
A Stem-like Patient-Derived Ovarian Cancer Model of Platinum Resistance Reveals Dissociation of Stemness and Resistance
 and
and 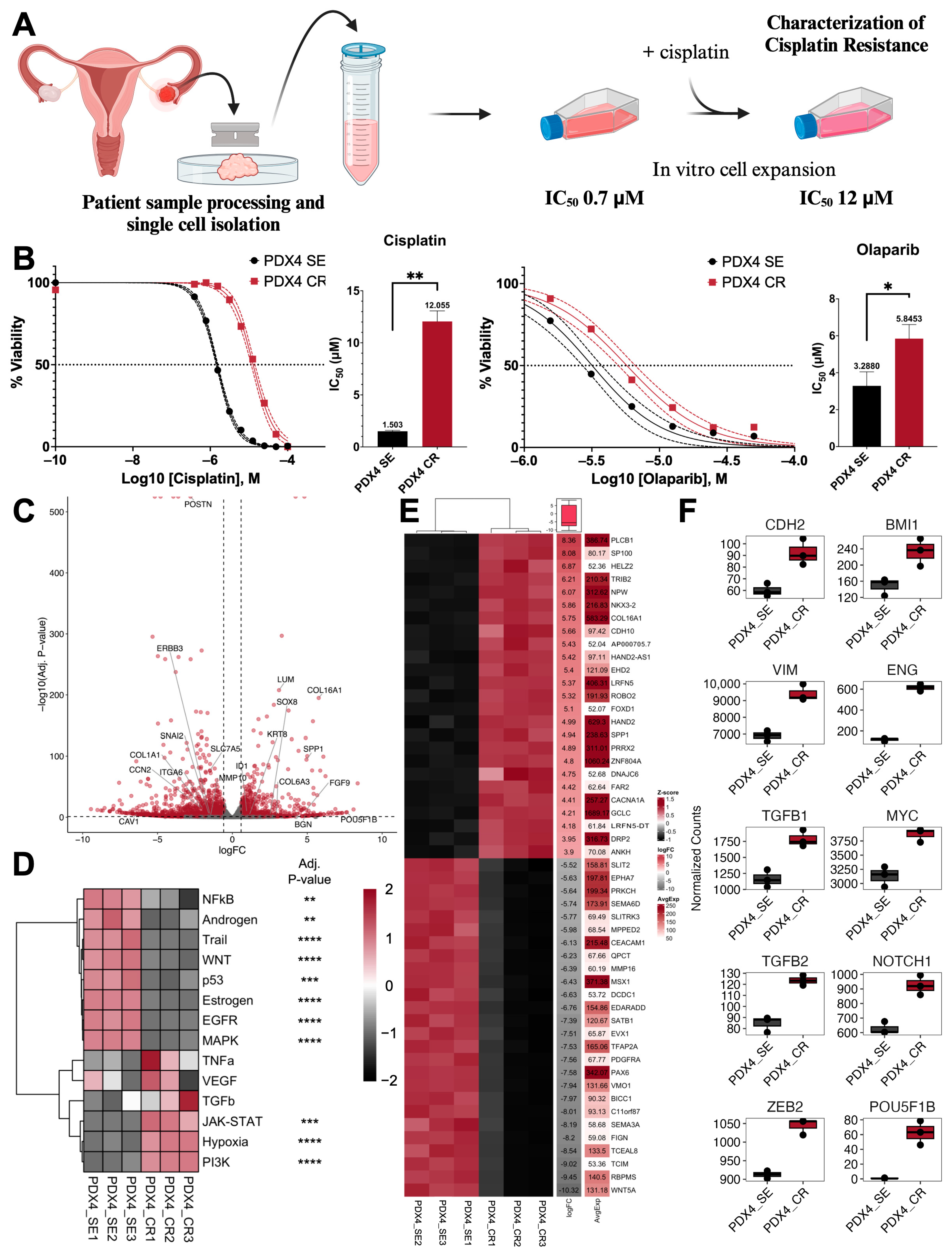{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
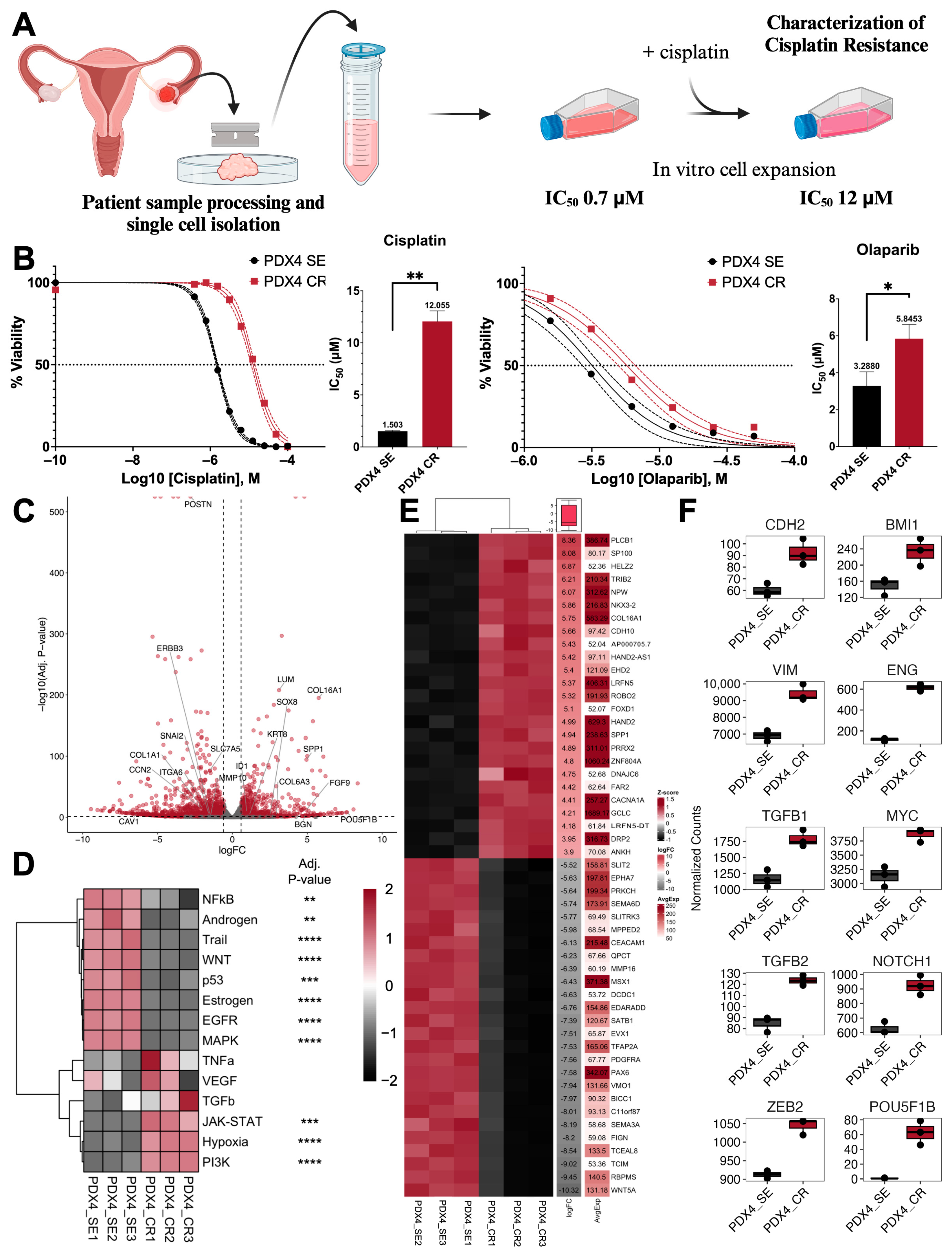
2.1. Generation of Cisplati-Resistant Cell Line
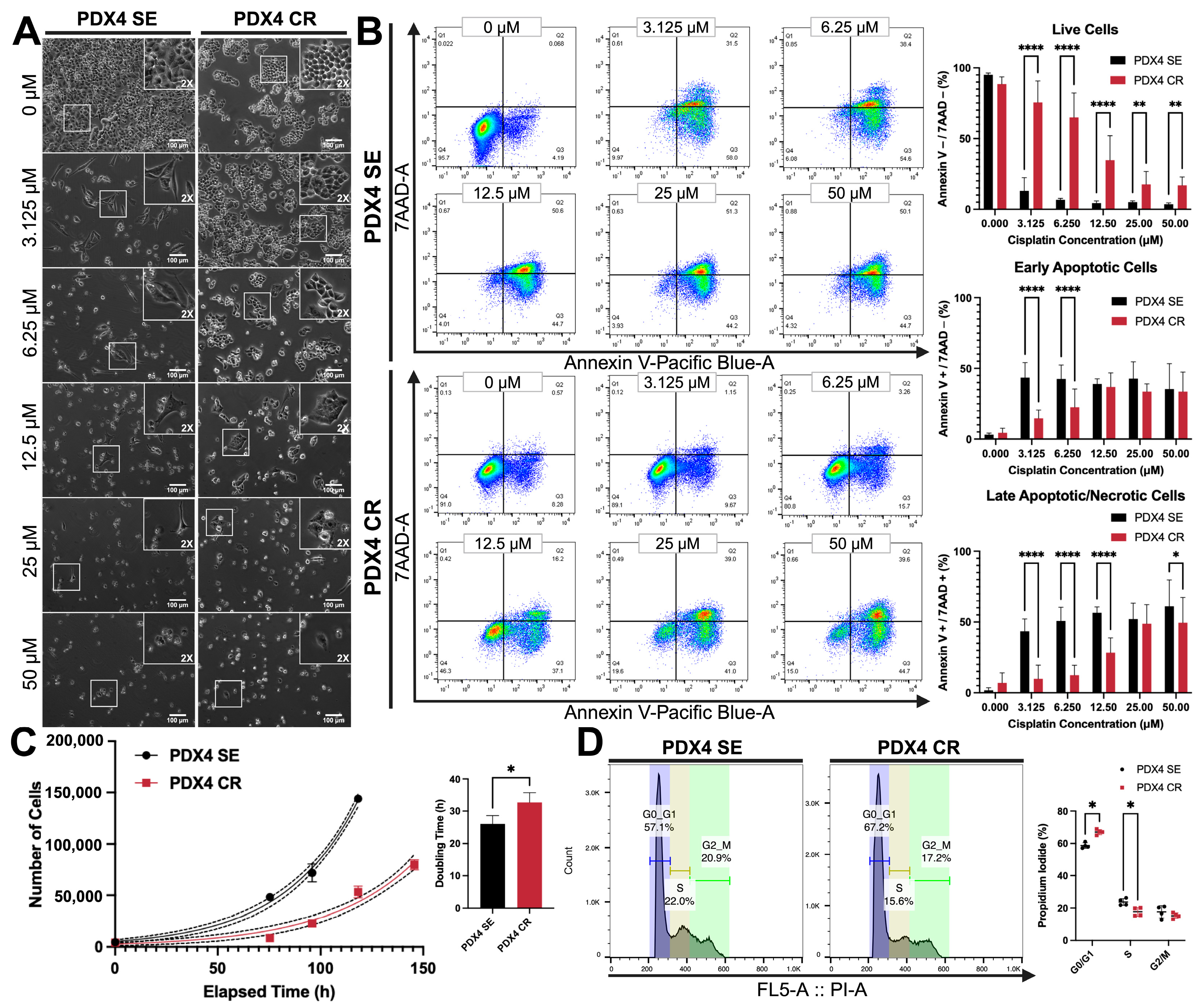
2.2. Growth and Proliferation Characteristics after the Acquisition of Cisplatin Resistance
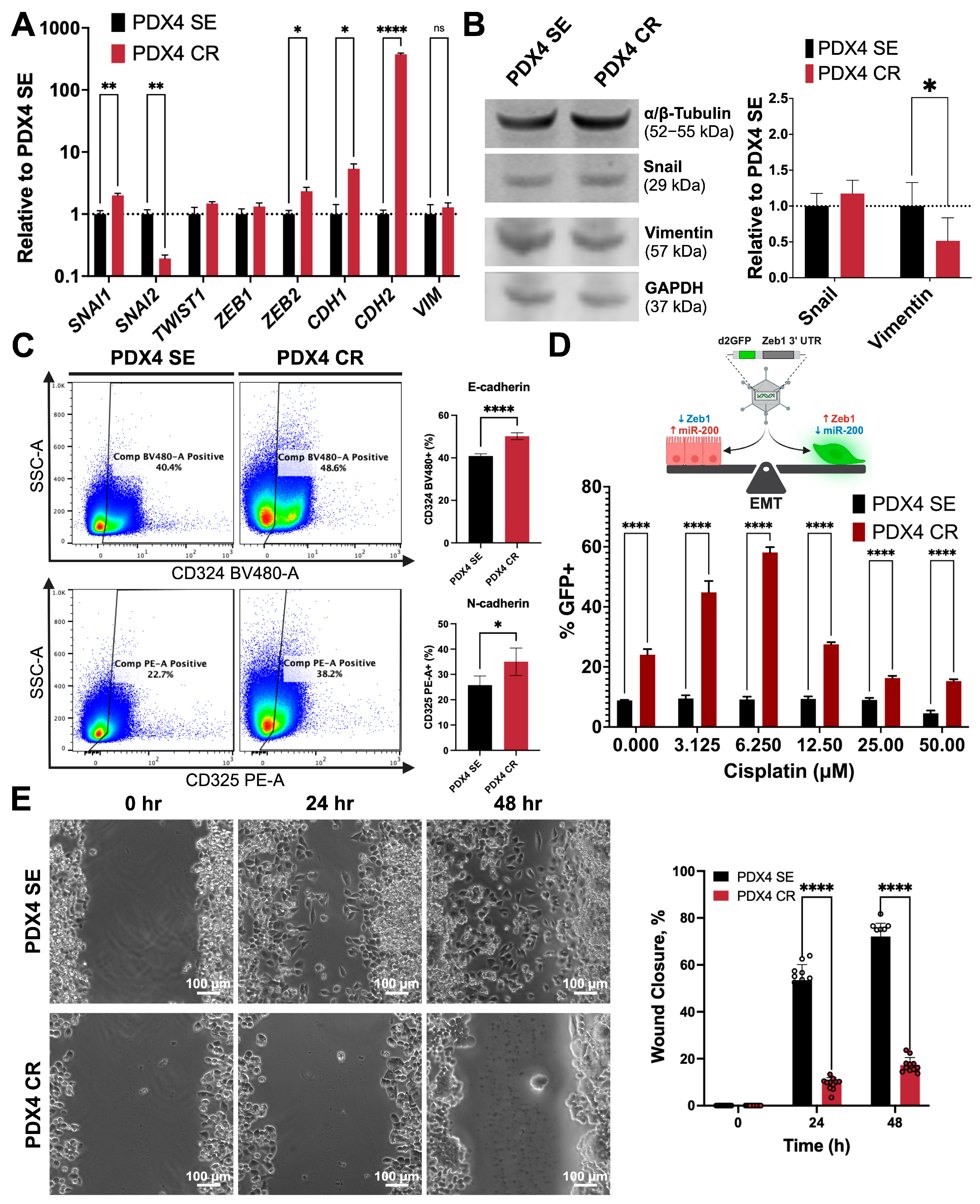
2.3. Defining Epithelial–Mesenchymal Transition Status
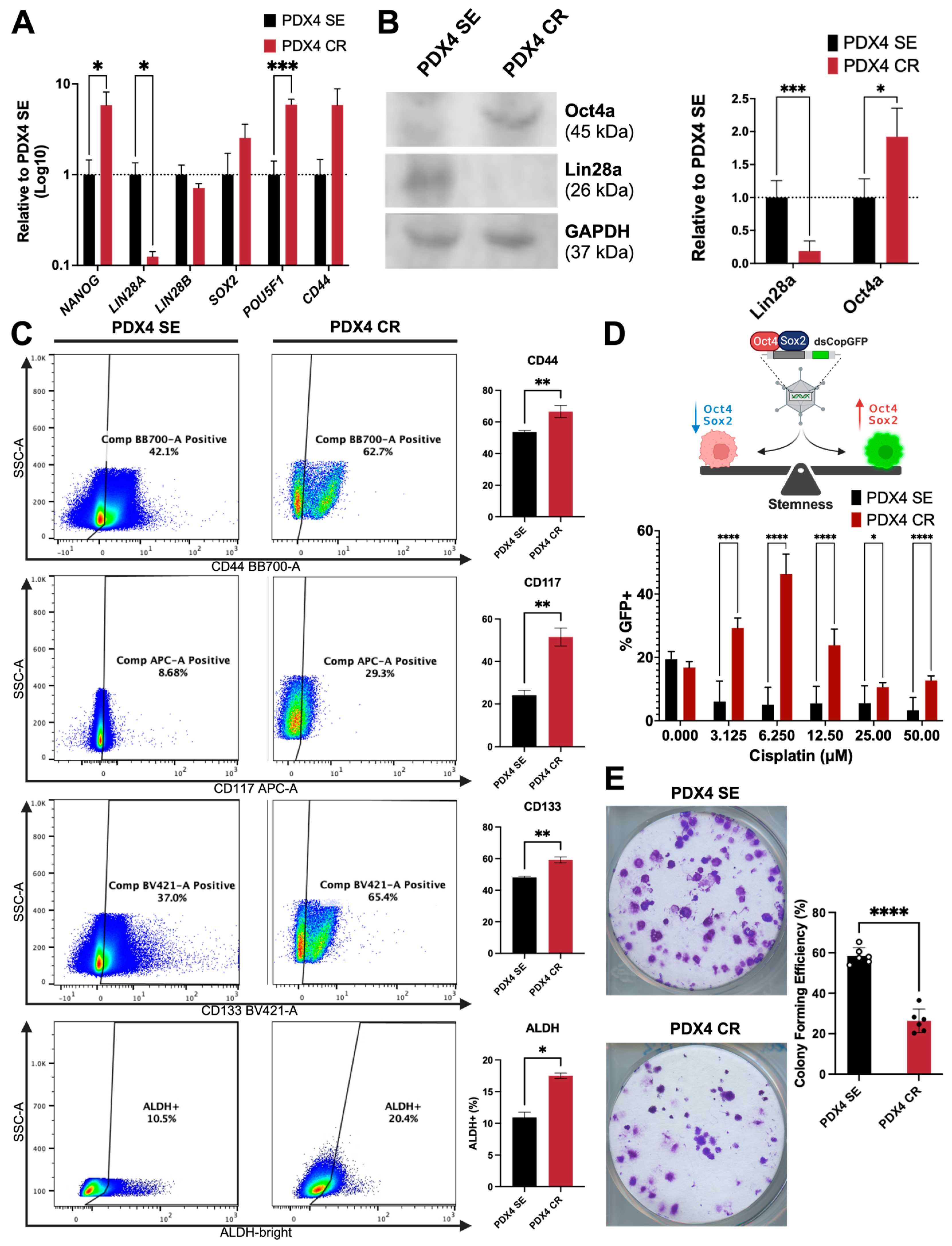
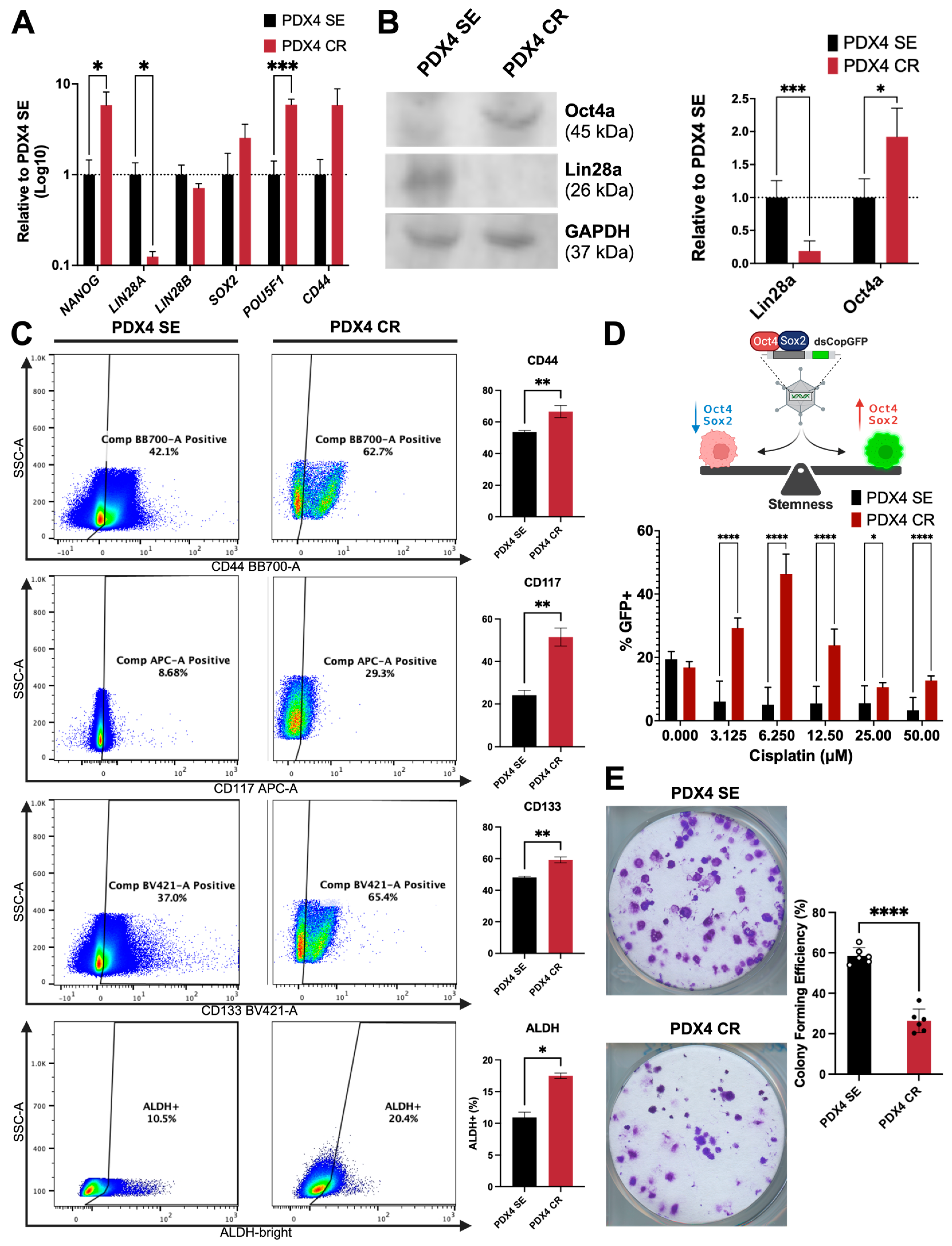
2.4. Stemness and Self-Renewal Characteristics
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Generation of Cisplatin Resistant Cell Line
4.3. Cell Viability Assays
4.4. Library Preparation and RNA Sequencing
4.5. Bioinformatics Analysis
4.6. Pathway Analysis
4.7. Microscope Imaging
4.8. Flow Cytometry
4.9. Proliferation Assay
4.10. Quantitative Reverse Transcription PCR (RT-qPCR)
4.11. Protein Extraction and Quantification
4.12. Electrophoresis and Immunoblotting
4.13. GFP Reporters
4.14. Scratch Migration Assay
4.15. Colony Formation Assay
4.16. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Veneziani, A.C.; Gonzalez-Ochoa, E.; Alqaisi, H.; Madariaga, A.; Bhat, G.; Rouzbahman, M.; Sneha, S.; Oza, A.M. Heterogeneity and treatment landscape of ovarian carcinoma. Nat. Rev. Clin. Oncol. 2023, 20, 820–842. [Google Scholar] [CrossRef] [PubMed]
- Hohn, A.K.; Brambs, C.E.; Hiller, G.G.R.; May, D.; Schmoeckel, E.; Horn, L.C. 2020 WHO Classification of Female Genital Tumors. Geburtshilfe Frauenheilkd 2021, 81, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Conant, A.; Curow, C.; Alexander, A.; Ioffe, Y.; Unternaehrer, J.J. Role of epithelial-mesenchymal transition factor SNAI1 and its targets in ovarian cancer aggressiveness. J. Cancer Metastasis Treat. 2023, 9, 25. [Google Scholar] [CrossRef]
- Tan, T.Z.; Miow, Q.H.; Huang, R.Y.; Wong, M.K.; Ye, J.; Lau, J.A.; Wu, M.C.; Bin Abdul Hadi, L.H.; Soong, R.; Choolani, M.; et al. Functional genomics identifies five distinct molecular subtypes with clinical relevance and pathways for growth control in epithelial ovarian cancer. EMBO Mol. Med. 2013, 5, 1051–1066. [Google Scholar] [CrossRef]
- Englisz, A.; Smycz-Kubanska, M.; Mielczarek-Palacz, A. Evaluation of the Potential Diagnostic Utility of the Determination of Selected Immunological and Molecular Parameters in Patients with Ovarian Cancer. Diagnostics 2023, 13, 1714. [Google Scholar] [CrossRef]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.K.; Alvarez, R.D.; Bakkum-Gamez, J.N.; Barroilhet, L.; Behbakht, K.; Berchuck, A.; Chen, L.M.; Cristea, M.; DeRosa, M.; Eisenhauer, E.L.; et al. Ovarian Cancer, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2021, 19, 191–226. [Google Scholar] [CrossRef]
- Kurnit, K.C.; Fleming, G.F.; Lengyel, E. Updates and New Options in Advanced Epithelial Ovarian Cancer Treatment. Obstet. Gynecol. 2021, 137, 108–121. [Google Scholar] [CrossRef]
- Yang, L.; Xie, H.J.; Li, Y.Y.; Wang, X.; Liu, X.X.; Mai, J. Molecular mechanisms of platinum-based chemotherapy resistance in ovarian cancer (Review). Oncol. Rep. 2022, 47, 82. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, G.S. Role of autophagy in cisplatin resistance in ovarian cancer cells. J. Biol. Chem. 2014, 289, 17163–17173. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Jaramillo, M.C.; Zhang, Z.; Zheng, Y.; Yao, M.; Zhang, D.D.; Yi, X. Induction of autophagy contributes to cisplatin resistance in human ovarian cancer cells. Mol. Med. Rep. 2015, 11, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Cui, M.; Liu, K. Therapeutic strategies to overcome cisplatin resistance in ovarian cancer. Eur. J. Med. Chem. 2022, 232, 114205. [Google Scholar] [CrossRef] [PubMed]
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Loret, N.; Denys, H.; Tummers, P.; Berx, G. The Role of Epithelial-to-Mesenchymal Plasticity in Ovarian Cancer Progression and Therapy Resistance. Cancers 2019, 11, 838. [Google Scholar] [CrossRef] [PubMed]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.; Weberpals, J.; et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012, 12, 91. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Ha, J.; Kang, E.; Cho, S. The role of epithelial-mesenchymal transition-regulating transcription factors in anti-cancer drug resistance. Arch. Pharm. Res. 2021, 44, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Hojo, N.; Huisken, A.L.; Wang, H.; Chirshev, E.; Kim, N.S.; Nguyen, S.M.; Campos, H.; Glackin, C.A.; Ioffe, Y.J.; Unternaehrer, J.J. Snail knockdown reverses stemness and inhibits tumour growth in ovarian cancer. Sci. Rep. 2018, 8, 8704. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef]
- Korch, C.; Spillman, M.A.; Jackson, T.A.; Jacobsen, B.M.; Murphy, S.K.; Lessey, B.A.; Jordan, V.C.; Bradford, A.P. DNA profiling analysis of endometrial and ovarian cell lines reveals misidentification, redundancy and contamination. Gynecol. Oncol. 2012, 127, 241–248. [Google Scholar] [CrossRef]
- Sonego, M.; Pellizzari, I.; Dall’Acqua, A.; Pivetta, E.; Lorenzon, I.; Benevol, S.; Bomben, R.; Spessotto, P.; Sorio, R.; Gattei, V.; et al. Common biological phenotypes characterize the acquisition of platinum-resistance in epithelial ovarian cancer cells. Sci. Rep. 2017, 7, 7104. [Google Scholar] [CrossRef]
- Parker, R.J.; Eastman, A.; Bostick-Bruton, F.; Reed, E. Acquired cisplatin resistance in human ovarian cancer cells is associated with enhanced repair of cisplatin-DNA lesions and reduced drug accumulation. J. Clin. Investig. 1991, 87, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.D.; Pan, L.Y.; Yuan, Y.; Lang, J.H.; Mao, N. Identification of platinum-resistance associated proteins through proteomic analysis of human ovarian cancer cells and their platinum-resistant sublines. J. Proteome Res. 2007, 6, 772–780. [Google Scholar] [CrossRef]
- Han, L.Y.; Landen, C.N.; Trevino, J.G.; Halder, J.; Lin, Y.G.; Kamat, A.A.; Kim, T.J.; Merritt, W.M.; Coleman, R.L.; Gershenson, D.M.; et al. Antiangiogenic and antitumor effects of SRC inhibition in ovarian carcinoma. Cancer Res. 2006, 66, 8633–8639. [Google Scholar] [CrossRef]
- Perego, P.; Giarola, M.; Righetti, S.C.; Supino, R.; Caserini, C.; Delia, D.; Pierotti, M.A.; Miyashita, T.; Reed, J.C.; Zunino, F. Association between cisplatin resistance and mutation of p53 gene and reduced bax expression in ovarian carcinoma cell systems. Cancer Res. 1996, 56, 556–562. [Google Scholar] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Papp, E.; Hallberg, D.; Konecny, G.E.; Bruhm, D.C.; Adleff, V.; Noe, M.; Kagiampakis, I.; Palsgrove, D.; Conklin, D.; Kinose, Y.; et al. Integrated Genomic, Epigenomic, and Expression Analyses of Ovarian Cancer Cell Lines. Cell Rep. 2018, 25, 2617–2633. [Google Scholar] [CrossRef]
- McCabe, A.; Zaheed, O.; McDade, S.S.; Dean, K. Investigating the suitability of in vitro cell lines as models for the major subtypes of epithelial ovarian cancer. Front. Cell Dev. Biol. 2023, 11, 1104514. [Google Scholar] [CrossRef]
- Barnes, B.M.; Nelson, L.; Tighe, A.; Burghel, G.J.; Lin, I.H.; Desai, S.; McGrail, J.C.; Morgan, R.D.; Taylor, S.S. Distinct transcriptional programs stratify ovarian cancer cell lines into the five major histological subtypes. Genome Med. 2021, 13, 140. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Jacquemont, C.; Chandramohan, K.V.; Couch, F.J.; Langdon, S.P.; Wurz, K.; Higgins, J.; Villegas, E.; Taniguchi, T. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009, 69, 6381–6386. [Google Scholar] [CrossRef]
- Chirshev, E.; Hojo, N.; Bertucci, A.; Sanderman, L.; Nguyen, A.; Wang, H.; Suzuki, T.; Brito, E.; Martinez, S.R.; Castanon, C.; et al. Epithelial/mesenchymal heterogeneity of high-grade serous ovarian carcinoma samples correlates with miRNA let-7 levels and predicts tumor growth and metastasis. Mol. Oncol. 2020, 14, 2796–2813. [Google Scholar] [CrossRef] [PubMed]
- Chirshev, E.; Suzuki, T.; Wang, H.; Nguyen, A.; Hojo, N.; Sanderman, L.; Mirshahidi, S.; Ioffe, Y.J.; Unternaehrer, J.J. Let-7i Reduces Aggressive Phenotype and Induces BRCAness in Ovarian Cancer Cells. Cancers 2021, 13, 4617. [Google Scholar] [CrossRef]
- Eoh, K.J.; Kim, H.M.; Lee, J.Y.; Kim, S.; Kim, S.W.; Kim, Y.T.; Nam, E.J. Mutation landscape of germline and somatic BRCA1/2 in patients with high-grade serous ovarian cancer. BMC Cancer 2020, 20, 204. [Google Scholar] [CrossRef]
- Ansaloni, L.; Coccolini, F.; Morosi, L.; Ballerini, A.; Ceresoli, M.; Grosso, G.; Bertoli, P.; Busci, L.M.; Lotti, M.; Cambria, F.; et al. Pharmacokinetics of concomitant cisplatin and paclitaxel administered by hyperthermic intraperitoneal chemotherapy to patients with peritoneal carcinomatosis from epithelial ovarian cancer. Br. J. Cancer 2015, 112, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Himmelstein, K.J.; Patton, T.F.; Belt, R.J.; Taylor, S.; Repta, A.J.; Sternson, L.A. Clinical kinetics on intact cisplatin and some related species. Clin. Pharmacol. Ther. 1981, 29, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Panteix, G.; Beaujard, A.; Garbit, F.; Chaduiron-Faye, C.; Guillaumont, M.; Gilly, F.; Baltassat, P.; Bressolle, F. Population pharmacokinetics of cisplatin in patients with advanced ovarian cancer during intraperitoneal hyperthermia chemotherapy. Anticancer Res. 2002, 22, 1329–1336. [Google Scholar] [PubMed]
- Huang, D.; Savage, S.R.; Calinawan, A.P.; Lin, C.; Zhang, B.; Wang, P.; Starr, T.K.; Birrer, M.J.; Paulovich, A.G. A highly annotated database of genes associated with platinum resistance in cancer. Oncogene 2021, 40, 6395–6405. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Deshmukh, A.P.; den Hollander, P.; Addanki, S.; Kuburich, N.A.; Kudaravalli, S.; Joseph, R.; Chang, J.T.; Soundararajan, R.; Mani, S.A. EMTome: A resource for pan-cancer analysis of epithelial-mesenchymal transition genes and signatures. Br. J. Cancer 2021, 124, 259–269. [Google Scholar] [CrossRef]
- Firdous, S.; Ghosh, A.; Saha, S. BCSCdb: A database of biomarkers of cancer stem cells. Database 2022, 2022, baac082. [Google Scholar] [CrossRef] [PubMed]
- Chebouti, I.; Kasimir-Bauer, S.; Buderath, P.; Wimberger, P.; Hauch, S.; Kimmig, R.; Kuhlmann, J.D. EMT-like circulating tumor cells in ovarian cancer patients are enriched by platinum-based chemotherapy. Oncotarget 2017, 8, 48820–48831. [Google Scholar] [CrossRef] [PubMed]
- Marchini, S.; Fruscio, R.; Clivio, L.; Beltrame, L.; Porcu, L.; Fuso Nerini, I.; Cavalieri, D.; Chiorino, G.; Cattoretti, G.; Mangioni, C.; et al. Resistance to platinum-based chemotherapy is associated with epithelial to mesenchymal transition in epithelial ovarian cancer. Eur. J. Cancer 2013, 49, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Chung, V.Y.; Thiery, J.P. Targeting pathways contributing to epithelial-mesenchymal transition (EMT) in epithelial ovarian cancer. Curr. Drug Targets 2012, 13, 1649–1653. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, M.; Ise, N.; Omi, K.; Goishi, K.; Higashiyama, S. Cisplatin influences acquisition of resistance to molecular-targeted agents through epithelial-mesenchymal transition-like changes. Cancer Sci. 2013, 104, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Latifi, A.; Abubaker, K.; Castrechini, N.; Ward, A.C.; Liongue, C.; Dobill, F.; Kumar, J.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J. Cell Biochem. 2011, 112, 2850–2864. [Google Scholar] [CrossRef] [PubMed]
- Toneff, M.J.; Sreekumar, A.; Tinnirello, A.; Hollander, P.D.; Habib, S.; Li, S.; Ellis, M.J.; Xin, L.; Mani, S.A.; Rosen, J.M. The Z-cad dual fluorescent sensor detects dynamic changes between the epithelial and mesenchymal cellular states. BMC Biol. 2016, 14, 47. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.M.; Weinberg, R.A.; Lees, J.A.; Guen, V.J. Emerging Mechanisms by which EMT Programs Control Stemness. Trends Cancer 2020, 6, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.A.; Bai, S.; McLean, K.; Yang, K.; Griffith, K.; Thomas, D.; Ginestier, C.; Johnston, C.; Kueck, A.; Reynolds, R.K.; et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 2011, 71, 3991–4001. [Google Scholar] [CrossRef]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef]
- Tang, B.; Raviv, A.; Esposito, D.; Flanders, K.C.; Daniel, C.; Nghiem, B.T.; Garfield, S.; Lim, L.; Mannan, P.; Robles, A.I.; et al. A flexible reporter system for direct observation and isolation of cancer stem cells. Stem Cell Rep. 2015, 4, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Shonibare, Z.; Monavarian, M.; Arend, R.C.; Lee, N.Y.; Inman, G.J.; Mythreye, K. TGFbeta signaling networks in ovarian cancer progression and plasticity. Clin. Exp. Metastasis 2021, 38, 139–161. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, H.S.; Zhou, B.H.; Li, C.L.; Zhang, F.; Wang, X.F.; Zhang, G.; Bu, X.Z.; Cai, S.H.; Du, J. Epithelial-mesenchymal transition (EMT) induced by TNF-alpha requires AKT/GSK-3beta-mediated stabilization of snail in colorectal cancer. PLoS ONE 2013, 8, e56664. [Google Scholar] [CrossRef]
- Joseph, J.V.; Conroy, S.; Pavlov, K.; Sontakke, P.; Tomar, T.; Eggens-Meijer, E.; Balasubramaniyan, V.; Wagemakers, M.; den Dunnen, W.F.; Kruyt, F.A. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1alpha-ZEB1 axis. Cancer Lett. 2015, 359, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Zhang, X.; Paladino, D.; Sengupta, B.; Ahmad, S.; Holloway, R.W.; Ingersoll, S.B.; Turkson, J. Hyperactive EGF receptor, Jaks and Stat3 signaling promote enhanced colony-forming ability, motility and migration of cisplatin-resistant ovarian cancer cells. Oncogene 2012, 31, 2309–2322. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lin, G.; Chen, K.; Liang, R.; Wan, F.; Zhang, C.; Tian, G.; Zhu, X. VEGF promotes migration and invasion by regulating EMT and MMPs in nasopharyngeal carcinoma. J. Cancer 2020, 11, 7291–7301. [Google Scholar] [CrossRef] [PubMed]
- Asad, M.; Wong, M.K.; Tan, T.Z.; Choolani, M.; Low, J.; Mori, S.; Virshup, D.; Thiery, J.P.; Huang, R.Y. FZD7 drives in vitro aggressiveness in Stem-A subtype of ovarian cancer via regulation of non-canonical Wnt/PCP pathway. Cell Death Dis. 2014, 5, e1346. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, C.; Sun, Y.; Guo, J.; Liu, S.; Cheng, Z. Identification of an Autophagy-Related Signature for Prognosis and Immunotherapy Response Prediction in Ovarian Cancer. Biomolecules 2023, 13, 339. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Qin, J.; Zhang, Y.; Cheng, X.; Wang, X.; Lu, W.; Xie, X.; Zhang, S. Autophagy maintains the stemness of ovarian cancer stem cells by FOXA2. J. Exp. Clin. Cancer Res. 2017, 36, 171. [Google Scholar] [CrossRef]
- Liu, X.; Han, E.K.; Anderson, M.; Shi, Y.; Semizarov, D.; Wang, G.; McGonigal, T.; Roberts, L.; Lasko, L.; Palma, J.; et al. Acquired resistance to combination treatment with temozolomide and ABT-888 is mediated by both base excision repair and homologous recombination DNA repair pathways. Mol. Cancer Res. 2009, 7, 1686–1692. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Rehman, F.L.; Bajrami, I.; Brough, R.; Wallberg, F.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Chen, L.; Campbell, J.; et al. A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of olaparib toxicity. PLoS ONE 2013, 8, e61520. [Google Scholar] [CrossRef] [PubMed]
- McPhee, T.R.; McDonald, P.C.; Oloumi, A.; Dedhar, S. Integrin-linked kinase regulates E-cadherin expression through PARP-1. Dev. Dyn. 2008, 237, 2737–2747. [Google Scholar] [CrossRef]
- Mariano, G.; Ricciardi, M.R.; Trisciuoglio, D.; Zampieri, M.; Ciccarone, F.; Guastafierro, T.; Calabrese, R.; Valentini, E.; Tafuri, A.; Del Bufalo, D.; et al. PARP inhibitor ABT-888 affects response of MDA-MB-231 cells to doxorubicin treatment, targeting Snail expression. Oncotarget 2015, 6, 15008–15021. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.I.; Gonzalez-Flores, A.; Dantzer, F.; Collard, J.; de Herreros, A.G.; Oliver, F.J. Poly(ADP-ribose)-dependent regulation of Snail1 protein stability. Oncogene 2011, 30, 4365–4372. [Google Scholar] [CrossRef] [PubMed]
- Doege, C.A.; Inoue, K.; Yamashita, T.; Rhee, D.B.; Travis, S.; Fujita, R.; Guarnieri, P.; Bhagat, G.; Vanti, W.B.; Shih, A.; et al. Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature 2012, 488, 652–655. [Google Scholar] [CrossRef]
- Lai, Y.S.; Chang, C.W.; Pawlik, K.M.; Zhou, D.; Renfrow, M.B.; Townes, T.M. SRY (sex determining region Y)-box2 (Sox2)/poly ADP-ribose polymerase 1 (Parp1) complexes regulate pluripotency. Proc. Natl. Acad. Sci. USA 2012, 109, 3772–3777. [Google Scholar] [CrossRef]
- Long, W.; Zhao, W.; Ning, B.; Huang, J.; Chu, J.; Li, L.; Ma, Q.; Xing, C.; Wang, H.Y.; Liu, Q.; et al. PHF20 collaborates with PARP1 to promote stemness and aggressiveness of neuroblastoma cells through activation of SOX2 and OCT4. J. Mol. Cell Biol. 2018, 10, 147–160. [Google Scholar] [CrossRef]
- Jiang, B.H.; Tseng, W.L.; Li, H.Y.; Wang, M.L.; Chang, Y.L.; Sung, Y.J.; Chiou, S.H. Poly(ADP-Ribose) Polymerase 1: Cellular Pluripotency, Reprogramming, and Tumorogenesis. Int. J. Mol. Sci. 2015, 16, 15531–15545. [Google Scholar] [CrossRef]
- Fu, G.; Wu, Y.; Zhao, G.; Chen, X.; Xu, Z.; Sun, J.; Tian, J.; Cheng, Z.; Shi, Y.; Jin, B. Activation of cGAS-STING Signal to Inhibit the Proliferation of Bladder Cancer: The Immune Effect of Cisplatin. Cells 2022, 11, 3011. [Google Scholar] [CrossRef]
- Lan, Q.; Peyvandi, S.; Duffey, N.; Huang, Y.T.; Barras, D.; Held, W.; Richard, F.; Delorenzi, M.; Sotiriou, C.; Desmedt, C.; et al. Type I interferon/IRF7 axis instigates chemotherapy-induced immunological dormancy in breast cancer. Oncogene 2019, 38, 2814–2829. [Google Scholar] [CrossRef]
- Musella, M.; Galassi, C.; Manduca, N.; Sistigu, A. The Yin and Yang of Type I IFNs in Cancer Promotion and Immune Activation. Biology 2021, 10, 856. [Google Scholar] [CrossRef]
- Liu, F.R.; Jiang, M.J.; Mei, Z.; Lin, C.J.; Tian, L. cGAS-STING signalings potentiate tumor progression via sustaining cancer stemness. Transl. Oncol. 2022, 20, 101404. [Google Scholar] [CrossRef]
- Grabosch, S.; Bulatovic, M.; Zeng, F.; Ma, T.; Zhang, L.; Ross, M.; Brozick, J.; Fang, Y.; Tseng, G.; Kim, E.; et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene 2019, 38, 2380–2393. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 24 March 2023).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Schneider, V.A.; Graves-Lindsay, T.; Howe, K.; Bouk, N.; Chen, H.C.; Kitts, P.A.; Murphy, T.D.; Pruitt, K.D.; Thibaud-Nissen, F.; Albracht, D.; et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017, 27, 849–864. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Smedley, D.; Haider, S.; Ballester, B.; Holland, R.; London, D.; Thorisson, G.; Kasprzyk, A. BioMart--biological queries made easy. BMC Genomics 2009, 10, 22. [Google Scholar] [CrossRef]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, T.; Conant, A.; Jung, Y.; Bax, R.; Antonissen, A.; Chen, W.; Yu, G.; Ioffe, Y.J.; Wang, C.; Unternaehrer, J.J. A Stem-like Patient-Derived Ovarian Cancer Model of Platinum Resistance Reveals Dissociation of Stemness and Resistance. Int. J. Mol. Sci. 2024, 25, 3843. https://doi.org/10.3390/ijms25073843
Suzuki T, Conant A, Jung Y, Bax R, Antonissen A, Chen W, Yu G, Ioffe YJ, Wang C, Unternaehrer JJ. A Stem-like Patient-Derived Ovarian Cancer Model of Platinum Resistance Reveals Dissociation of Stemness and Resistance. International Journal of Molecular Sciences. 2024; 25(7):3843. https://doi.org/10.3390/ijms25073843
Chicago/Turabian StyleSuzuki, Tise, Ashlyn Conant, Yeonkyu Jung, Ryan Bax, Ashley Antonissen, Wanqiu Chen, Gary Yu, Yevgeniya J. Ioffe, Charles Wang, and Juli J. Unternaehrer. 2024. "A Stem-like Patient-Derived Ovarian Cancer Model of Platinum Resistance Reveals Dissociation of Stemness and Resistance" International Journal of Molecular Sciences 25, no. 7: 3843. https://doi.org/10.3390/ijms25073843
APA StyleSuzuki, T., Conant, A., Jung, Y., Bax, R., Antonissen, A., Chen, W., Yu, G., Ioffe, Y. J., Wang, C., & Unternaehrer, J. J. (2024). A Stem-like Patient-Derived Ovarian Cancer Model of Platinum Resistance Reveals Dissociation of Stemness and Resistance. International Journal of Molecular Sciences, 25(7), 3843. https://doi.org/10.3390/ijms25073843