
Synthesis of Estrone Heterodimers and Evaluation of Their In Vitro Antiproliferative Activity
 , ,
, ,
Abstract
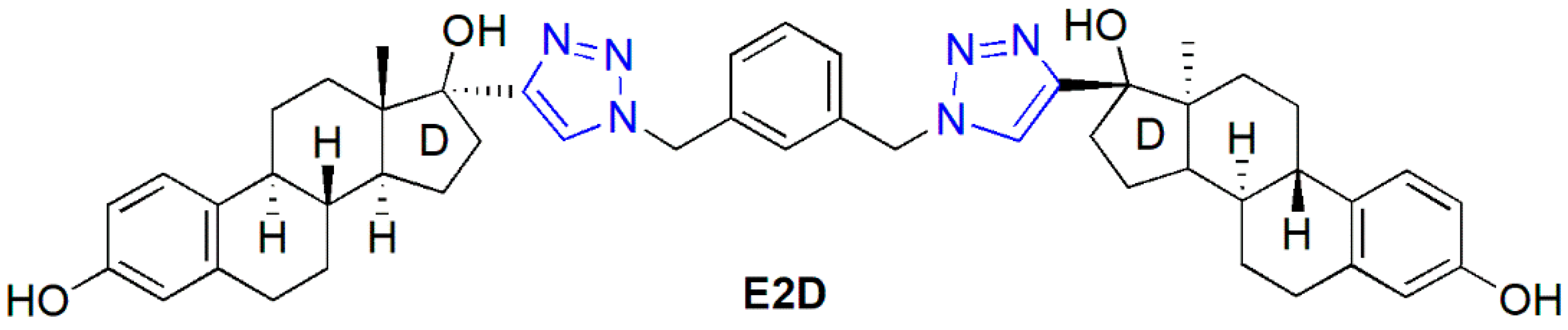
1. Introduction
2. Results
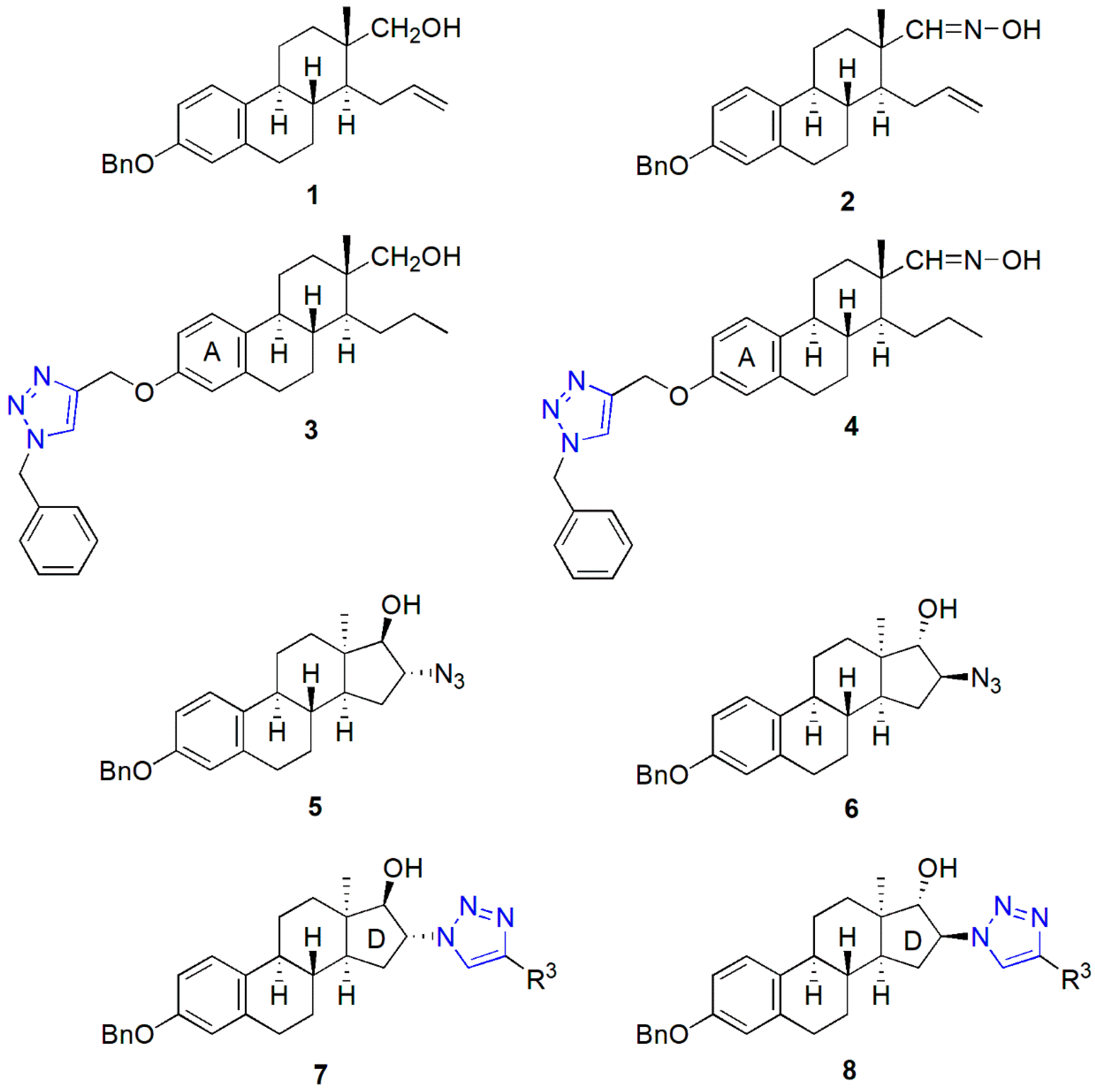
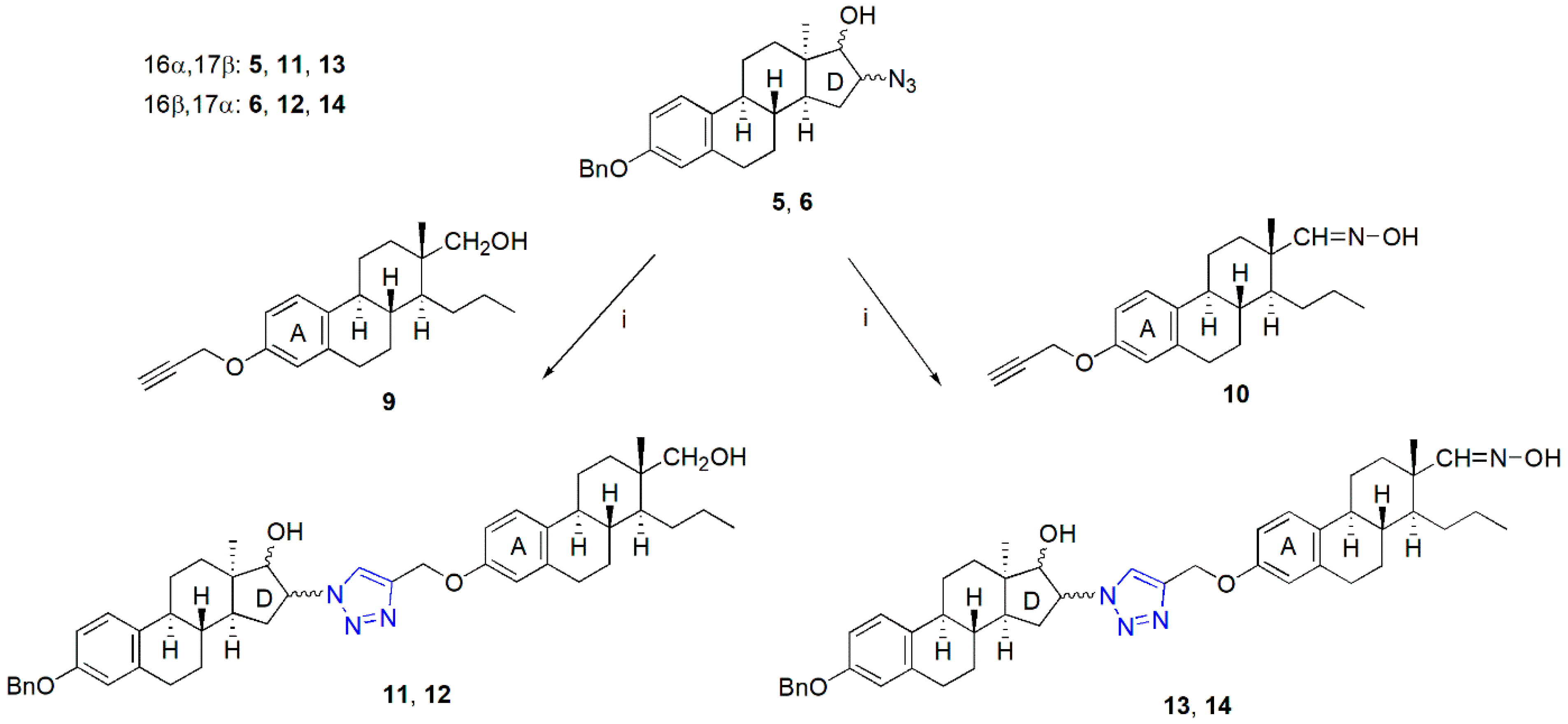
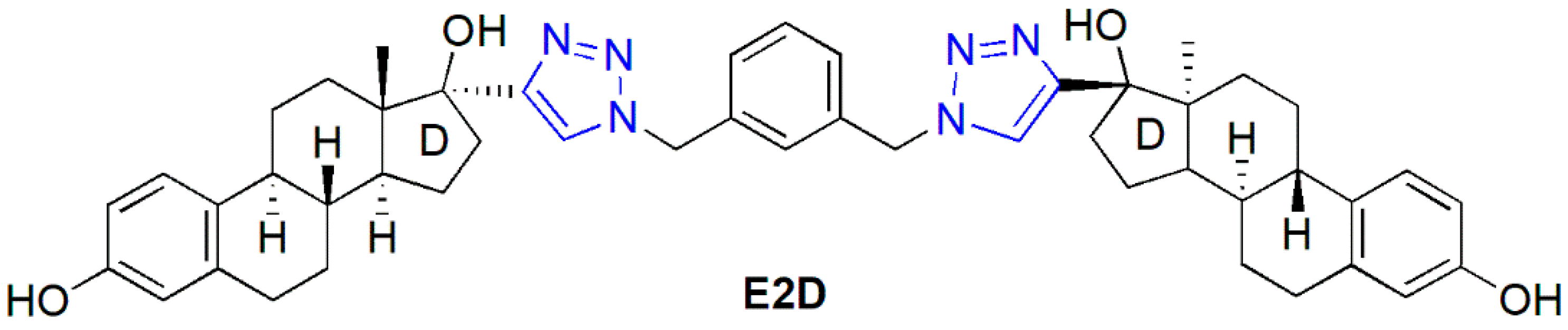
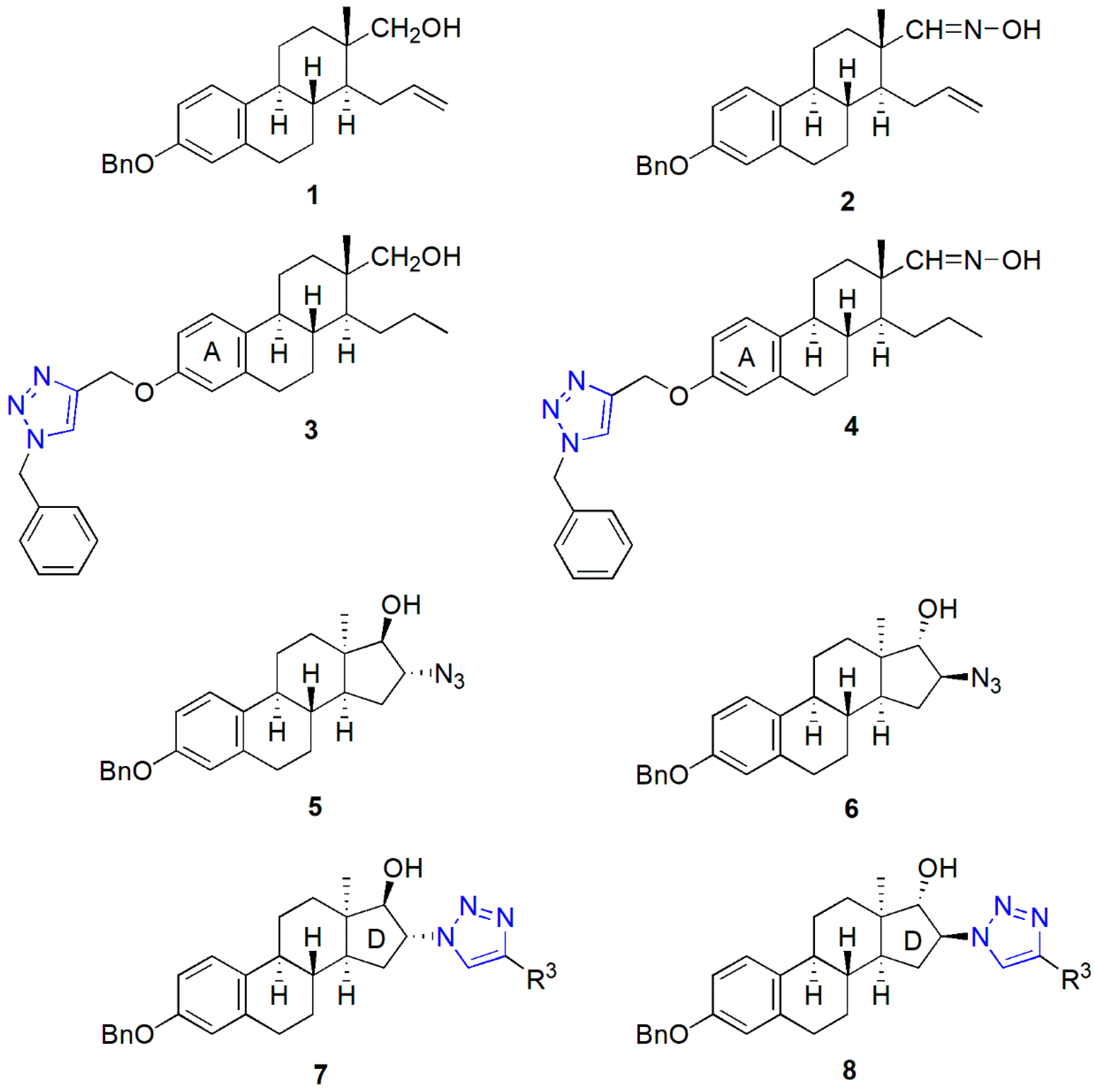
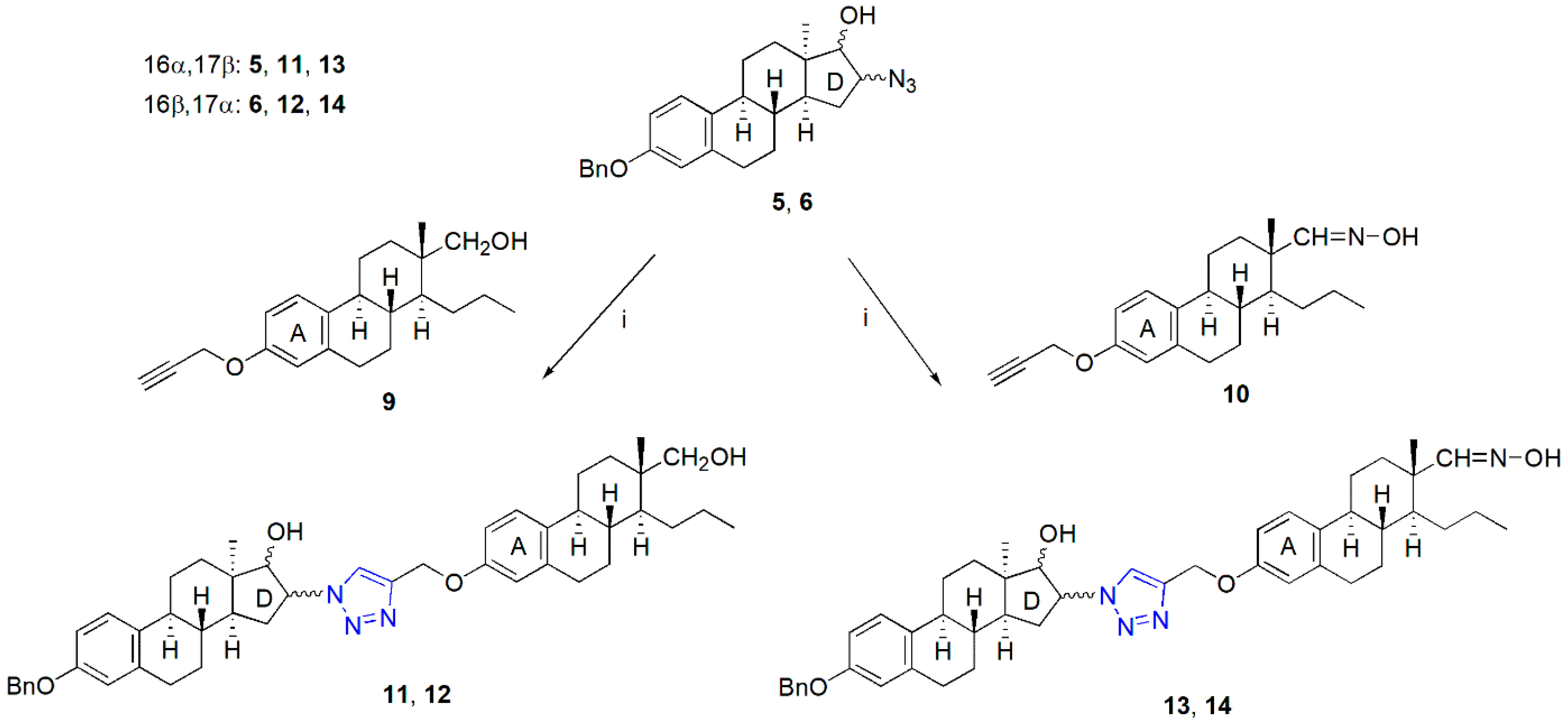
2.1. Synthesis of the Heterodimers (11–14)
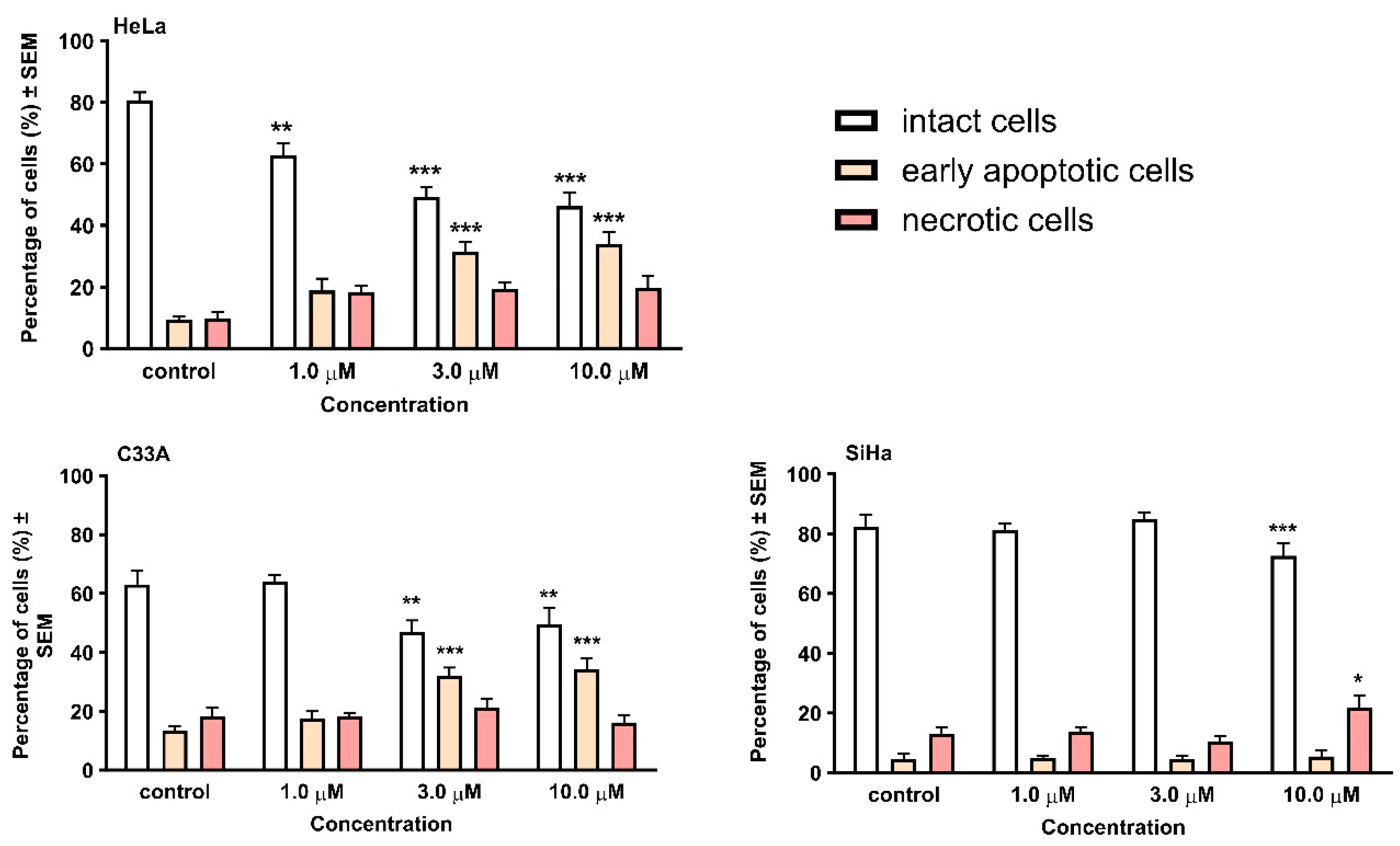
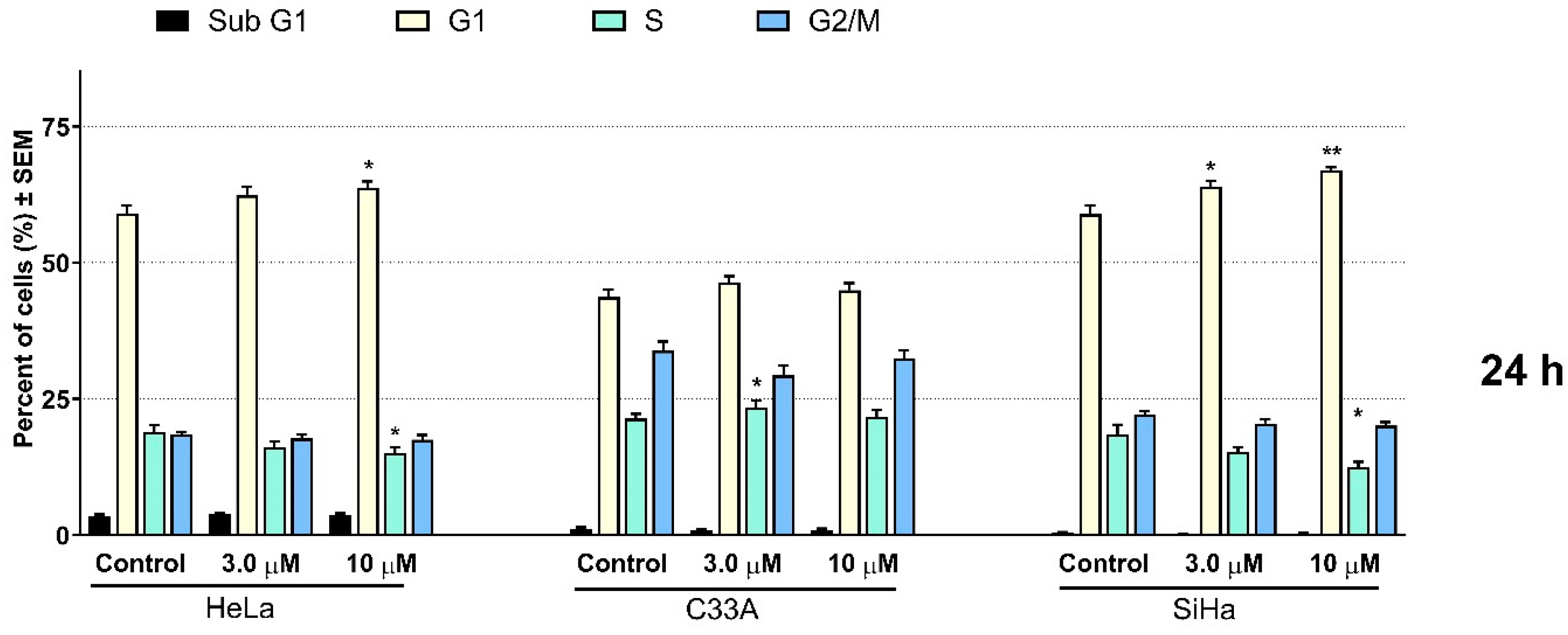
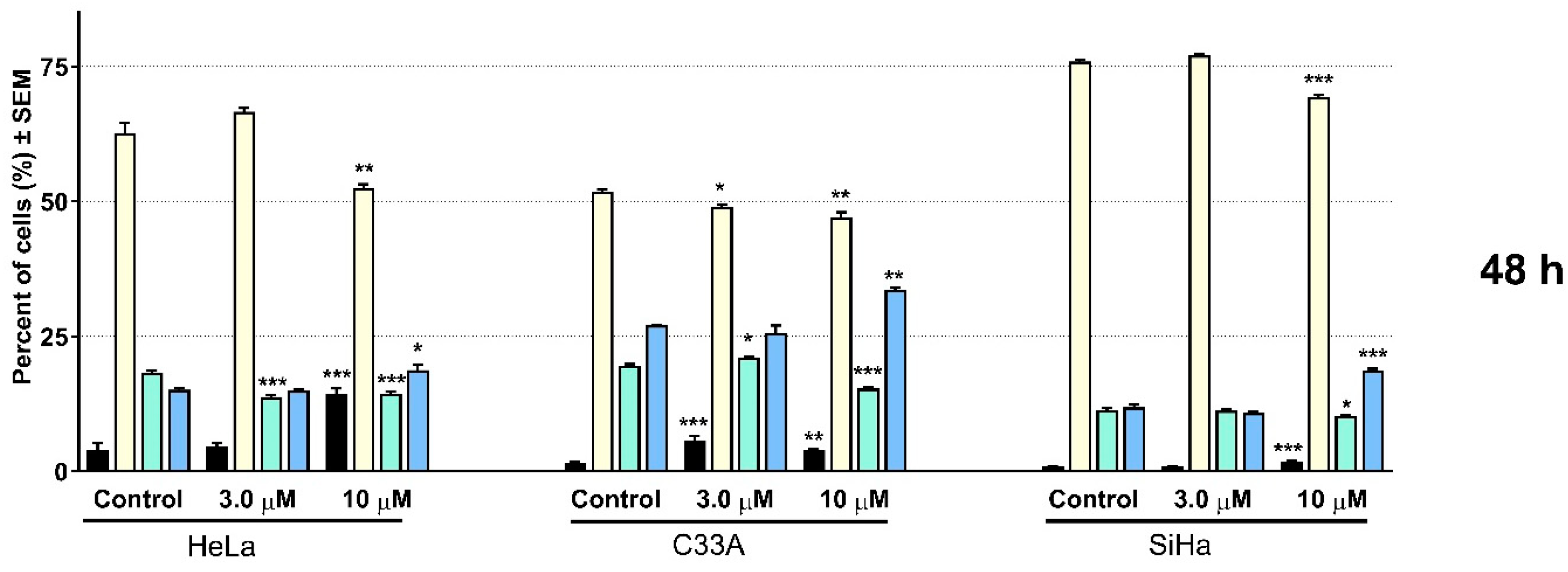
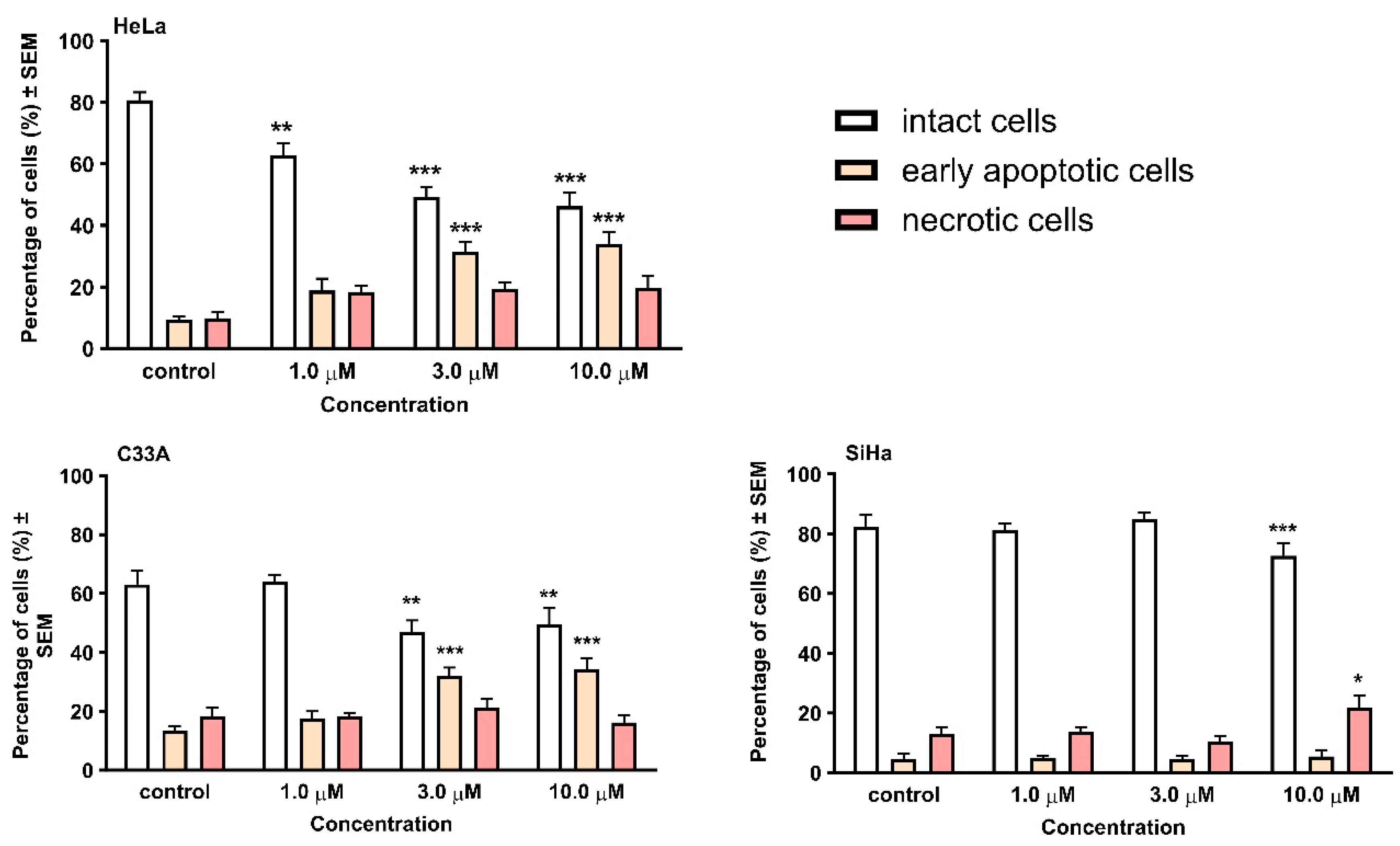
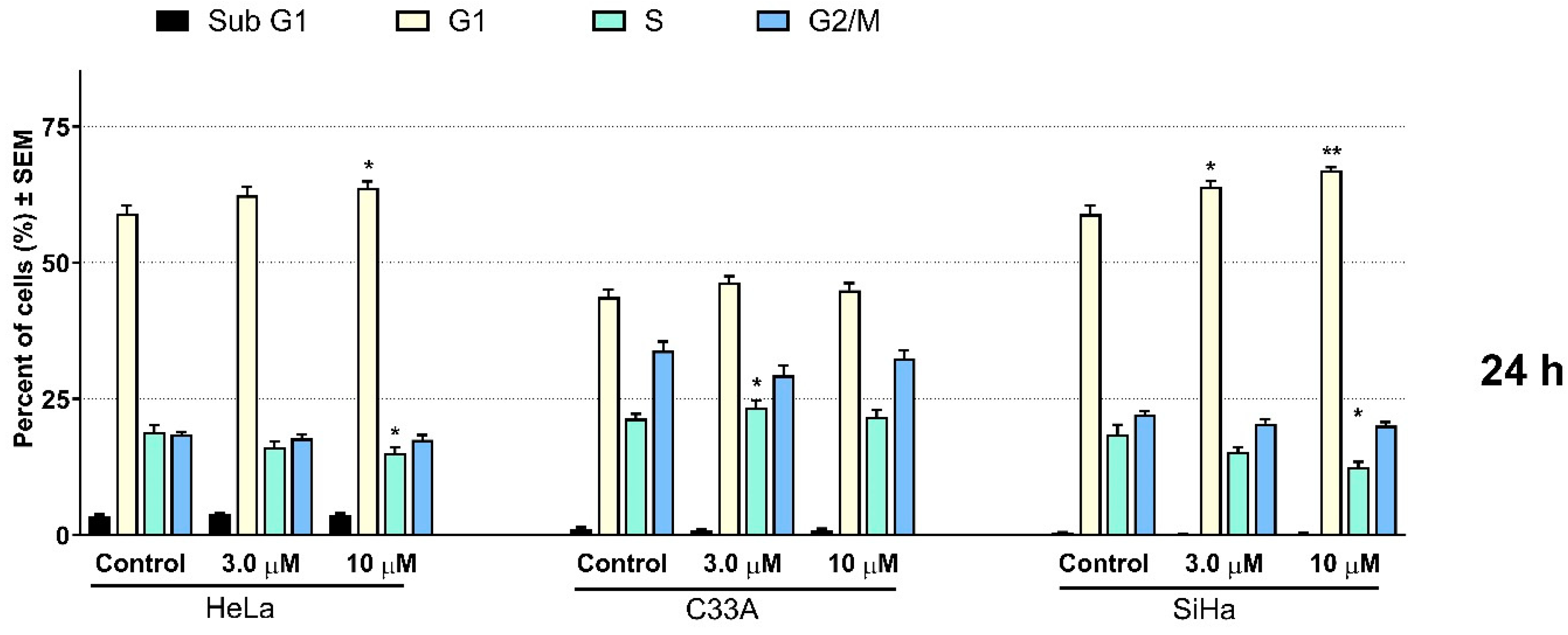
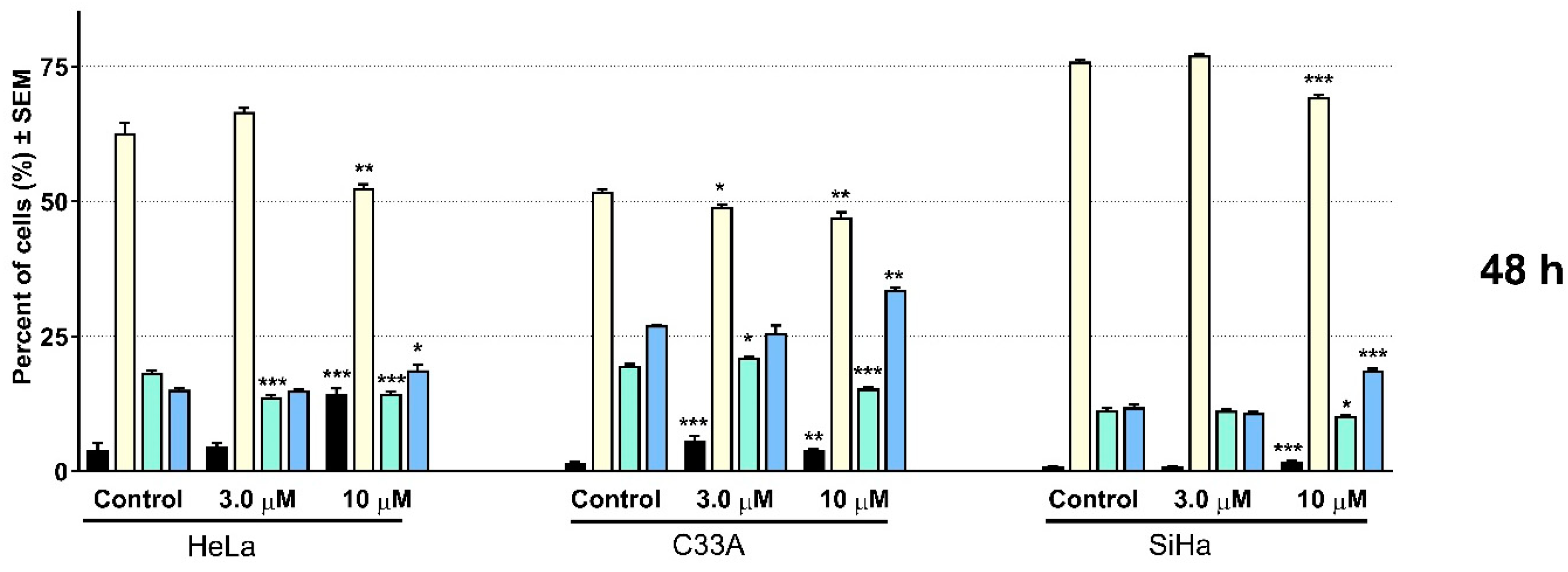
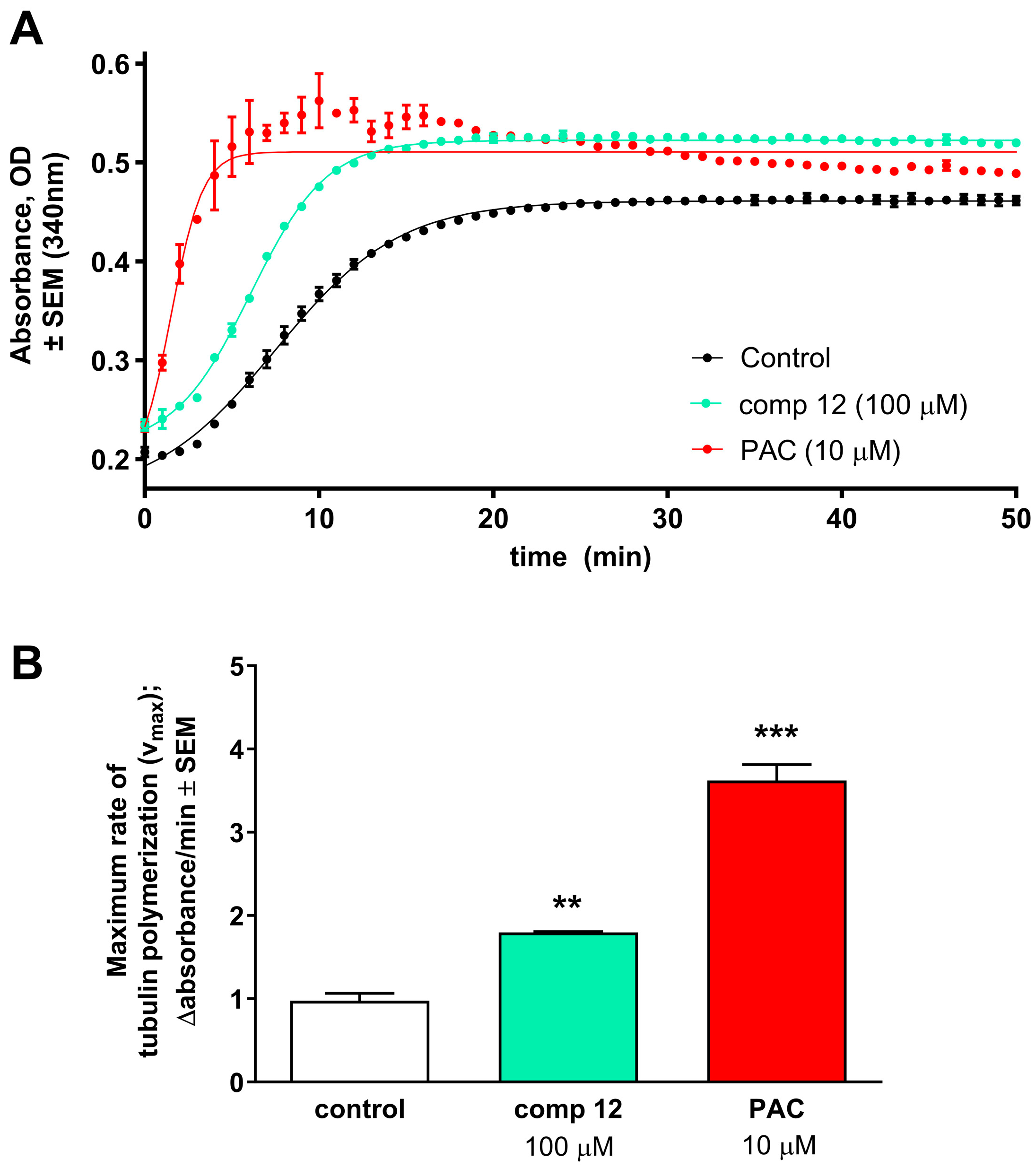
2.2. Pharmacology
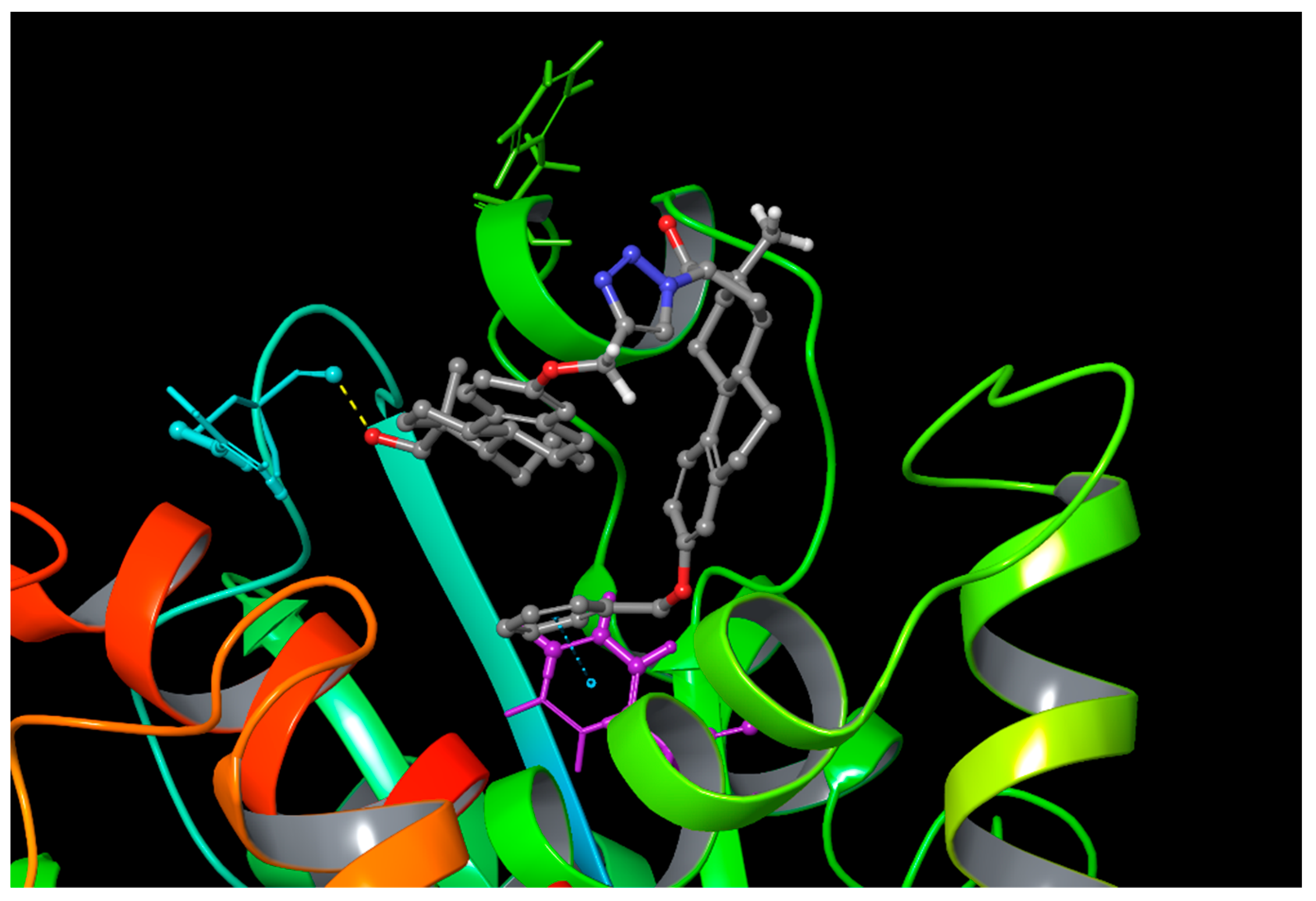
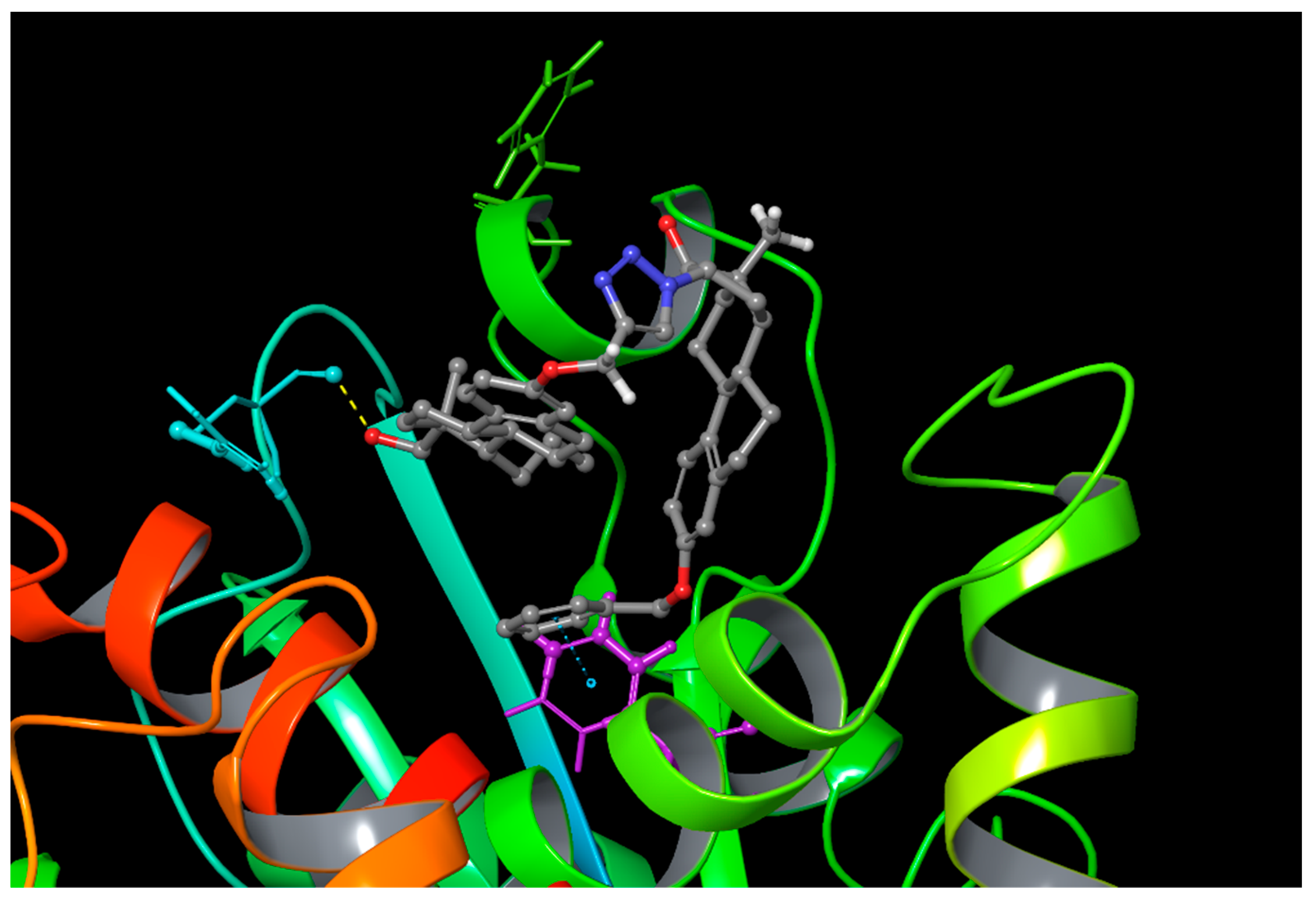
2.3. Computational Simulations
3. Discussion
4. Methods and Materials
4.1. Chemistry
General Procedure for the Synthesis of Heterodimers (11–14)
4.2. Pharmacology
4.2.1. Antiproliferative (MTT) Assay
4.2.2. Cell Cycle Analysis by Flow Cytometry
4.2.3. Hoechst 33258—Propidium Iodide (HOPI) Double Staining
4.2.4. Tubulin Polymerization Assay
4.3. Computational Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Tietze, L.F.; Bell, H.P.; Chandrasekhar, S. Natural product hybrids as new leads for drug discovery. Angew. Chem. Int. Ed. Engl. 2003, 42, 3996–4028. [Google Scholar] [CrossRef]
- Nahar, L.; Sarker, S.D. A review on steroid dimers: 2011–2019. Steroids 2020, 164, 108736–108767. [Google Scholar] [CrossRef] [PubMed]
- Nahar, L.; Sarker, S.D. Steroid Dimers: Chemistry and Applications in Drug Design and Delivery; Wiley & Sons: Chichester, UK, 2012. [Google Scholar]
- Nahar, L.; Sarker, S.D.; Turner, A.B. A review on synthetic and natural steroid dimers: 1997–2006. Curr. Med. Chem. 2007, 14, 1349–1370. [Google Scholar] [CrossRef]
- Yu, B.; Qi, P.P.; Shi, X.J.; Huang, R.; Guo, H.; Zheng, Y.C.; Yu, D.Q.; Liu, H.M. Efficient synthesis of new antiproliferative steroidal hybrids using the molecular hybridization approach. Eur. J. Med. Chem. 2016, 117, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Shi, X.J.; Zheng, T.F.; Fang, Y.; Zhang, E.; Yu, D.Q.; Liu, H.M. A novel [1,2,4] triazolo [1,5-α] pyrimidine-based phenyl-linked steroid dimer: Synthesis and its cytotoxic activity. Eur. J. Med. Chem. 2013, 69, 323–330. [Google Scholar] [CrossRef]
- Pettit, G.R.; Xu, J.P.; Chapuis, J.C.; Melody, N. The cephalostatins. 24. Isolation, structure, and cancer cell growth inhibition of cephalostatin 20. J. Nat. Prod. 2015, 78, 1446–1450. [Google Scholar] [CrossRef]
- Tahtamouni, L.H.; Nawasreh, M.M.; Al-Mazaydeh, Z.A.; Al-Khateeb, R.A.; Abdellatif, R.N.; Bawadi, R.M.; Bamburg, J.R.; Yasin, S.R. Cephalostatin 1 analogues activate apoptosis via the endoplasmic reticulum stress signaling pathway. Eur. J. Pharmacol. 2018, 818, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Jurášek, M.; Černohorská, M.; Řehulka, J.; Spiwok, V.; Sulimenko, T.; Dráberová, E.; Darmostuk, M.; Gurská, S.; Frydrych, I.; Buriánová, R.; et al. Estradiol dimer inhibits tubulin polymerization and microtubule dynamics. J. Steroid Biochem. Mol. Biol. 2018, 183, 68–79. [Google Scholar] [CrossRef]
- Jurášek, M.; Řehulka, J.; Hrubá, L.; Ivanová, A.; Gurská, S.; Mokshyna, O.; Trousil, P.; Huml, L.; Polishchuk, P.; Hajdúch, M.; et al. Triazole-based estradiol dimers prepared via CuAAC from 17α-ethinyl estradiol with five-atom linkers causing G2/M arrest and tubulin inhibition. Bioorg. Chem. 2023, 131, 106334–106347. [Google Scholar] [CrossRef]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef]
- Naaz, F.; Haider, M.R.; Shafi, S.; Yar, M.S. Anti-tubulin agents of natural origin: Targeting taxol; vinca; and colchicine binding domains. Eur. J. Med. Chem. 2019, 171, 310–331. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.N.; Zheng, L.L.; Wang, D.; Liang, X.X.; Gao, F.; Zhou, X.L. Recent advances in microtubule-stabilizing agents. Eur. J. Med. Chem. 2018, 143, 806–828. [Google Scholar] [CrossRef] [PubMed]
- Field, J.J.; Dıaz, J.F.; Miller, J.H. The binding sites of microtubule stabilizing agents. Chem. Biol. 2013, 20, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sun, H.; Xu, S.; Zhu, Z.; Xu, J. Tubulin inhibitors targeting the colchicine binding site: A perspective of privileged structures. Future Med. Chem. 2017, 9, 1765–1794. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic-Santa, S.; Petrovic, J.; Andric, S.; Kovacevic, R.; Durendic, E.; Sakac, M.; Lazar, D.; Stankovic, S. Synthesis, structure, and screening of estrogenic and antiestrogenic activity of new 3,17-substituted-16,17-seco-estratriene derivatives. Bioorg. Chem. 2003, 31, 475–484. [Google Scholar] [CrossRef]
- Yaremenko, F.G.; Khvat, A.V. A new one-pot synthesis of 17-oxo-13α-steroids of the androstane series from their 13β-analogues. Mendeleev Commun. 1994, 187, 187–188. [Google Scholar]
- Schönecker, B.; Lange, C.; Kötteritzsch, M.; Günther, W.; Weston, J.; Anders, E.; Görls, H. Conformational design for 13α-steroids. J. Org. Chem. 2000, 65, 5487–5497. [Google Scholar] [CrossRef]
- Ayan, D.; Roy, J.; Maltais, R.; Poirier, D. Impact of estradiol structural modifications (18-methyl and/or 17-hydroxy inversion of configuration) on the in vitro and in vivo estrogenic activity. J. Steroid Biochem. Mol. Biol. 2011, 127, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Bózsity, N.; Minorics, R.; Szabó, J.; Mernyák, E.; Schneider, G.; Wölfling, J.; Wang, H.C.; Wu, C.C.; Ocsovszki, I.; Zupkó, I. Mechanism of antiproliferative action of a new d-secoestrone-triazole derivative in cervical cancer cells and its effect on cancer cell motility. J. Steroid Biochem. Mol. Biol. 2017, 165, 247–257. [Google Scholar] [CrossRef]
- Mernyák, E.; Fiser, G.; Szabó, J.; Bodnár, B.; Schneider, G.; Kovács, I.; Ocsovszki, I.; Zupkó, I.; Wölfling, J. Synthesis and in vitro antiproliferative evaluation of D-secooxime derivatives of 13β- and 13α-estrone. Steroids 2014, 89, 47–55. [Google Scholar] [CrossRef]
- Szabó, J.; Jerkovics, N.; Schneider, G.; Wölfling, J.; Bózsity, N.; Minorics, R.; Zupkó, I.; Mernyák, E. Synthesis and in Vitro Antiproliferative Evaluation of C-13 Epimers of Triazolyl-D-Secoestrone Alcohols: The First Potent 13α-d-Secoestrone Derivative. Molecules 2016, 12, 611. [Google Scholar] [CrossRef]
- Mernyák, E.; Kovács, I.; Minorics, R.; Sere, P.; Czégány, D.; Sinka, I.; Wölfling, J.; Schneider, G.; Újfaludi, Z.; Boros, I.; et al. Synthesis of trans-16-triazolyl-13α-methyl-17-estradiol diastereomers and the effects of structural modifications on their in vitro antiproliferative activities. J. Steroid Biochem. Mol. Biol. 2015, 150, 123–134. [Google Scholar] [CrossRef]
- Mallmann, P.; Beckmann, M.W.; Emons, G. Innovations in cervical and endometrial cancer. Geburtshilfe Und Frauenheilkd. 2013, 73, 908–910. [Google Scholar] [CrossRef]
- Berghe, T.; Vanlangenakker, N.; Parthoens, E.; Deckers, W.; Devos, M.; Festjens, N.; Guerin, C.J.; Brunk, U.T.; Declercq, W.; Vandenabeele, P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010, 17, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Stander, A.; Joubert, F.; Joubert, A. Docking, synthesis, and in vitro evaluation of antimitotic estrone analogs. Chem. Biol. Drug. Des. 2011, 77, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Minorics, R.; Bózsity, N.; Molnár, J.; Wölfling, J.; Mernyák, E.; Schneider, G.; Ocsovszki, I.; Zupkó, I. A molecular understanding of D-homoestrone-induced G2/M cell cycle arrest in HeLa human cervical carcinoma cells. J. Cell Mol. Med. 2015, 19, 2365–2374. [Google Scholar] [CrossRef]
- Peyrat, J.F.; Brion, J.D.; Alami, M. Synthetic 2-methoxyestradiol derivatives: Structure-activity relationships. Curr. Med. Chem. 2012, 19, 4142–4156. [Google Scholar] [CrossRef]
- Bhalla, K.N. Microtubule-targeted anticancer agents and apoptosis. Oncogene 2003, 22, 9075–9086. [Google Scholar] [CrossRef] [PubMed]
- Vermes, I.; Haanen, C.; Reutelingsperger, C. Flow cytometry of apoptotic cell death. J. Immunol. Methods 2000, 243, 167–190. [Google Scholar] [CrossRef] [PubMed]
- Fritzer-Szekeres, M.; Savinc, I.; Horvath, Z.; Saiko, P.; Pemberger, M.; Graser, G.; Bernhaus, A.; Ozsvar-Kozma, M.; Grusch, M.; Jaeger, W.; et al. Biochemical effects of piceatannol in human HL-60 promyelocytic leukemia cells-synergism with Ara-c. Int. J. Oncol. 2008, 33, 887–892. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Schrödinger Release 2023-4: Maestro; Schrödinger, LLC.: New York, NY, USA, 2023.
- Liu, P.; Byungchan, K.; Friesner, R.A.; Berne, B.J. Replica exchange with solute tempering: A method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. USA 2005, 102, 13749–13754. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2023-4: Glide; Schrödinger, LLC.: New York, NY, USA, 2023.
- Schrödinger Release 2023-4: Desmond Molecular Dynamics System; Maestro-Desmond Interoperability Tools; Shaw DE Research/Schrödinger: New York, NY, USA, 2023.
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug. Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conc. | Inhibition, % ± SEM | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 11 | 12 | 13 | 14 | Cisplatin | |||||||||
| Cell Lines | µM | Mean | SEM | Mean | SEM | IC50 (µM) | Mean | SEM | Mean | SEM | Mean | SEM | IC50 (µM) |
| HeLa | 10 | 3.06 | 0.33 | 94.20 | 0.18 | 1.71 | 11.20 | 2.67 | 43.33 | 2.93 | 42.60 | 2.30 | 12.40 |
| 30 | 4.17 | 1.39 | 96.98 | 0.16 | 11.97 | 0.94 | 48.03 | 1.65 | 99.90 | 0.30 | |||
| SiHa | 10 | 4.97 | 1.99 | 86.91 | 0.78 | 0.97 | 9.82 | 3.08 | 45.61 | 2.12 | 88.60 | 0.50 | 7.80 |
| 30 | 16.42 | 2.14 | 93.43 | 0.72 | 28.67 | 0.85 | 75.29 | 1.92 | 90.20 | 1.80 | |||
| C33A | 10 | 39.84 | 2.78 | 69.99 | 1.57 | 2.99 | 28.03 | 1.16 | 52.76 | 1.13 | 83.80 | 0.80 | 3.70 |
| 30 | 43.27 | 2.32 | 96.46 | 0.21 | 41.84 | 1.28 | 54.21 | 2.20 | 94.00 | 0.60 | |||
| A2780 | 10 | 24.40 | 1.00 | 78.21 | 2.96 | 3.44 | 27.32 | 2.02 | 33.44 | 1.11 | 83.60 | 1.20 | 1.30 |
| 30 | 28.35 | 1.49 | 92.65 | 1.05 | 35.35 | 3.03 | 27.09 | 0.81 | 95.00 | 0.30 | |||
| MCF-7 | 10 | 7.93 | 3.54 | 88.68 | 1.24 | 1.80 | 1.40 | 3.69 | 55.78 | 2.57 | 66.90 | 1.80 | 5.80 |
| 30 | 6.12 | 3.05 | 89.95 | 0.93 | 46.56 | 4.02 | 58.14 | 1.89 | 96.80 | 0.40 | |||
| T-47D | 10 | 10.90 | 2.81 | 75.42 | 0.52 | 1.04 | 17.57 | 1.99 | 58.85 | 2.55 | 51.00 | 2.00 | 9.80 |
| 30 | 11.46 | 0.90 | 87.13 | 1.53 | 53.64 | 2.59 | 58.76 | 2.15 | 57.90 | 1.50 | |||
| MDA-MB-231 | 10 | 19.34 | 1.42 | 90.00 | 0.95 | 2.04 | 32.05 | 3.92 | 24.45 | 2.44 | 20.84 | 0.81 | 19.10 |
| 30 | 23.65 | 2.16 | 93.40 | 1.06 | 38.37 | 2.58 | 27.98 | 2.86 | 74.47 | 1.20 | |||
| MDA-MB-361 | 10 | 5.67 | 1.26 | 82.42 | 3.01 | 2.07 | 0.45 | 5.85 | 60.32 | 4.62 | 67.50 | 1.00 | 3.70 |
| 30 | 5.27 | 3.51 | 83.99 | 1.04 | 40.25 | 5.37 | 66.86 | 3.65 | 87.80 | 1.10 | |||
| MRC-5 | 10 | n.d. | n.d. | 85.19 | 1.23 | 1.24 | n.d. | n.d. | n.d. | n.d. | 72.30 | 2.30 | 4.51 |
| 30 | n.d. | n.d. | 89.55 | 0.53 | n.d. | n.d. | n.d. | n.d. | 70.70 | 1.30 | |||
| NIH/3T3 | 10 | n.d. | n.d. | 41.39 | 2.76 | 15.02 | n.d. | n.d. | n.d. | n.d. | 76.74 | 1.26 | 1.26 |
| 30 | n.d. | n.d. | 68.64 | 3.74 | n.d. | n.d. | n.d. | n.d. | 96.90 | 0.25 | |||
| Tumor Selectivity Index | ||||
|---|---|---|---|---|
| NIH/3T3 | MRC-5 | |||
| Cell Lines | comp 12 | cispl | comp 12 | cispl |
| HeLa | 8.78 | 0.38 | 0.73 | 0.36 |
| SiHa | 15.55 | 0.61 | 1.28 | 0.58 |
| C33A | 5.02 | 1.28 | 0.41 | 1.22 |
| A2780 | 4.37 | 3.64 | 0.36 | 3.47 |
| MCF-7 | 8.35 | 0.82 | 0.69 | 0.78 |
| T-47D | 14.47 | 0.48 | 1.20 | 0.46 |
| MDA-MB-231 | 7.38 | 0.25 | 0.61 | 0.24 |
| MDA-MB-361 | 7.26 | 1.28 | 0.60 | 1.22 |
| MRC-5 | -- | -- | 1.00 | 1.00 |
| NIH/3T3 | 1.00 | 1.00 | -- | -- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bózsity, N.; Nagy, V.; Szabó, J.; Pálházi, B.; Kele, Z.; Resch, V.; Paragi, G.; Zupkó, I.; Minorics, R.; Mernyák, E. Synthesis of Estrone Heterodimers and Evaluation of Their In Vitro Antiproliferative Activity. Int. J. Mol. Sci. 2024, 25, 4274. https://doi.org/10.3390/ijms25084274
Bózsity N, Nagy V, Szabó J, Pálházi B, Kele Z, Resch V, Paragi G, Zupkó I, Minorics R, Mernyák E. Synthesis of Estrone Heterodimers and Evaluation of Their In Vitro Antiproliferative Activity. International Journal of Molecular Sciences. 2024; 25(8):4274. https://doi.org/10.3390/ijms25084274
Chicago/Turabian StyleBózsity, Noémi, Viktória Nagy, Johanna Szabó, Balázs Pálházi, Zoltán Kele, Vivien Resch, Gábor Paragi, István Zupkó, Renáta Minorics, and Erzsébet Mernyák. 2024. "Synthesis of Estrone Heterodimers and Evaluation of Their In Vitro Antiproliferative Activity" International Journal of Molecular Sciences 25, no. 8: 4274. https://doi.org/10.3390/ijms25084274
APA StyleBózsity, N., Nagy, V., Szabó, J., Pálházi, B., Kele, Z., Resch, V., Paragi, G., Zupkó, I., Minorics, R., & Mernyák, E. (2024). Synthesis of Estrone Heterodimers and Evaluation of Their In Vitro Antiproliferative Activity. International Journal of Molecular Sciences, 25(8), 4274. https://doi.org/10.3390/ijms25084274