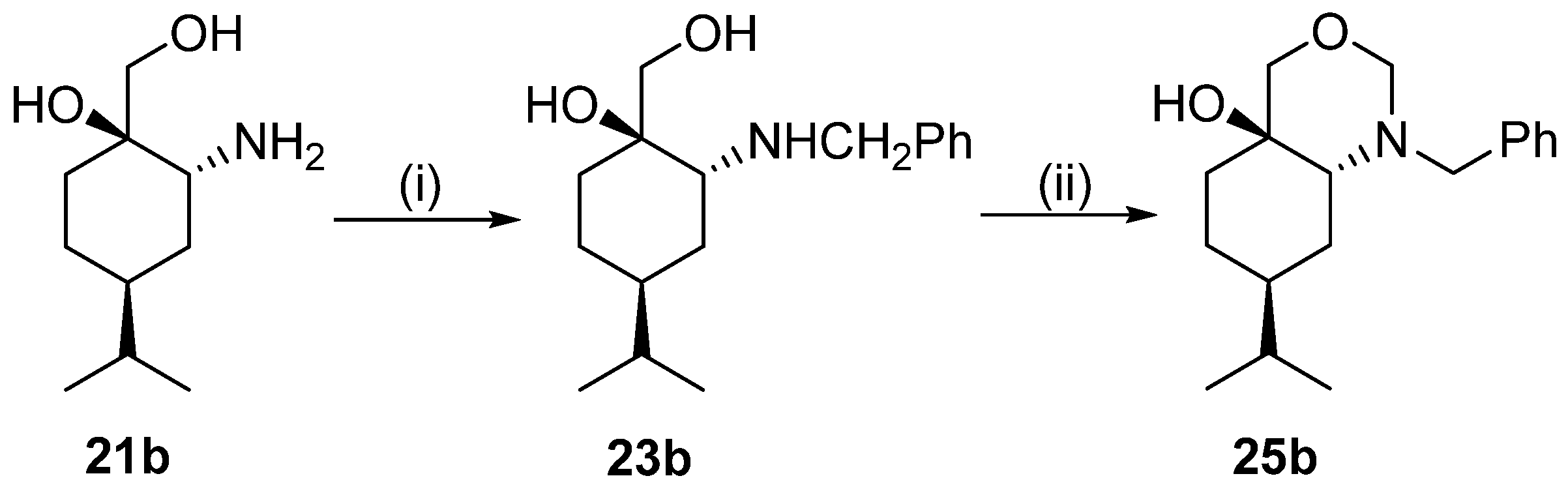
General procedure for preparation of N-benzyl derivatives 23a and 23b
To a solution of primer aminodiol base form 21a or 21b (0.19 g, 1.0 mmol) in dry EtOH (20 mL), 0.11 g (1.05 mmol) benzaldehyde was added, and the reaction mixture was stirred at room temperature. When the reaction was completed (monitored by means of TLC, 1 h), the solvent was concentrated and the residue was dissolved in dry EtOH (20 mL) and stirred for 1 h, and then NaBH4 (0.11 g, 3.0 mmol) was added in small portions. When the reaction was completed (indicated by TLC), the solvent was concentrated to dryness; then, H2O (15 mL) was poured into the residue and extracted with DCM (3 × 25 mL). After drying (Na2SO4), filtration, and solvent evaporation, the crude product was purified via column chromatography by applying a CHCl3/MeOH 9:1 mixture.
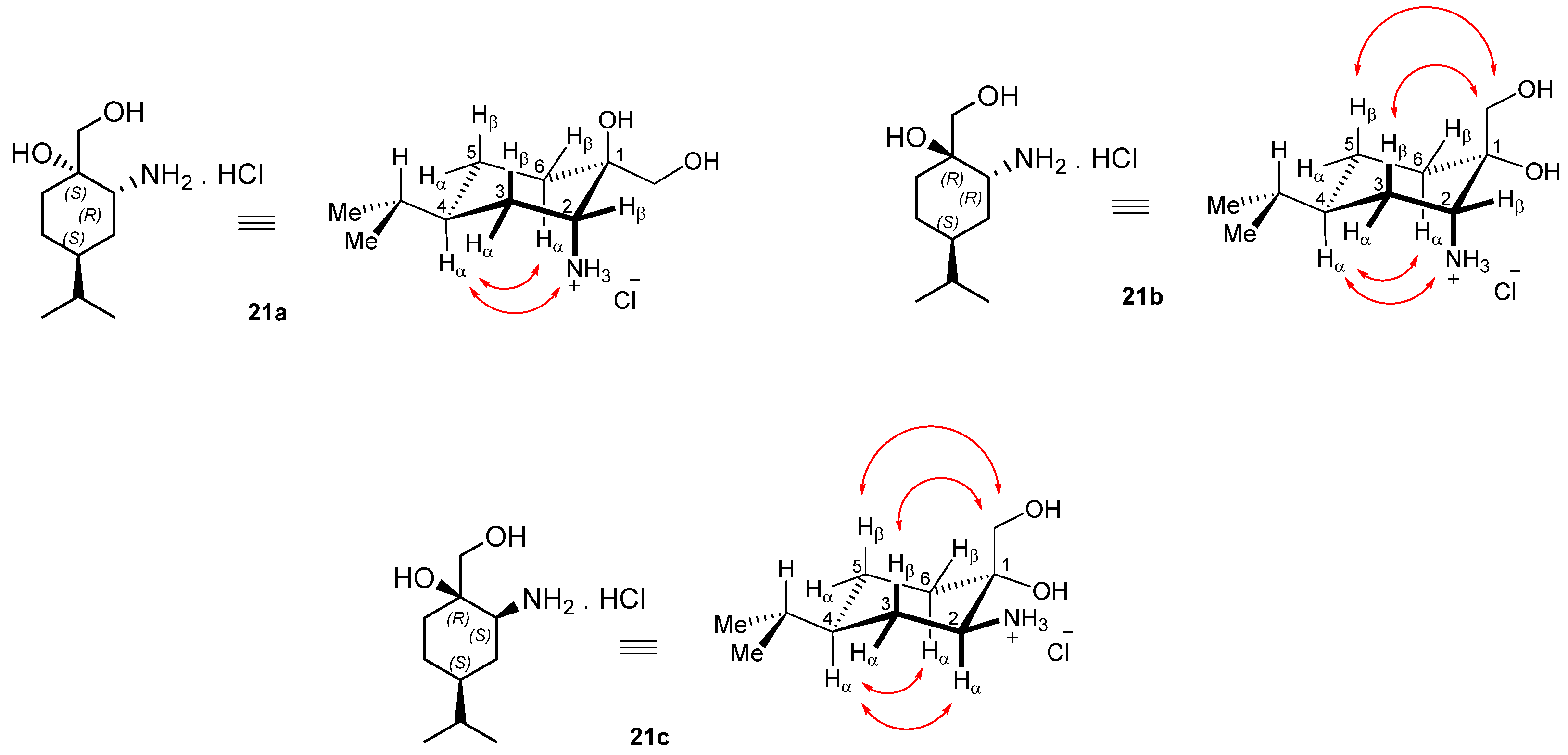
Prepared from 21a according to the general method. Yield: 0.150 g (54%); light yellow oil; [α = −44 (c 0.405, MeOH); 1H NMR (DMSO-d6, 500.2 MHz): δ = 0.78 (3H, d, J = 6.8 Hz, CHMe2, 0.79 (3H, d, J = 6.8 Hz, CHMe2, 0.79–0.86 (1H, m, H5α), 1.02–1.13 (1H, m, CHMe2, 1.34–1.52 (5H, m H3α, H4, H5β, CH2(6)) 1.56–1.63 (1H, m H3β), 2.02 (1H, br s, OH), 2.60–2.66 (1H, m, H2), 3.30 (1H, d, J = 11.1 Hz, overlap with H2O sign, CH2OH), 3.39 (1H, d, J = 11.2 Hz, CH2OH, 3.60 (1H, d, J = 13.4 Hz, CH2Ph), 3.76 (1H, d, J = 13.2 Hz, CH2Ph), 4.15 (1H, s CH2OH, 4.58 (1H, br s, NH), 7.21–7.24 (1H, m, CHAr), 7.29–7.34 (4H, m, 4 × CHAr); 13C NMR (125.8 MHz, DMSO-d6): δ = 20.6 (CHMe2), 20.7 (CHMe2), 25.4 (C5), 29.0 (C3), 30.3 (CHMe2), 30.4 (C6), 37.0 (C4), 51.6 (CH2Ph), 56.6 (C2), 66.5 (CH2OH), 73.0 (C1), 127.0 (CHAr), 128.4 (2 × CHAr), 128.6 (2 × CHAr), 141.9 (CqAr); HR-MS (ESI): m/z calcd for C17H28NO2 [M + H]+: 278.21146; found: 278.21096.
Prepared from 21b according to the general method. Yield: 0.246 g (89%); light yellow oil; [α = −76 (c 0.245, MeOH); 1H NMR (DMSO-d6, 500.2 MHz): δ = 0.83 (6H, d, J = 5.6 Hz, 2 × CH2Me2) 1.22–1.42 (6H, m, H5α, CH2(3), H4, CH2(5)), 1.48–1.55 (1H, m, H5β), 1.59–1.67 (1H, m, H6α), 2.55–2.58 (1H, m, H6β), 3.29 (2H, dd, J = 10.8, 14.9 Hz CH2OH), 3.61 (1H, d, J = 13.8 Hz, CH2Ph), 3.77 (1H, d, J = 13.8 Hz, CH2Ph), 3.90 (1H, s, OH), 7.20–7.23 (1H, m, CHAr), 729–7.33 (4H, m, 4 × CHAr); 13C NMR (125.8 MHz, DMSO-d6): δ = 20.2 (CH2Me2), 20.3 (CH2Me2), 23.9 (C5), 27.4 (C3), 29.6 (CHMe2), 32.5, (C6), 36.4 (C4), 51.4 (CH2Ph), 59.2 (C2), 70.0 (CH2OH), 71.7 (C1), 127.0 (CHAr), 128.5 (2 × CHAr), 128.6 (2 × CHAr), 141.6 (CqAr); HR-MS (ESI): m/z calcd for C17H28NO2 [M + H]+: 278.21146; found: 278.21073.
To a suspension of LiAlH4 (0.18 g, 4.7 mmol) in dry THF (5 mL), a solution of compound 19a (0.45 g, 1.57 mmol) in dry THF (5 mL) was added dropwise and stirred for 2 h, and then the excess of LiAlH4 was quenched with a mixture of H2O (0.36 mL) and THF (5 mL) at 0 °C. The suspension was stirred for 1 h at room temperature and then filtered. The inorganic residue was washed with THF (3 × 30 mL), and then the organic layer was dried, filtered, and concentrated to dryness. As its hydrochloride salt, the crude product was crystallised using a solution of HCl (10%, in EtOH/Et2O).
Yield: 0.243 g (65%); white crystals m.p.: 155–157 °C; [α = −35 (c 0.265, MeOH); 1H NMR (DMSO-d6, 500.2 MHz): δ = 0.85 (3H, d, J = 7.1 Hz), 0.86 (3H, d, J = 7.1 Hz), 1.25–1.51 (5H, m), 1.63–1.77 (3H, m), 2.54 (3H, s), 3.07 (1H, br s), 3.38 (1H, d, J = 12.1 Hz), 3.49 (1H, d, J = 11.9 Hz), 4.89 (1H, s), 5.28 (1H, br s), 8.51 (2H, br s,); 13C NMR (125.8 MHz, DMSO-d6): δ = 20.1, 20.2, 23.3, 25.4, 29.3, 31.2, 32.6, 36.5, 60.8, 66.5, 70.8; HR-MS (ESI): m/z calcd for C11H24NO2 [M + H]+: 202.18016; found: 202.17983.
General method for ring closure of compounds 22, 23a, and 23b with formaldehyde
To a solution of aminodiol 22, 23a, and 23b (0.58 mmol) in Et2O (10 mL), an aqueous solution of formaldehyde (40%, 5 mL) was added, and the mixture was stirred at room temperature for 1 h. An aqueous solution of NaOH (5%) was added to the reaction mixture to make it alkaline and extracted with Et2O (3 × 20 mL). The combined organic phase was dried (Na2SO4), filtered, and evaporated in vacuo. The crude products were purified by column chromatography (toluene/EtOH 4:1).
Prepared from 22 according to the general method. Yield: 0.081 g (66%); brown oil; [α = −38 (c 0.280, MeOH); 1H NMR (DMSO-d6, 500.2 MHz): δ = 0.83 (6H, d, J = 6.5 Hz), 1.21–1.27 (1H, m), 1.30–1.41 (4H, m), 1.44–1.52 (1H, m), 1.62–1.68 (1H, m), 1.87–1.91 (1H, m), 1.97 (3H, s), 3.10 (1H, d, J = 10.5 Hz,), 3.40 (1H, d, J = 11.3 Hz), 3.41 (1H, d, J = 7.3 Hz), 4.27 (1H, d, J = 7.4 Hz), 4.41 (1H, s); 13C NMR (125.8 MHz, DMSO-d6): δ = 20.0, 20.2, 24.9, 26.1, 31.6, 32.4, 35.8, 36.1, 65.6, 67.7, 77.3, 87.6; HR-MS (ESI): m/z calcd for C12H24NO2 [M + H]+: 214.18016; found: 214.17979.
Prepared from 23a according to the general method. Yield: 0.149 g (89%); yellowish-brown transparent oil; [α = −45 (c 0.255, MeOH); 1H NMR (DMSO-d6, 500.2 MHz): δ = 0.77 (3H, d, J = 7.4 Hz, CHMe2), 0.79 (3H, d, J = 7.4 Hz, CHMe2), 1.22–1.50 (CH2(3, m, CH2(3), H7, CH2(5)), 1.52–1.60 (1H, m, H8α), 1.80–1.87 (1H, m H8β), 2.16–2.27 (2H, m H8a), 3.08 (1H, d, J = 14.3 Hz, H4α), 3.12 (1H, d, J = 10.6 Hz CH2Ph), 3.42 (1H, d, J = 10.5 Hz CH2Ph), 3.53 (1H, d, J = 7.8 Hz H2), 3.86 (1H, d, J = 14.3 Hz H4β), 4.20 (1H, d, J = 7.7 Hz H2), 4.47 (1H, s, OH), 7.21–7.35 (5H, m, 5 × CHAr); 13C NMR (125.8 MHz, DMSO-d6): δ = 20.0 (CH2Me2), 20.2 (CH2Me2), 25.0 (C5), 26.1 (C6), 31.6 (CHMe2), 32.1 (C5), 36.4 (C7), 52.0 (CH2Ph), 65.4 (C8a), 66.0 (C4a), 77.3 (C4), 85.2 (C2), 127.3 (CHAr), 128.7 (2 × CHAr), 128.8 (2 × CHAr), 139.2 (CqAr); HR-MS (ESI): m/z calcd for C18H28NO2 [M + H]+: 290.21146; found: 290.21082.
Prepared from 23b according to the general method. Yield: 0.134 g (80%); yellowish-brown transparent oil; [α = −21 (c 0.250, MeOH); 1H NMR (DMSO-d6, 500.2 MHz): δ = 0.79 (3H, d, J = 7.8 Hz), 0.83 (3H, d, J = 7.8 Hz), 1.20–1.53 (5H, m), 1.56–1.66 (1H, m), 1.79–1.87 (1H, m), 2.16–2.29 (2H, m), 3.09 (1H, d, J = 14.3 Hz), 3.12 (1H, d, J = 10.6 Hz), 3.42 (1H, d, J = 10.5 Hz), 3.53 (1H, d, J = 7.8 Hz), 3.86 (1H, d, J = 14.3 Hz), 4.20 (1H, d, J = 7.7 Hz), 4.47 (1H, s), 7.21–7.35 (5H, m); 13C NMR (125.8 MHz, DMSO-d6): δ = 20.0, 20.2, 25.0, 26.1, 31.6, 32.1, 36.4, 52.0, 65.4, 66.0, 77.3, 85.2, 127.3, 128.7, 128.8, 139.2; HR-MS (ESI): m/z calcd for C18H28NO2 [M + H]+: 290.21146; found: 290.21123.
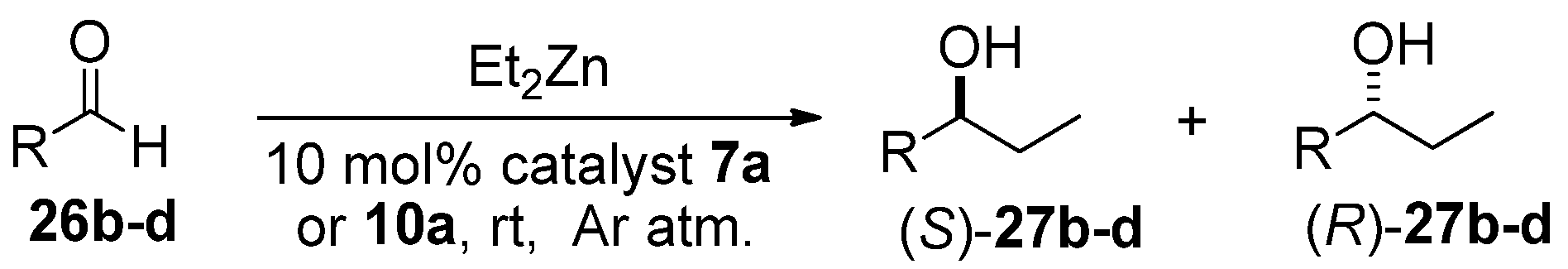
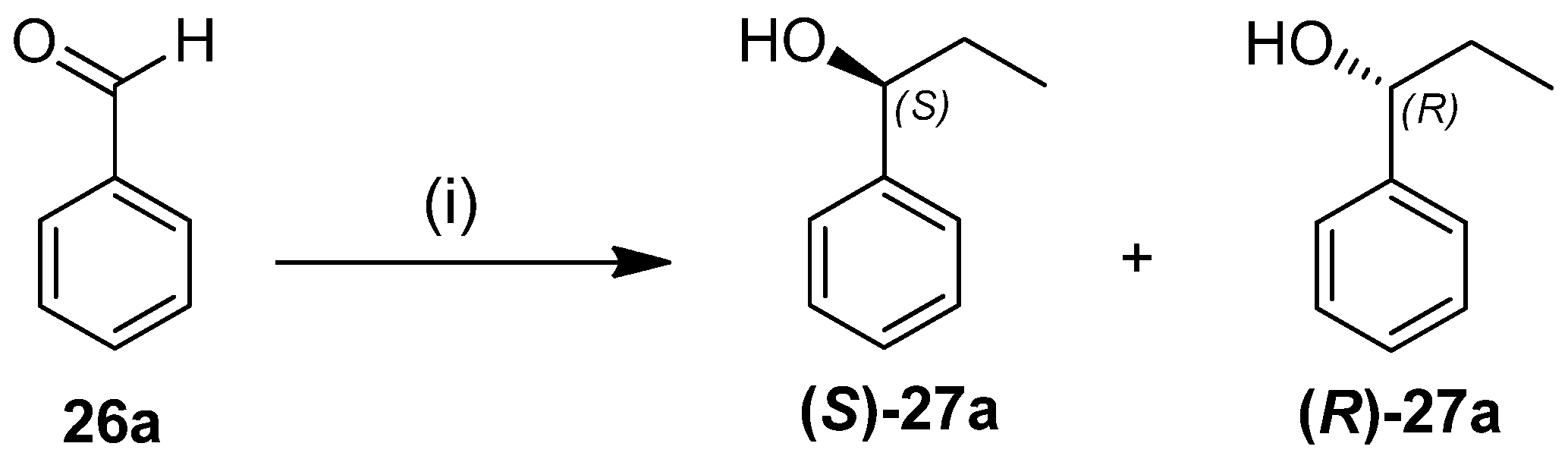
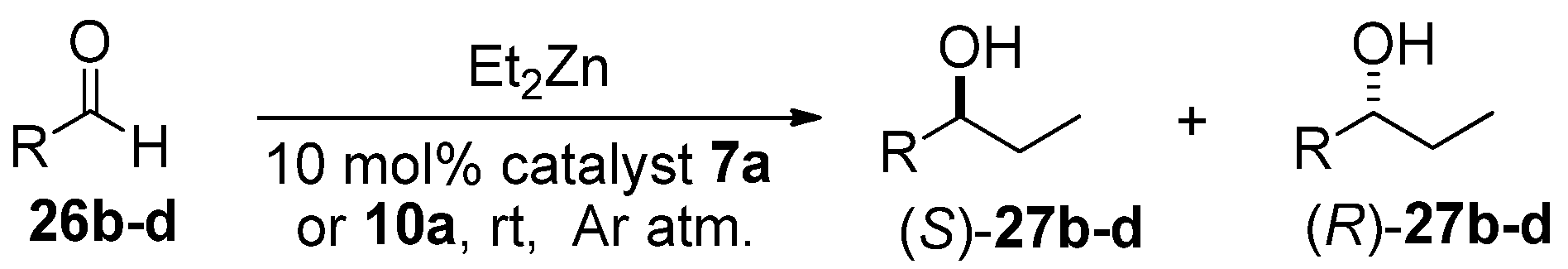
General procedure for the reaction of benzaldehyde with diethylzinc in the presence of chiral catalysts.
A solution of Et2Zn in n-hexane (1M, 4.5 mL) was added to the appropriate catalyst (10 mol%) under an Ar atmosphere at room temperature. The reaction mixture was stirred for 20 min at room temperature, and then benzaldehyde (0.156 g, 153 μL, 1.5 mmol) was added. The mixture was stirred for a further 20 h at room temperature, then quenched with a saturated solution of NH4Cl (50 mL) and extracted with EtOAc (2 × 30 mL). The combined organic layer was dried (Na2SO4), filtered, and evaporated. The ee values and absolute configurations of the obtained secondary alcohols were determined by chiral-phase GC by using a CHIRASIL-DEX CB column, at 90 °C, after O-acetylation in an AcO2/4-dimethylaminopyridine/pyridine system.
Identification of
27b–
d was achieved by chiral HPLC analysis on a Chiralcel OD-H column and the data are as follows: 1-(4-tolyl)-1-propanol
27b V(
n-hexane)/
V(2-propanol) = 95:5, 0.5 mL/min,
tR1 = 16.0 min for
R-isomer,
tR2 = 22.2 min for
S-isomer. 1-(4-methoxyphenyl)-1-propanol
27c;
V(
n-hexane)/
V(2-propanol) = 95:5, 0.7 mL/min, 210 nm,
tR1 = 15.9 min for
R-isomer,
tR2 = 18.0 min for
S-isomer. 1-(3-Methoxyphenyl)-1-propanol
27d;
V(
n-hexane)/
V(2-propanol) = 98:2, 0.4 mL/min, 210 nm,
tR1 = 74.9 min for
R-isomer,
tR2 = 77.8 min for
S-isomer (
Figures S157–S162).
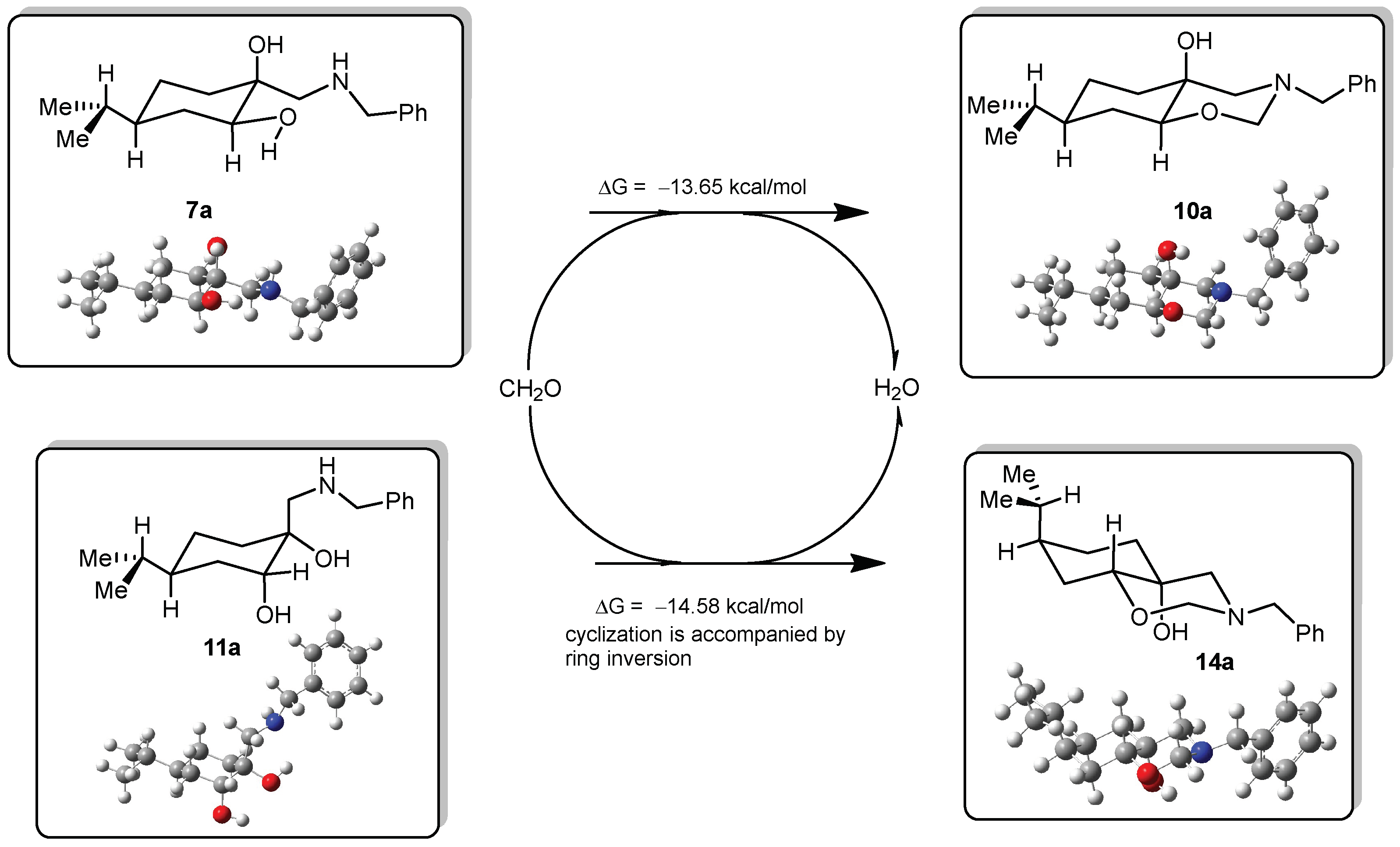
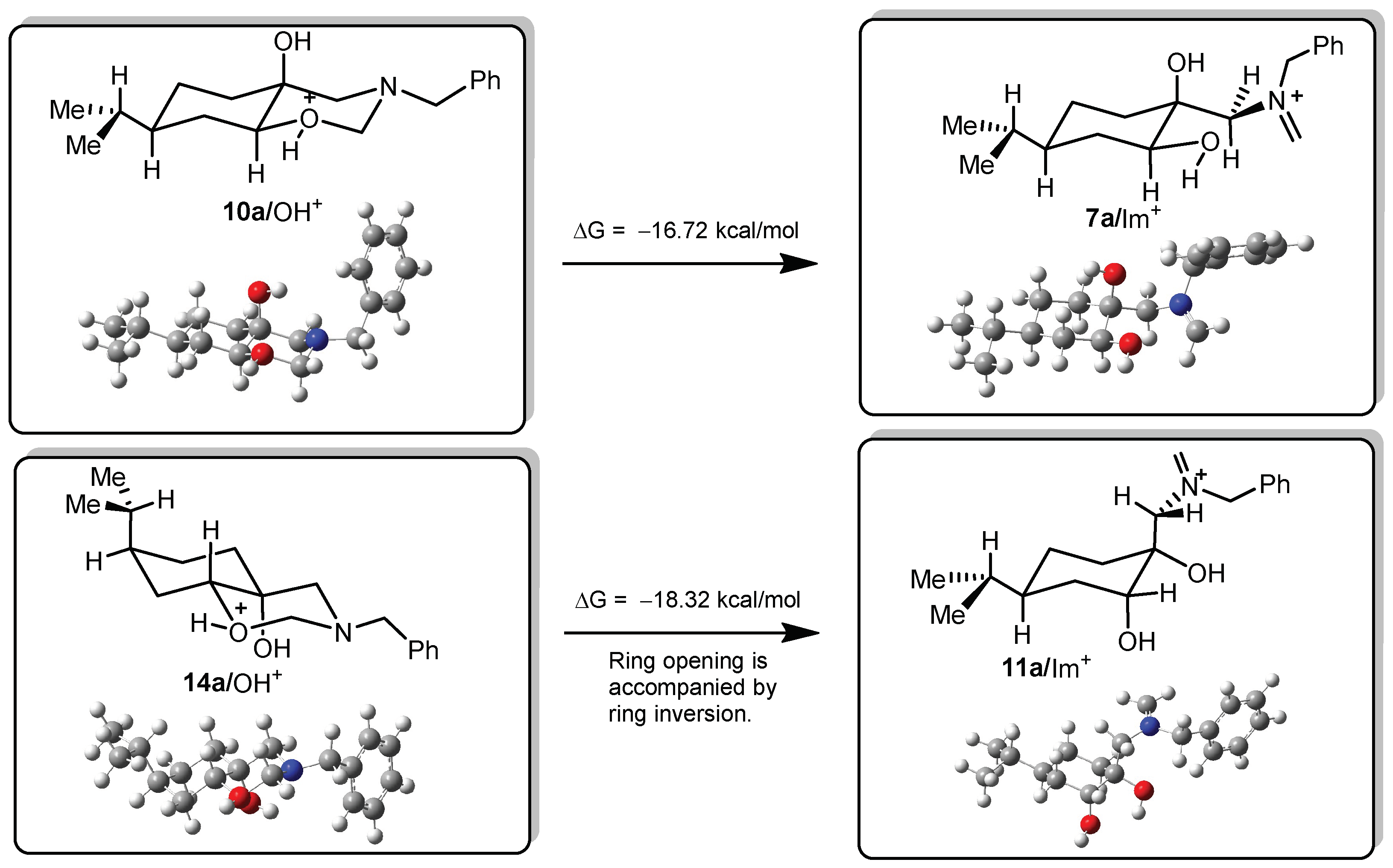
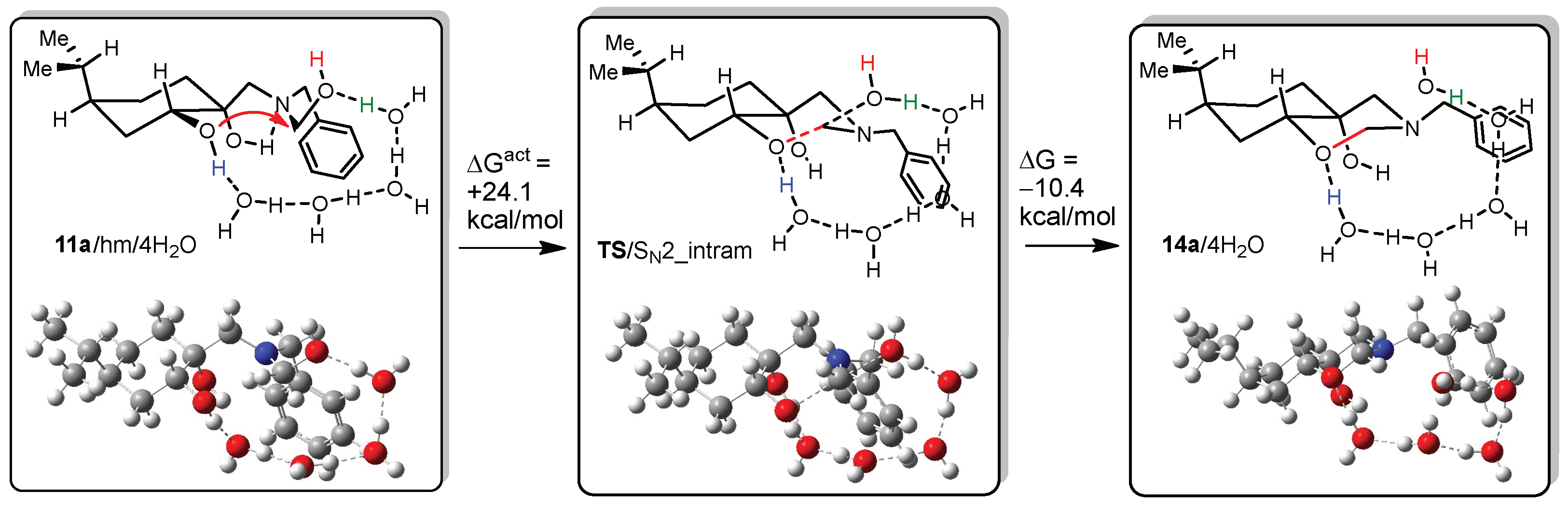
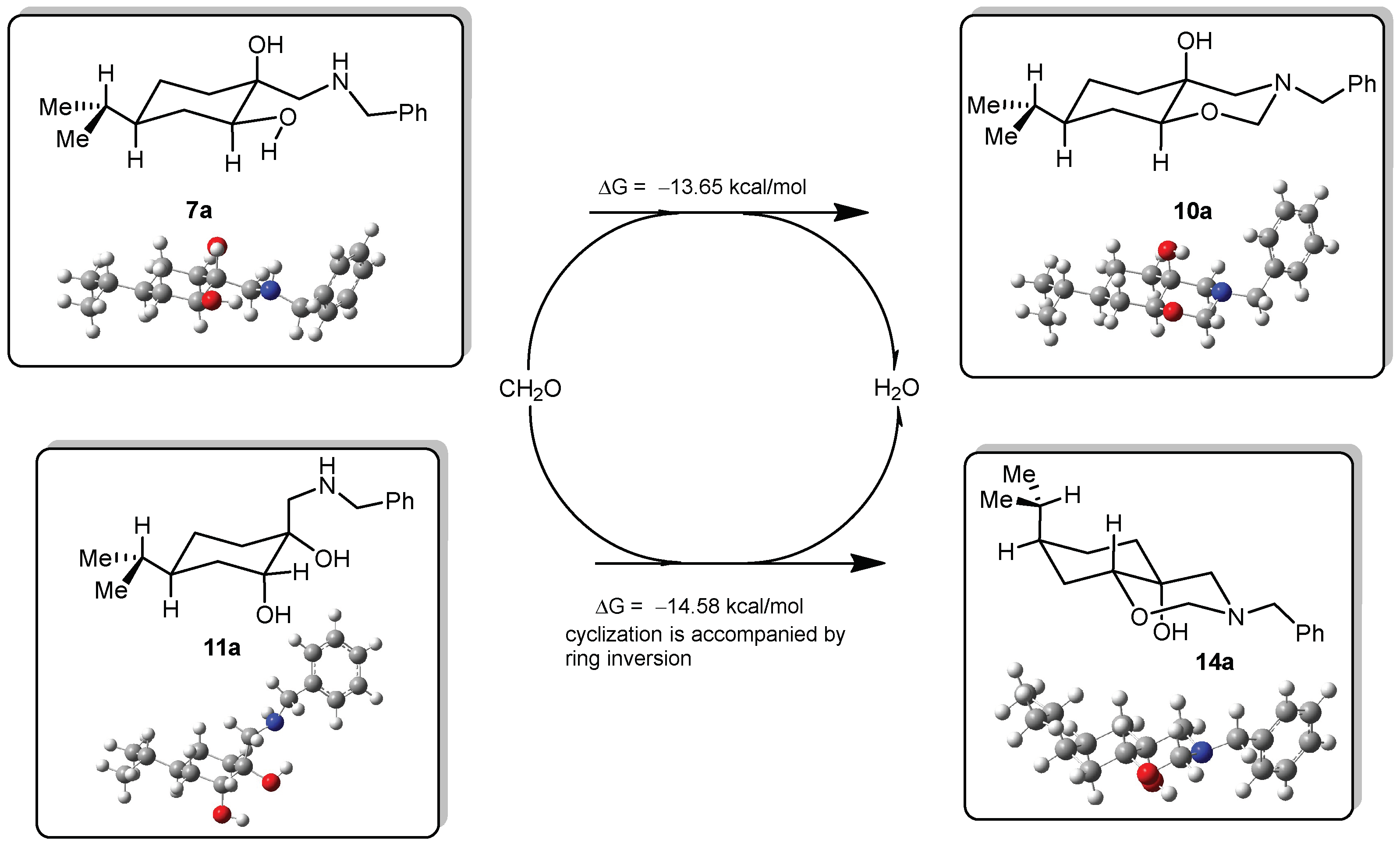
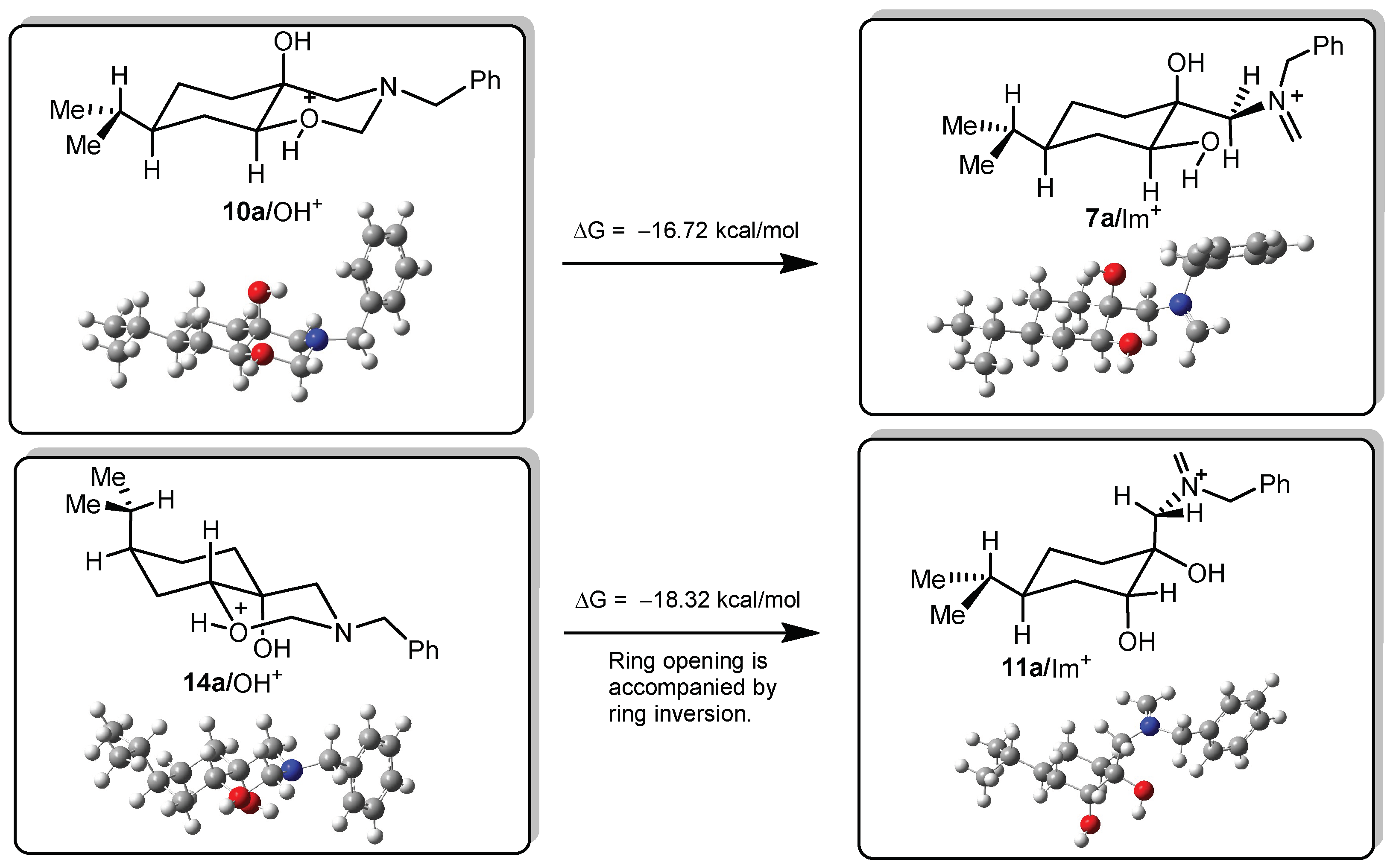
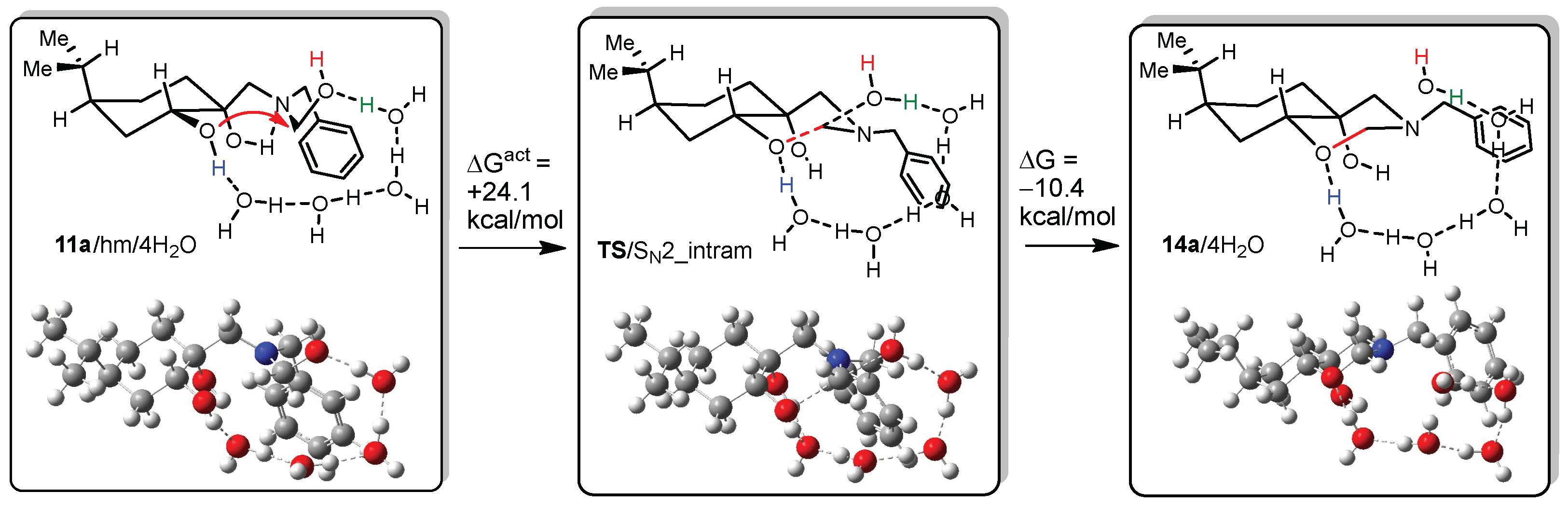
DFT calculations on compounds 10a and 14a
All DFT calculations were carried out by Gaussian 09 Revision A.02 software (Gaussian Incorporation, Pittsburgh, PA, USA), package [Gaussian 09], using the M06-2X global hybrid DFT functional [
38] and 6-31+G(d,p) basis set [
39]. Structural optimisations and subsequent frequency calculations were supported by the IEFPCM solvent model [
40] parameterised with the dielectric constant of water (ε = 78.4) to represent the approximate polarity of the experimental reaction conditions. The Gibbs free-energy values of optimised structures were obtained by correcting the computed total energy with zero-point vibrational energy (ZPE) and the calculated thermal corrections. The optimised structures are available from the authors.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}