
Treatment of Double-Refractory Chronic Lymphocytic Leukemia—An Unmet Clinical Need


Abstract
1. Introduction
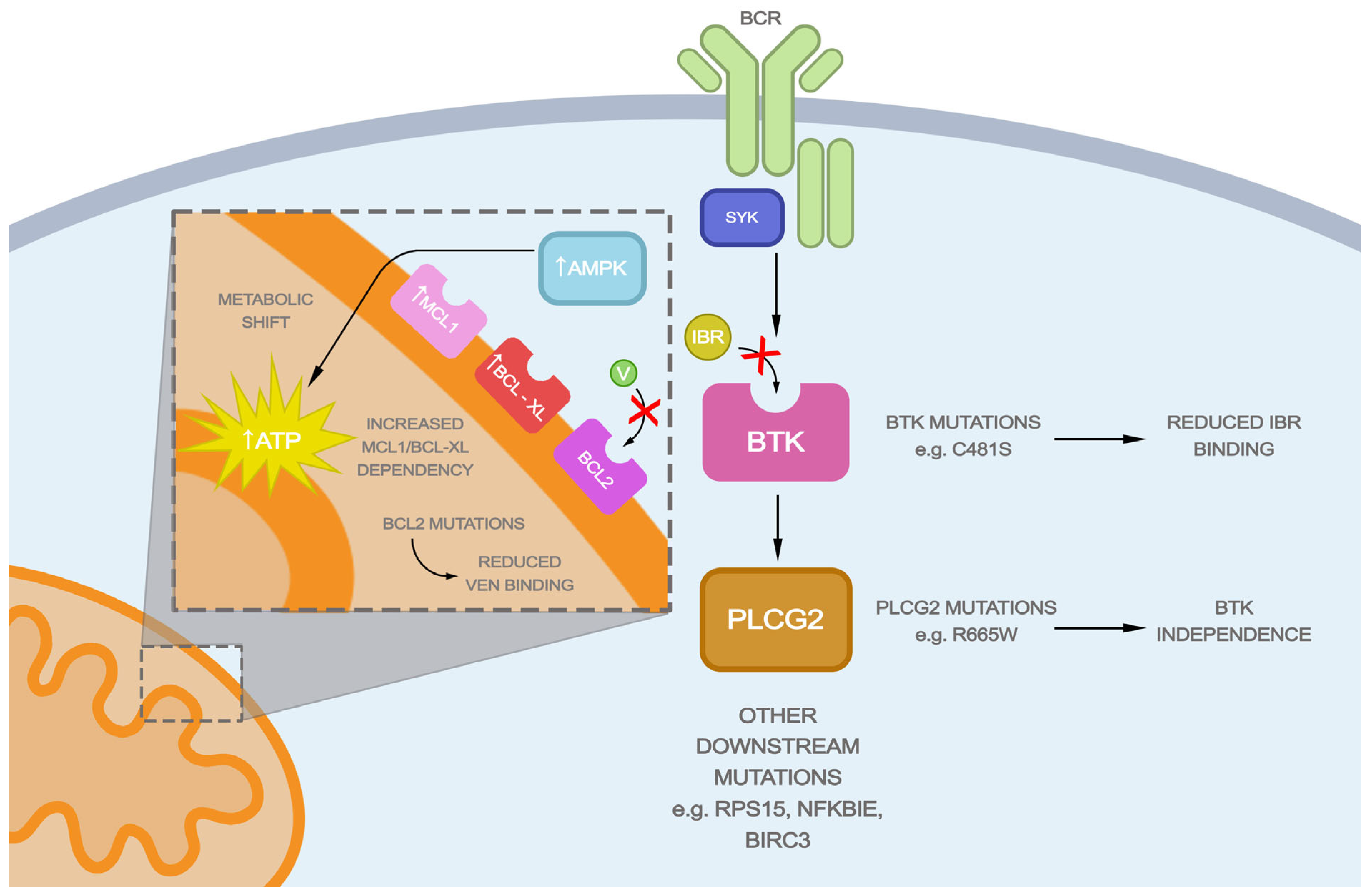
2. Mechanism of Action of BTK Inhibitors and CLL Resistance to Ibrutinib
3. Mechanism of Action of BCL2 Inhibitors and CLL Resistance to Venetoclax
4. BTK Inhibitor and BCL2 Inhibitor Combination
5. Regimens after Ibrutinib and Venetoclax Failure
5.1. Non-covalent BTK Inhibitors
5.2. Other BCL2 Inhibitors and MCL-1 Inhibitors
5.3. Phosphoinositide 3-Kinase Inhibitors (PI3Kis)
5.4. BTK Degraders
5.5. Bispecific T-Cell Engagers
5.6. Chimeric Antigen Receptor-Positive T (CAR-T) and NK (CAR-NK) Cell Therapy
5.7. Allogenic Hematopoietic Stem Cell Transplantation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Kreuzer, K.A.; Soosapilla, A.; Spacek, M.; Stehlikova, O.; Gambell, P.; McIver-Brown, N.; Villamor, N.; Psarra, K.; Arroz, M.; et al. Reproducible diagnosis of chronic lymphocytic leukemia by flow cytometry: An European Research Initiative on CLL (ERIC) & European Society for Clinical Cell Analysis (ESCCA) Harmonisation project. Cytom. Part B Clin. Cytom. 2018, 94, 121–128. [Google Scholar] [CrossRef]
- Miranda-Filho, A.; Piñeros, M.; Ferlay, J.; Soerjomataram, I.; Monnereau, A.; Bray, F. Epidemiological patterns of leukaemia in 184 countries: A population-based study. Lancet Haematol. 2018, 5, e14–e24. [Google Scholar] [CrossRef]
- National Cancer Institute. Bethesda, MD. SEER Cancer Stat Facts: Chronic Lymphocytic Leukemia. Available online: https://seer.cancer.gov/statfacts/html/clyl.html (accessed on 16 January 2024).
- Alrawashdh, N.; Sweasy, J.; Erstad, B.; McBride, A.; Persky, D.O.; Abraham, I. Survival trends in chronic lymphocytic leukemia across treatment eras: US SEER database analysis (1985–2017). Ann. Hematol. 2021, 100, 2501–2512. [Google Scholar] [CrossRef] [PubMed]
- Samples, L.; Ujjani, C.S.; Khajaviyan, S.; Lynch, R.C.; Shakib-Azar, M.; Crider, C.; Fredericks, M.; Louie, S.; Poh, C.; Smith, S.D.; et al. Clinical Outcomes in Patients Treated with Both Covalent Btkis and Venetoclax and the Significance of “Double-Refractory” Status in Patients with CLL/SLL. In Proceedings of the 65th ASH Annual Meeting & Exposition, San Diego, CA, USA, 9–12 December 2023. [Google Scholar]
- Mohamed, A.J.; Yu, L.; Bäckesjö, C.M.; Vargas, L.; Faryal, R.; Aints, A.; Christensson, B.; Berglöf, A.; Vihinen, M.; Nore, B.F.; et al. Bruton’s tyrosine kinase (Btk): Function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 2009, 228, 58–73. [Google Scholar] [CrossRef]
- Wen, T.; Wang, J.; Shi, Y.; Qian, H.; Liu, P. Inhibitors targeting Bruton’s tyrosine kinase in cancers: Drug development advances. Leukemia 2021, 35, 312–332. [Google Scholar] [CrossRef]
- Kil, L.P.; de Bruijn, M.J.; van Hulst, J.A.; Langerak, A.W.; Yuvaraj, S.; Hendriks, R.W. Bruton’s tyrosine kinase mediated signaling enhances leukemogenesis in a mouse model for chronic lymphocytic leukemia. Am. J. Blood Res. 2013, 3, 71–83. [Google Scholar]
- Woyach, J.A.; Bojnik, E.; Ruppert, A.S.; Stefanovski, M.R.; Goettl, V.M.; Smucker, K.A.; Smith, L.L.; Dubovsky, J.A.; Towns, W.H.; MacMurray, J.; et al. Bruton’s tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood 2014, 123, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.E.; Gordon, A.L.; Hertlein, E.; Ramanunni, A.; Zhang, X.; Jaglowski, S.; Flynn, J.; Jones, J.; Blum, K.A.; Buggy, J.J.; et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood 2011, 117, 6287–6296. [Google Scholar] [CrossRef]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef]
- de Gorter, D.J.; Beuling, E.A.; Kersseboom, R.; Middendorp, S.; van Gils, J.M.; Hendriks, R.W.; Pals, S.T.; Spaargaren, M. Bruton’s tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity 2007, 26, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Ponader, S.; Chen, S.S.; Buggy, J.J.; Balakrishnan, K.; Gandhi, V.; Wierda, W.G.; Keating, M.J.; O’Brien, S.; Chiorazzi, N.; Burger, J.A. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood 2012, 119, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Robak, T.; Witkowska, M.; Smolewski, P. The Role of Bruton’s Kinase Inhibitors in Chronic Lymphocytic Leukemia: Current Status and Future Directions. Cancers 2022, 14, 771. [Google Scholar] [CrossRef] [PubMed]
- Lovell, A.R.; Jammal, N.; Bose, P. Selecting the optimal BTK inhibitor therapy in CLL: Rationale and practical considerations. Ther. Adv. Hematol. 2022, 13, 20406207221116577. [Google Scholar] [CrossRef] [PubMed]
- Majeranowski, A.; Lebiedziński, F.; Okrój, M.; Osowski, J.; Mital, A. Zanubrutinib: A novel therapeutic option for the treatment of B-cell neoplasms. Acta Haematol. Pol. 2023, 54, 53–64. [Google Scholar] [CrossRef]
- Xu, W.; Zhou, K.; Wang, T.; Yang, S.; Liu, L.; Hu, Y.; Zhang, W.; Ding, K.; Zhou, J.; Gao, S.; et al. Orelabrutinib in relapsed or refractory chronic lymphocytic leukemia/small lymphocytic lymphoma patients: Multi-center, single-arm, open-label, phase 2 study. Am. J. Hematol. 2023, 98, 571–579. [Google Scholar] [CrossRef]
- Brown, J.R.; Harb, W.A.; Hill, B.T.; Gabrilove, J.; Sharman, J.P.; Schreeder, M.T.; Barr, P.M.; Foran, J.M.; Miller, T.P.; Burger, J.A.; et al. Phase I study of single-agent CC-292, a highly selective Bruton’s tyrosine kinase inhibitor, in relapsed/refractory chronic lymphocytic leukemia. Haematologica 2016, 101, e295–e298. [Google Scholar] [CrossRef] [PubMed]
- Walter, H.S.; Rule, S.A.; Dyer, M.J.; Karlin, L.; Jones, C.; Cazin, B.; Quittet, P.; Shah, N.; Hutchinson, C.V.; Honda, H.; et al. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory mature B-cell malignancies. Blood 2016, 127, 411–419. [Google Scholar] [CrossRef]
- Szmit, S.; Hus, I.; Giannopoulos, K.; Jamroziak, K.; Robak, T. Recommendations on cardiac safety during ibrutinib therapy. Acta. Haematol. Pol. 2023, 54, 3–5. [Google Scholar]
- Estupiñán, H.Y.; Berglöf, A.; Zain, R.; Smith, C.I.E. Comparative Analysis of BTK Inhibitors and Mechanisms Underlying Adverse Effects. Front. Cell Dev. Biol. 2021, 9, 630942. [Google Scholar] [CrossRef]
- Robak, P.; Witkowska, M.; Wolska-Washer, A.; Robak, T. The preclinical discovery and development of orelabrutinib as a novel treatment option for B-cell lymphoid malignancies. Expert Opin. Drug Discov. 2023, 18, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Mato, A.R.; Shah, N.N.; Jurczak, W.; Cheah, C.Y.; Pagel, J.M.; Woyach, J.A.; Fakhri, B.; Eyre, T.A.; Lamanna, N.; Patel, M.R.; et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): A phase 1/2 study. Lancet 2021, 397, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Naeem, A.; Utro, F.; Wang, Q.; Cha, J.; Vihinen, M.; Martindale, S.; Zhou, Y.; Ren, Y.; Tyekucheva, S.; Kim, A.S.; et al. Pirtobrutinib targets BTK C481S in ibrutinib-resistant CLL but second-site BTK mutations lead to resistance. Blood Adv. 2023, 7, 1929–1943. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Mi, X.; Thompson, M.C.; Montoya, S.; Notti, R.Q.; Afaghani, J.; Durham, B.H.; Penson, A.; Witkowski, M.T.; Lu, S.X.; et al. Mechanisms of Resistance to Noncovalent Bruton’s Tyrosine Kinase Inhibitors. N. Engl. J. Med. 2022, 386, 735–743. [Google Scholar] [CrossRef]
- Mato, A.R.; Woyach, J.A.; Brown, J.R.; Ghia, P.; Patel, K.; Eyre, T.A.; Munir, T.; Lech-Maranda, E.; Lamanna, N.; Tam, C.S.; et al. Pirtobrutinib after a Covalent BTK Inhibitor in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2023, 389, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.; Flinn, I.W.; Awan, F.T.; Eradat, H.; Brander, D.; Tees, M.; Parikh, S.A.; Phillips, T.; Wang, W.; Reddy, N.M.; et al. P682: Nemtabrutinib (MK-1026), a non-covalent inhibitor of wild-type and C481S mutated Bruton Tyrosine Kinase for B-cell malignancies: Efficacy and safety of the Phase 2 dose expansion BELLWAVE-001 study. HemaSphere 2022, 6, 578–579. [Google Scholar] [CrossRef]
- Allan, J.N.; Pinilla-Ibarz, J.; Gladstone, D.E.; Patel, K.; Sharman, J.P.; Wierda, W.G.; Choi, M.Y.; O’Brien, S.M.; Shadman, M.; Davids, M.S.; et al. Phase Ib dose-escalation study of the selective, non-covalent, reversible Bruton’s tyrosine kinase inhibitor vecabrutinib in B-cell malignancies. Haematologica 2021, 107, 984–987. [Google Scholar] [CrossRef]
- Byrd, J.C.; Smith, S.; Wagner-Johnston, N.; Sharman, J.; Chen, A.I.; Advani, R.; Augustson, B.; Marlton, P.; Renee Commerford, S.; Okrah, K.; et al. First-in-human phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell NHL and CLL. Oncotarget 2018, 9, 13023–13035. [Google Scholar] [CrossRef]
- Reiff, S.D.; Muhowski, E.M.; Guinn, D.; Lehman, A.; Fabian, C.A.; Cheney, C.; Mantel, R.; Smith, L.; Johnson, A.J.; Young, W.B.; et al. Noncovalent inhibition of C481S Bruton tyrosine kinase by GDC-0853: A new treatment strategy for ibrutinib-resistant CLL. Blood 2018, 132, 1039–1049. [Google Scholar] [CrossRef]
- Byrd, J.C.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Kay, N.E.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N. Engl. J. Med. 2014, 371, 213–223. [Google Scholar] [CrossRef]
- Byrd, J.C.; Hillmen, P.; O’Brien, S.; Barrientos, J.C.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; Barr, P.M.; et al. Long-term follow-up of the RESONATE phase 3 trial of ibrutinib vs ofatumumab. Blood 2019, 133, 2031–2042. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Tedeschi, A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Bairey, O.; Hillmen, P.; Bartlett, N.L.; Li, J.; et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2015, 373, 2425–2437. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Tedeschi, A.; Bairey, O.; Hillmen, P.; Coutre, S.E.; Devereux, S.; et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia 2020, 34, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Barr, P.M.; Owen, C.; Robak, T.; Tedeschi, A.; Bairey, O.; Burger, J.A.; Hillmen, P.; Coutre, S.E.; Dearden, C.; Grosicki, S.; et al. Up to 8-year follow-up from RESONATE-2: First-line ibrutinib treatment for patients with chronic lymphocytic leukemia. Blood Adv. 2022, 6, 3440–3450. [Google Scholar] [CrossRef] [PubMed]
- Lampson, B.L.; Brown, J.R. Are BTK and PLCG2 mutations necessary and sufficient for ibrutinib resistance in chronic lymphocytic leukemia? Expert Rev. Hematol. 2018, 11, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Ruppert, A.S.; Guinn, D.; Lehman, A.; Blachly, J.S.; Lozanski, A.; Heerema, N.A.; Zhao, W.; Coleman, J.; Jones, D.; et al. BTK(C481S)-Mediated Resistance to Ibrutinib in Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2017, 35, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Furman, R.R.; Liu, T.M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.; Steggerda, S.M.; Versele, M.; et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef]
- Liu, T.M.; Woyach, J.A.; Zhong, Y.; Lozanski, A.; Lozanski, G.; Dong, S.; Strattan, E.; Lehman, A.; Zhang, X.; Jones, J.A.; et al. Hypermorphic mutation of phospholipase C, γ2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation. Blood 2015, 126, 61–68. [Google Scholar] [CrossRef]
- Burger, J.A.; Landau, D.A.; Taylor-Weiner, A.; Bozic, I.; Zhang, H.; Sarosiek, K.; Wang, L.; Stewart, C.; Fan, J.; Hoellenriegel, J.; et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat. Commun. 2016, 7, 11589. [Google Scholar] [CrossRef]
- Gángó, A.; Alpár, D.; Galik, B.; Marosvári, D.; Kiss, R.; Fésüs, V.; Aczél, D.; Eyüpoglu, E.; Nagy, N.; Nagy, Á.; et al. Dissection of subclonal evolution by temporal mutation profiling in chronic lymphocytic leukemia patients treated with ibrutinib. Int. J. Cancer 2020, 146, 85–93. [Google Scholar] [CrossRef]
- Kanagal-Shamanna, R.; Jain, P.; Patel, K.P.; Routbort, M.; Bueso-Ramos, C.; Alhalouli, T.; Khoury, J.D.; Luthra, R.; Ferrajoli, A.; Keating, M.; et al. Targeted multigene deep sequencing of Bruton tyrosine kinase inhibitor-resistant chronic lymphocytic leukemia with disease progression and Richter transformation. Cancer 2019, 125, 559–574. [Google Scholar] [CrossRef]
- Cosson, A.; Chapiro, E.; Bougacha, N.; Lambert, J.; Herbi, L.; Cung, H.A.; Algrin, C.; Keren, B.; Damm, F.; Gabillaud, C.; et al. Gain in the short arm of chromosome 2 (2p+) induces gene overexpression and drug resistance in chronic lymphocytic leukemia: Analysis of the central role of XPO1. Leukemia 2017, 31, 1625–1629. [Google Scholar] [CrossRef]
- Kadri, S.; Lee, J.; Fitzpatrick, C.; Galanina, N.; Sukhanova, M.; Venkataraman, G.; Sharma, S.; Long, B.; Petras, K.; Theissen, M.; et al. Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Adv. 2017, 1, 715–727. [Google Scholar] [CrossRef]
- Bonfiglio, S.; Sutton, L.A.; Ljungström, V.; Capasso, A.; Pandzic, T.; Weström, S.; Foroughi-Asl, H.; Skaftason, A.; Gellerbring, A.; Lyander, A.; et al. BTK and PLCG2 remain unmutated in one-third of patients with CLL relapsing on ibrutinib. Blood Adv. 2023, 7, 2794–2806. [Google Scholar] [CrossRef]
- Kitada, S.; Andersen, J.; Akar, S.; Zapata, J.M.; Takayama, S.; Krajewski, S.; Wang, H.G.; Zhang, X.; Bullrich, F.; Croce, C.M.; et al. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: Correlations with In vitro and In vivo chemoresponses. Blood 1998, 91, 3379–3389. [Google Scholar] [CrossRef] [PubMed]
- Letai, A.G. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat. Rev. Cancer 2008, 8, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Roberts, A.W.; Colman, P.M.; Adams, J.M. Targeting BCL-2-like Proteins to Kill Cancer Cells. Trends Cancer 2016, 2, 443–460. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Birkinshaw, R.W.; Gong, J.N.; Luo, C.S.; Lio, D.; White, C.A.; Anderson, M.A.; Blombery, P.; Lessene, G.; Majewski, I.J.; Thijssen, R.; et al. Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat. Commun. 2019, 10, 2385. [Google Scholar] [CrossRef] [PubMed]
- Tausch, E.; Close, W.; Dolnik, A.; Bloehdorn, J.; Chyla, B.; Bullinger, L.; Döhner, H.; Mertens, D.; Stilgenbauer, S. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica 2019, 104, e434–e437. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Anderson, M.A.; Gong, J.N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Herling, C.D.; Abedpour, N.; Weiss, J.; Schmitt, A.; Jachimowicz, R.D.; Merkel, O.; Cartolano, M.; Oberbeck, S.; Mayer, P.; Berg, V.; et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat. Commun. 2018, 9, 727. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, G.S.; Al-Harbi, S.; Mazumder, S.; Hill, B.T.; Smith, M.R.; Bodo, J.; Hsi, E.D.; Almasan, A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death. Dis. 2015, 6, e1593. [Google Scholar] [CrossRef]
- Guièze, R.; Liu, V.M.; Rosebrock, D.; Jourdain, A.A.; Hernández-Sánchez, M.; Martinez Zurita, A.; Sun, J.; Ten Hacken, E.; Baranowski, K.; Thompson, P.A.; et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 2019, 36, 369–384.e313. [Google Scholar] [CrossRef]
- Jayappa, K.D.; Portell, C.A.; Gordon, V.L.; Capaldo, B.J.; Bekiranov, S.; Axelrod, M.J.; Brett, L.K.; Wulfkuhle, J.D.; Gallagher, R.I.; Petricoin, E.F.; et al. Microenvironmental agonists generate de novo phenotypic resistance to combined ibrutinib plus venetoclax in CLL and MCL. Blood Adv. 2017, 1, 933–946. [Google Scholar] [CrossRef]
- Haselager, M.V.; Kielbassa, K.; Ter Burg, J.; Bax, D.J.C.; Fernandes, S.M.; Borst, J.; Tam, C.; Forconi, F.; Chiodin, G.; Brown, J.R.; et al. Changes in Bcl-2 members after ibrutinib or venetoclax uncover functional hierarchy in determining resistance to venetoclax in CLL. Blood 2020, 136, 2918–2926. [Google Scholar] [CrossRef] [PubMed]
- Tessoulin, B.; Papin, A.; Gomez-Bougie, P.; Bellanger, C.; Amiot, M.; Pellat-Deceunynck, C.; Chiron, D. BCL2-Family Dysregulation in B-Cell Malignancies: From Gene Expression Regulation to a Targeted Therapy Biomarker. Front. Oncol. 2018, 8, 645. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Cervantes-Gomez, F.; Lamothe, B.; Woyach, J.A.; Wierda, W.G.; Keating, M.J.; Balakrishnan, K.; Gandhi, V. Pharmacological and Protein Profiling Suggests Venetoclax (ABT-199) as Optimal Partner with Ibrutinib in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2015, 21, 3705–3715. [Google Scholar] [CrossRef]
- Deng, J.; Isik, E.; Fernandes, S.M.; Brown, J.R.; Letai, A.; Davids, M.S. Bruton’s tyrosine kinase inhibition increases BCL-2 dependence and enhances sensitivity to venetoclax in chronic lymphocytic leukemia. Leukemia 2017, 31, 2075–2084. [Google Scholar] [CrossRef]
- Kater, A.P.; Slinger, E.; Cretenet, G.; Martens, A.W.; Balasubramanian, S.; Leverson, J.D.; Eldering, E. Combined ibrutinib and venetoclax treatment vs single agents in the TCL1 mouse model of chronic lymphocytic leukemia. Blood Adv. 2021, 5, 5410–5414. [Google Scholar] [CrossRef]
- Bojarczuk, K.; Sasi, B.K.; Gobessi, S.; Innocenti, I.; Pozzato, G.; Laurenti, L.; Efremov, D.G. BCR signaling inhibitors differ in their ability to overcome Mcl-1-mediated resistance of CLL B cells to ABT-199. Blood 2016, 127, 3192–3201. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.E.; Niemann, C.U.; Farooqui, M.; Jones, J.; Mustafa, R.Z.; Lipsky, A.; Saba, N.; Martyr, S.; Soto, S.; Valdez, J.; et al. Ibrutinib-induced lymphocytosis in patients with chronic lymphocytic leukemia: Correlative analyses from a phase II study. Leukemia 2014, 28, 2188–2196. [Google Scholar] [CrossRef] [PubMed]
- Kurtova, A.V.; Balakrishnan, K.; Chen, R.; Ding, W.; Schnabl, S.; Quiroga, M.P.; Sivina, M.; Wierda, W.G.; Estrov, Z.; Keating, M.J.; et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: Development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood 2009, 114, 4441–4450. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M.; Butterworth, M.; Majid, A.; Walewska, R.J.; Sun, X.M.; Dyer, M.J.; Cohen, G.M. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood 2009, 113, 4403–4413. [Google Scholar] [CrossRef] [PubMed]
- Kielbassa, K.; Haselager, M.V.; Bax, D.J.C.; van Driel, B.F.; Dubois, J.; Levin, M.D.; Kersting, S.; Svanberg, R.; Niemann, C.U.; Kater, A.P.; et al. Ibrutinib sensitizes CLL cells to venetoclax by interrupting TLR9-induced CD40 upregulation and protein translation. Leukemia 2023, 37, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Herishanu, Y.; Pérez-Galán, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011, 117, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Petlickovski, A.; Laurenti, L.; Li, X.; Marietti, S.; Chiusolo, P.; Sica, S.; Leone, G.; Efremov, D.G. Sustained signaling through the B-cell receptor induces Mcl-1 and promotes survival of chronic lymphocytic leukemia B cells. Blood 2005, 105, 4820–4827. [Google Scholar] [CrossRef]
- Timofeeva, N.; Gandhi, V. Ibrutinib combinations in CLL therapy: Scientific rationale and clinical results. Blood Cancer J. 2021, 11, 79. [Google Scholar] [CrossRef]
- Jain, N.; Keating, M.; Thompson, P.; Ferrajoli, A.; Burger, J.; Borthakur, G.; Takahashi, K.; Estrov, Z.; Fowler, N.; Kadia, T.; et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. N. Engl. J. Med. 2019, 380, 2095–2103. [Google Scholar] [CrossRef]
- Wierda, W.G.; Allan, J.N.; Siddiqi, T.; Kipps, T.J.; Opat, S.; Tedeschi, A.; Badoux, X.C.; Kuss, B.J.; Jackson, S.; Moreno, C.; et al. Ibrutinib Plus Venetoclax for First-Line Treatment of Chronic Lymphocytic Leukemia: Primary Analysis Results From the Minimal Residual Disease Cohort of the Randomized Phase II CAPTIVATE Study. J. Clin. Oncol. 2021, 39, 3853–3865. [Google Scholar] [CrossRef]
- Tam, C.S.; Allan, J.N.; Siddiqi, T.; Kipps, T.J.; Jacobs, R.; Opat, S.; Barr, P.M.; Tedeschi, A.; Trentin, L.; Bannerji, R.; et al. Fixed-duration ibrutinib plus venetoclax for first-line treatment of CLL: Primary analysis of the CAPTIVATE FD cohort. Blood 2022, 139, 3278–3289. [Google Scholar] [CrossRef]
- Moreno, C.; Solman, I.G.; Tam, C.S.; Grigg, A.; Scarfò, L.; Kipps, T.J.; Srinivasan, S.; Mali, R.S.; Zhou, C.; Dean, J.P.; et al. Immune restoration with ibrutinib plus venetoclax in first-line chronic lymphocytic leukemia: The phase 2 CAPTIVATE study. Blood Adv. 2023, 7, 5294–5303. [Google Scholar] [CrossRef]
- Hillmen, P.; Rawstron, A.C.; Brock, K.; Muñoz-Vicente, S.; Yates, F.J.; Bishop, R.; Boucher, R.; MacDonald, D.; Fegan, C.; McCaig, A.; et al. Ibrutinib Plus Venetoclax in Relapsed/Refractory Chronic Lymphocytic Leukemia: The CLARITY Study. J. Clin. Oncol. 2019, 37, 2722–2729. [Google Scholar] [CrossRef]
- Levin, M.D.; Kater, A.P.; Mattsson, M.; Kersting, S.; Ranti, J.; Thi Tuyet Tran, H.; Nasserinejad, K.; Niemann, C.U. Protocol description of the HOVON 141/VISION trial: A prospective, multicentre, randomised phase II trial of ibrutinib plus venetoclax in patients with creatinine clearance ≥30 mL/min who have relapsed or refractory chronic lymphocytic leukaemia (RR-CLL) with or without TP53 aberrations. BMJ Open 2020, 10, e039168. [Google Scholar] [CrossRef] [PubMed]
- Kater, A.P.; Levin, M.D.; Dubois, J.; Kersting, S.; Enggaard, L.; Veldhuis, G.J.; Mous, R.; Mellink, C.H.M.; van der Kevie-Kersemaekers, A.F.; Dobber, J.A.; et al. Minimal residual disease-guided stop and start of venetoclax plus ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia (HOVON141/VISION): Primary analysis of an open-label, randomised, phase 2 trial. Lancet Oncol. 2022, 23, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Hyak, J.M.; Huang, Y.; Rogers, K.A.; Bhat, S.A.; Grever, M.R.; Byrd, J.C.; Kittai, A.S.; Jones, D.; Miller, C.R.; Woyach, J.A. Combined BCL2 and BTK inhibition in CLL demonstrates efficacy after monotherapy with both classes. Blood Adv. 2022, 6, 5124–5127. [Google Scholar] [CrossRef] [PubMed]
- Hampel, P.J.; Rabe, K.G.; Call, T.G.; Ding, W.; Leis, J.F.; Kenderian, S.S.; Muchtar, E.; Wang, Y.; Koehler, A.B.; Parrondo, R.; et al. Combined ibrutinib and venetoclax for treatment of patients with ibrutinib-resistant or double-refractory chronic lymphocytic leukaemia. Br. J. Haematol. 2022, 199, 239–244. [Google Scholar] [CrossRef]
- Muhowski, E.M.; Ravikrishnan, J.; Gordon, B.; Yu, L.; Misra, S.; Walker, B.; Eathiraj, S.; Sampath, D.; Rogers, K.A.; Byrd, J.C.; et al. Preclinical evaluation of combination nemtabrutinib and venetoclax in chronic lymphocytic leukemia. J. Hematol. Oncol. 2022, 15, 166. [Google Scholar] [CrossRef] [PubMed]
- Guièze, R.; Cheah, C.Y.; Tam, C.S.; Lasica, M.; Verner, E.; Browett, P.J.; Anderson, M.A.; Hilger, J.; Fang, Y.; Simpson, D.; et al. BGB-11417 (Bcl-2 Inhibitor) Monotherapy or Combination with Zanubrutinib in CLL/SLL Patients: Preliminary Phase 1 Data. Available online: https://www.beigenemedical.com/CongressDocuments/Guièze_BGB-11417-101_SFH_Presentation_2023.pdf (accessed on 16 January 2024).
- Davids, M.S.; Chanan-Khan, A.; Mudenda, B.; Nogaieva, L.; Kriachok, I.; Usenko, H.; Ivanov, V.; Kyselova, O.; Perekhrestenko, T.; Muzhychuk, I.; et al. Lisaftoclax (APG-2575) Safety and Activity As Monotherapy or Combined with Acalabrutinib or Rituximab in Patients (pts) with Treatment-Naïve, Relapsed or Refractory Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (R/R CLL/SLL): Initial Data from a Phase 2 Global Study. Blood 2022, 140, 2326–2328. [Google Scholar] [CrossRef]
- Kwiatek, M.; Subramanian Guru Murthy, G.; Hoffmann, M.; Roeker, L.; Tessoulin, B.; Danilov, A.; Alencar, A.J.; Shah, N.N.; Ghesquieres, H.; Le Gouill, S.; et al. P636: A first-in-human Phase 1 study of oral LOXO-338, a seleective BCL2 inhibitor, in patients with advanced hematologic malignancies. Hemasphere 2023, 7, e216785f. [Google Scholar] [CrossRef]
- Brown, J.R.; Byrd, J.C.; Coutre, S.E.; Benson, D.M.; Flinn, I.W.; Wagner-Johnston, N.D.; Spurgeon, S.E.; Kahl, B.S.; Bello, C.; Webb, H.K.; et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110δ, for relapsed/refractory chronic lymphocytic leukemia. Blood 2014, 123, 3390–3397. [Google Scholar] [CrossRef]
- Flinn, I.W.; Hillmen, P.; Montillo, M.; Nagy, Z.; Illés, Á.; Etienne, G.; Delgado, J.; Kuss, B.J.; Tam, C.S.; Gasztonyi, Z.; et al. The phase 3 DUO trial: Duvelisib vs ofatumumab in relapsed and refractory CLL/SLL. Blood 2018, 132, 2446–2455. [Google Scholar] [CrossRef] [PubMed]
- Mato, A.R.; Ghosh, N.; Schuster, S.J.; Lamanna, N.; Pagel, J.M.; Flinn, I.W.; Barrientos, J.C.; Rai, K.R.; Reeves, J.A.; Cheson, B.D.; et al. Phase 2 study of the safety and efficacy of umbralisib in patients with CLL who are intolerant to BTK or PI3Kδ inhibitor therapy. Blood 2021, 137, 2817–2826. [Google Scholar] [CrossRef]
- Tam, C.S.; Cheah, C.; Stevens, D.A.; By, K.; Chen, X.; Tariq, B.; Vosganian, G.S.; Huang, J.; Alwan, M. P686: A Phase 1 first-in-human study of BGB-16673, a Bruton tyrosine kinase protein degrader, in patients (PTS) with B-cell malignancies (Trial in progress). HemaSphere 2022, 6, 582–583. [Google Scholar] [CrossRef]
- Linton, K.; Doorduijn, J.; El-Sharkawi, D.; Mous, R.; Forconi, F.; Lewis, D.; Gleeson, M.; Riches, J.; McKay, P.; Stevens, W.; et al. PB2331: An ongoing first-in-human Phase 1 Trial of NX-5948, an oral Bruton’s tyrosine kinase (BTK) degrader, in patients with relapsed/refractory B cell malignancies. Hemasphere 2023, 7, e29005fd. [Google Scholar] [CrossRef]
- Mato, A.R.; Wierda, W.G.; Ai, W.Z.; Flinn, I.W.; Tees, M.; Patel, M.R.; Patel, K.; O’Brien, S.; Bond, D.A.; Roeker, L.E.; et al. NX-2127-001, a First-in-Human Trial of NX-2127, a Bruton’s Tyrosine Kinase-Targeted Protein Degrader, in Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia and B-Cell Malignancies. Blood 2022, 140, 2329–2332. [Google Scholar] [CrossRef]
- Kater, A.P.; Christensen, J.H.; Bentzen, H.H.; Niemann, C.U.; Hutchings, M.; Chen, J.; Rios, M.; Palenski, T.; Li, T.; Mato, A.R. Subcutaneous Epcoritamab in Patients with Relapsed/Refractory Chronic Lymphocytic Leukemia: Preliminary Results from the Epcore CLL-1 Trial. Blood 2021, 138, 2627. [Google Scholar] [CrossRef]
- Patel, K.; Michot, J.-M.; Chanan-Khan, A.A.; Salles, G.A.; Cartron, G.; Peyrade, F.; Bouabdallah, R.; Reid, E.G.; Thomas, S.K.; Wierda, W.G.; et al. Preliminary Safety and Anti-Tumor Activity of XmAb13676, an Anti-CD20 x Anti-CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Non-Hodgkin’s Lymphoma and Chronic Lymphocytic Leukemia. Blood 2019, 134, 4079. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Cherian, S.; Chen, X.; Wood, B.; Lozanski, A.; Byrd, J.C.; Heimfeld, S.; et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With CD19-Specific Chimeric Antigen Receptor-Modified T Cells After Failure of Ibrutinib. J. Clin. Oncol. 2017, 35, 3010–3020. [Google Scholar] [CrossRef]
- Siddiqi, T.; Maloney, D.G.; Kenderian, S.S.; Brander, D.M.; Dorritie, K.; Soumerai, J.; Riedell, P.A.; Shah, N.N.; Nath, R.; Fakhri, B.; et al. Lisocabtagene maraleucel in chronic lymphocytic leukaemia and small lymphocytic lymphoma (TRANSCEND CLL 004): A multicentre, open-label, single-arm, phase 1–2 study. Lancet 2023, 402, 641–654. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Munir, T.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Barr, P.M.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Final analysis from RESONATE: Up to six years of follow-up on ibrutinib in patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma. Am. J. Hematol. 2019, 94, 1353–1363. [Google Scholar] [CrossRef]
- Bond, D.A.; Woyach, J.A. Targeting BTK in CLL: Beyond Ibrutinib. Curr. Hematol. Malig. Rep. 2019, 14, 197–205. [Google Scholar] [CrossRef]
- Lewis, K.L.; Cheah, C.Y. Non-Covalent BTK Inhibitors-The New BTKids on the Block for B-Cell Malignancies. J. Pers. Med. 2021, 11, 764. [Google Scholar] [CrossRef]
- Reiff, S.D.; Mantel, R.; Smith, L.L.; Greene, J.T.; Muhowski, E.M.; Fabian, C.A.; Goettl, V.M.; Tran, M.; Harrington, B.K.; Rogers, K.A.; et al. The BTK Inhibitor ARQ 531 Targets Ibrutinib-Resistant CLL and Richter Transformation. Cancer Discov. 2018, 8, 1300–1315. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Guo, Y.; Xue, H.; Liu, Y.; Guo, Y.; Wang, F.; Song, X.; Guo, Y.; Chen, S.; Xu, H.; et al. Preclinical characterization of BGB-11417, a potent and selective Bcl-2 inhibitor with superior antitumor activities in haematological tumor models. Cancer 2020, 80, 3077. [Google Scholar] [CrossRef]
- Cheah, C.Y.; Tam, C.S.; Lasica, M.; Verner, E.; Browett, P.J.; Anderson, M.A.; Hilger, J.; Fang, Y.; Simpson, D.; Opat, S. A Phase 1 Study with the Novel B-Cell Lymphoma 2 (Bcl-2) Inhibitor Bgb-11417 As Monotherapy or in Combination with Zanubrutinib (ZANU) in Patients (Pts) with CLL/SLL: Preliminary Data. Blood 2022, 140, 2321–2323. [Google Scholar] [CrossRef]
- Desai, P.; Lonial, S.; Cashen, A.; Kamdar, M.; Blachly, J.S.; Flinn, I.W.; O’brien, S.; Garcia, J.; Korde, N.; Moslehi, J.; et al. P579: Safety, tolerability, pharmacokinetics, and preliminary antitumor activity of AZD5991 in relapsed/refractory hematologic malignancies: A Phase 1 first-in-human study. Hemasphere 2023, 7, e98838e7. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Hus, I.; Puła, B.; Robak, T. PI3K Inhibitors for the Treatment of Chronic Lymphocytic Leukemia: Current Status and Future Perspectives. Cancers 2022, 14, 1571. [Google Scholar] [CrossRef]
- Mato, A.R.; Roeker, L.E.; Jacobs, R.; Hill, B.T.; Lamanna, N.; Brander, D.; Shadman, M.; Ujjani, C.S.; Yazdy, M.S.; Perini, G.F.; et al. Assessment of the Efficacy of Therapies Following Venetoclax Discontinuation in CLL Reveals BTK Inhibition as an Effective Strategy. Clin. Cancer Res. 2020, 26, 3589–3596. [Google Scholar] [CrossRef]
- Buhimschi, A.D.; Armstrong, H.A.; Toure, M.; Jaime-Figueroa, S.; Chen, T.L.; Lehman, A.M.; Woyach, J.A.; Johnson, A.J.; Byrd, J.C.; Crews, C.M. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 2018, 57, 3564–3575. [Google Scholar] [CrossRef] [PubMed]
- Shorer Arbel, Y.; Katz, B.Z.; Gabizon, R.; Shraga, A.; Bronstein, Y.; Kamdjou, T.; Globerson Levin, A.; Perry, C.; Avivi, I.; London, N.; et al. Proteolysis Targeting Chimeras for BTK Efficiently Inhibit B-Cell Receptor Signaling and Can Overcome Ibrutinib Resistance in CLL Cells. Front. Oncol. 2021, 11, 646971. [Google Scholar] [CrossRef]
- Iyer, P.; Wang, L. Emerging Therapies in CLL in the Era of Precision Medicine. Cancers 2023, 15, 1583. [Google Scholar] [CrossRef]
- Hutchings, M. The evolving therapy of DLBCL: Bispecific antibodies. Hematol. Oncol. 2023, 41, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Perutelli, F.; Jones, R.; Griggio, V.; Vitale, C.; Coscia, M. Immunotherapeutic Strategies in Chronic Lymphocytic Leukemia: Advances and Challenges. Front. Oncol. 2022, 12, 837531. [Google Scholar] [CrossRef] [PubMed]
- Alderuccio, J.P.; Mackrides, N.; Chapman, J.R.; Vega, F.; Lossos, I.S. Rapid complete response to blinatumomab as a successful bridge to allogeneic stem cell transplantation in a case of refractory Richter syndrome. Leuk. Lymphoma 2019, 60, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Mihályová, J.; Hradská, K.; Jelínek, T.; Motais, B.; Celichowski, P.; Hájek, R. Promising Immunotherapeutic Modalities for B-Cell Lymphoproliferative Disorders. Int. J. Mol. Sci. 2021, 22, 11470. [Google Scholar] [CrossRef] [PubMed]
- Mhibik, M.; Gaglione, E.M.; Eik, D.; Herrick, J.; Le, J.; Ahn, I.E.; Chiu, C.; Wielgos-Bonvallet, M.; Hiemstra, I.H.; Breij, E.C.W.; et al. Cytotoxicity of the CD3×CD20 bispecific antibody epcoritamab in CLL is increased by concurrent BTK or BCL-2 targeting. Blood Adv. 2023, 7, 4089–4101. [Google Scholar] [CrossRef] [PubMed]
- Eichhorst, B.; Eradat, H.; Niemann, C.U.; Oki, T.; Kuznetsova, A.; Rios, M.; Valentin, R.; Kater, A.P. Epcoritamab Monotherapy and Combinations in Relapsed or Refractory Chronic Lymphocytic Leukemia or Richter’s Syndrome: New escalation and expansion cohorts in EPCORE CLL-1. Hematol. Oncol. 2023, 41, 828–829. [Google Scholar] [CrossRef]
- Cai, W.; Dong, J.; Gallolu Kankanamalage, S.; Titong, A.; Shi, J.; Jia, Z.; Wang, B.; Huang, C.; Zhang, J.; Lin, J.; et al. Biological activity validation of a computationally designed Rituximab/CD3 T cell engager targeting CD20+ cancers with multiple mechanisms of action. Antib. Ther. 2021, 4, 228–241. [Google Scholar] [CrossRef]
- Cao, Y.; Marcucci, E.C.; Budde, L.E. Mosunetuzumab and lymphoma: Latest updates from 2022 ASH annual meeting. J. Hematol. Oncol. 2023, 16, 69. [Google Scholar] [CrossRef] [PubMed]
- Barrett, K.; Lu, K.; Jin, H.Y.; Millen, R.; Lefebure, M.; Jiang, Y. Abstract 1869: Preclinical evaluation of mosunetuzumab for the treatment of relapsed/refractory chronic lymphocytic leukemia. Cancer Res. 2023, 83, 1869. [Google Scholar] [CrossRef]
- Ribeiro, M.L.; Normant, E.; Garau, D.R.; Miskin, H.P.; Sportelli, P.; Weiss, M.S.; Bosch, F.; Roué, G. PS1310 The novel Bispecific CD47-CD19 antibody TG-1801 potentiates the activity of Ublituximab-Umbralisib (U2) Drug Combination in Preclinical Models of B-NHL. HemaSphere 2019, 3, 598. [Google Scholar] [CrossRef]
- Peters, F.S.; Strefford, J.C.; Eldering, E.; Kater, A.P. T-cell dysfunction in chronic lymphocytic leukemia from an epigenetic perspective. Haematologica 2021, 106, 1234–1243. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- Geyer, M.B.; Rivière, I.; Sénéchal, B.; Wang, X.; Wang, Y.; Purdon, T.J.; Hsu, M.; Devlin, S.M.; Palomba, M.L.; Halton, E.; et al. Safety and tolerability of conditioning chemotherapy followed by CD19-targeted CAR T cells for relapsed/refractory CLL. JCI Insight 2019, 4, e122627. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.V.; Gill, S.; Hexner, E.O.; Schuster, S.; Nasta, S.; Loren, A.; Svoboda, J.; Stadtmauer, E.; Landsburg, D.J.; Mato, A.; et al. Long-Term Outcomes From a Randomized Dose Optimization Study of Chimeric Antigen Receptor Modified T Cells in Relapsed Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2020, 38, 2862–2871. [Google Scholar] [CrossRef]
- Siddiqi, T.; Soumerai, J.D.; Dorritie, K.A.; Stephens, D.M.; Riedell, P.A.; Arnason, J.; Kipps, T.J.; Gillenwater, H.H.; Gong, L.; Yang, L.; et al. Phase 1 TRANSCEND CLL 004 study of lisocabtagene maraleucel in patients with relapsed/refractory CLL or SLL. Blood 2022, 139, 1794–1806. [Google Scholar] [CrossRef]
- Sośnia, O.; Puła, B. Chronic lymphocytic leukemia following venetoclax treatment failure. Acta Haematol. Pol. 2022, 53, 94–103. [Google Scholar] [CrossRef]
- Roeker, L.E.; Dreger, P.; Brown, J.R.; Lahoud, O.B.; Eyre, T.A.; Brander, D.M.; Skarbnik, A.; Coombs, C.C.; Kim, H.T.; Davids, M.; et al. Allogeneic stem cell transplantation for chronic lymphocytic leukemia in the era of novel agents. Blood Adv. 2020, 4, 3977–3989. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.T.; Shaughnessy, C.J.; Rai, S.C.; Reynolds, C.; Ho, V.T.; Cutler, C.; Koreth, J.; Gooptu, M.; Romee, R.; Nikiforow, S.; et al. Allogeneic hematopoietic cell transplantation after prior targeted therapy for high-risk chronic lymphocytic leukemia. Blood Adv. 2020, 4, 4113–4123. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| BTK Inhibitor | Binding and Selectivity | Approval Status | Clinical Indications | Safety | Refs. |
|---|---|---|---|---|---|
| Ibrutinib (IMBRUVICA, Janssen, Beerse, Belgium) | Covalent irreversible targeting BTK C481. Inhibits ITK, EGFR, CSK, ErbB2, and TEC | Approved by FDA and EMA | MCL, CLL, WM, MZL, and GVHD | Hypertension, bleeding, atrial fibrillation/atrial flutter | [21,22] |
| Acalabrutinib (ACP-196, Calquence®, AstraZeneca Pharmaceuticals, Cambridge, UK) | Covalent irreversible targeting BTK C481 with high selectivity, reduced off-target effects, no inhibition of EGFR or ITK | Approved by FDA and EMA | MCL, CLL | Atrial fibrillation/atrial flutter (risk lower than with ibrutinib) | [16,22] |
| Zanubrutinib (Brukinsa, BeiGene, Beijing, China) | Selective, covalent irreversible targeting BTK C481, reduced off-target effects | Approved by FDA and EMA | MZL, CLL, WM | Thrombocytopenia, neutropenia, and bruising | [16,17,22] |
| Orelabrutinib (ICP-022, HIBRUKA Biogen/Innocare Pharma, Cambridge, MA, USA/Beijing, China) | Covalent irreversible targeting BTK C481 more selectively than ibrutinib | Breakthrough Therapy Designation for RR MCL | MCL | Neutropenia, thrombocytopenia, upper respiratory tracts, and lung infections | [18,23] |
| Spebrutinib (CC-292, AVL-292, Avila Therapeutics/Celgene, Waltham, MA, USA/Summit, NJ, USA) | Covalent irreversible targeting BTK C481 with high affinity | Phase 1 study in RR CLL/SLL | - | Neutropenia, thrombocytopenia, diarrhea, fatigue, nausea, cough, pyrexia, and headache | [19] |
| Tirabrutinib (Velexbru®, ONO/GS-4059, Ono Pharmaceutical, Gilead Sciences, Osaka, Japan/Foster City, CA, USA) | Covalent irreversible very potent and specific BTKi targeting C481 with greater selectivity than ibrutinib | Phase 1 study in various B-cell malignancies | - | Anemia, neutropenia, thrombocytopenia, pyrexia | [20] |
| Pirtobrutinib (LOXO-305, Jaypirca, Eli Lilly, Indianapolis, IN, USA) | Non-covalent reversible highly selective, next-generation BTKi, blocks the ATP site of BTK through non-covalent, non-C481-dependent binding | FDA approval, Conditional Marketing Authorization of EMA | MCL, CLL/SLL | Infections, neutropenia, anemia, fatigue, pyrexia | [24,25,26,27] |
| Nemtabrutinib (MK1026, ARQ 531; ArQule, Inc./Merck Sharp and Dohme, Woburn, MA, USA/Rahway, NY, USA) | Non-covalent reversible highly selective BTKi | Phase 1/2 (NCT03162536) | - | Fatigue, constipation, dysgeusia, cough, nausea | [28] |
| Vecabrutinib (SNS-062, Viracta Therapeutics, Cardiff, NY, USA) | Non-covalent, reversible highly selective BTKi, no activity on EGFR | Phase 1b/2 (NCT03037645) | - | Fatigue, nausea, diarrhea, thrombocytopenia | [29] |
| Fenebrutinib (GDC-0853, Roche/Chugai Pharmaceutical, Tokyo, Japan/Basel, Switzerland) | Non-covalent reversible BTKi with strong inhibitory efficacy against a single (C481S) and double (T474S/C481S) BTK variant | Phase 1 study in RR B-cell NHL and CLL (NCT01991184) | - | Fatigue, nausea, diarrhea, thrombocytopenia, headache | [30,31] |
| Author/Reference | Phase | Previous Treatment | Therapeutic Intervention | ORR/CR | mPFS | Safety | |
|---|---|---|---|---|---|---|---|
| Non-covalent BTKi | Mato A.R. et al. [27] | I/II | Median of three prior therapies; 100% BTKi, 87.9% anti-CD20-Ab, 78.9% chemotherapy, 40.5% BCL2i, 18.2% PI3Ki, 5.7% CAR-T 2.4% allo-SCT | Pirtobrutinib | 73.3%/1.6% | 19.6 months | Most common AEs of grade ≥3: infections 28.1%, 26.8% neutropenia, 8.8% anemia; discontinuation of therapy due to AEs in 2.6% |
| Woyach J. et al. [28] | I/II | Median of four prior therapies; 84% BTKi | Nemtabrutinib | 57.9%/2.6% | NR | Most common AEs: 33% fatigue, 31% constipation, 25% dysgeusia, 25% cough, 25% nausea, 25% pyrexia; AEs of grade ≥3 occurred in 68% of participants, discontinuation of therapy due to AEs in 8% | |
| BCL2i | Guièze R. et al. [82] | I | Median of one prior therapy | Sonrotoclax (BGB-11417) +/− zanubrutinib | Monotherapy: 67%/33%; combination therapy: 95%/30% | NR | Most common AEs of grade ≥3 in monotherapy: 50% neutropenia, 25% thrombocytopenia, 12.5% pyrexia; most common AEs of grade ≥3 in combination therapy: 14.1% neutropenia, 1.4% thrombocytopenia, 1.4% diarrhea, 1.4% COVID-19 |
| Davids M.S. et al. [83] | II | Median of two prior therapies; 12% refractory to BTKi and/or BCL2i | Lisaftoclax (APG-2575) +/− acalabrutinib or rituximab | Monotherapy: 65%/NR; lisaftoclax + acalabrutinib: 98%/NR; lisaftoclax + rituximab: 87%/NR | NR | Most common AEs of grade ≥3 in any group: 26% neutropenia, 12% anemia, 5% thrombocytopenia | |
| Kwiatek M. et al. [84] | I | Median of three prior therapies; 68% BTKi | LOXO-338 | NR | NR | Most common AEs of grade ≥3: 15% anemia, 4% COVID-19 | |
| PI3Ki | Brown J.R. et al. [85] | I | Median of five prior therapies; 100% fludarabine, 96% rituximab, 87% alkylating agents | Idelalisib | 72%/- | 15.8 months | Most common AEs of grade ≥3: 42.6% neutropenia, 20.4% pneumonia, 16.7% thrombocytopenia, 11.1% anemia, 11.1% neutropenic fever |
| Flinn I.W. et al. [86] | III | Median of two prior therapies; 93% alkylating agent, 78% monoclonal antibody, 60% purine analog | Duvelisib | 73.8%/0.6% | 15.7 months | Most common AEs of grade ≥3: 30% neutropenia, 15% diarrhea, 14% pneumonia, 13% anemia, 12% colitis | |
| Mato A.R. et al. [87] | II | Median of two prior therapies; 86% BTKi, 14% PI3Ki | Umbralisib | 44%/4.2% | 23.5 months | Most common AEs of grade ≥3: 18% neutropenia, 14% leukocytosis, 12% thrombocytosis, 12% pneumonia, 8% diarrhea | |
| BTK degrader | Tam C et al. [88] | Ia/Ib | NR | BGB-16673 | NR | NR | NR |
| Linton K. et al. [89] | Ia/Ib | NR | NX-5948 | NR | NR | NR | |
| Mato A.R. et al. [90] | Ia/Ib | NR | NX-2127 | 33%/NR | NR | Most common AEs of grade ≥3: 35% neutropenia, 15% anemia, 4% hypertension | |
| BiTE | Kater A.P. et al. [91] | Ib/II | Median of four prior therapies; 100% BTKi | Epcoritamab (CD3xCD20 bispecific antibody) | NR | NR | Most common AEs: CRS (100%), fatigue (71%), injection-site reaction (43%), and nausea (43%); no episodes of grade ≥3 CRS were noted |
| Patel K. et al. [92] | I | NR | Plamotamab (XmAb13676) (CD3xCD20 bispecific antibody) | NR | NR | Among eight CLL patients there were five AEs of grade ≥3: anemia, thrombocytopenia, neutropenia, lymphopenia, CRS | |
| CAR-T | Turtle C.J. et al. [93] | I/II | Median of five prior therapies; 21% of patients were double-refractory; 100% chemoimmunotherapy, 100% ibrutinib, 25% venetoclax | CD4+ and CD8+ CD19-specific CAR-T cells | 74%/21% | 8.5 months | Most common AEs: 83% CRS, 33% neurotoxicity; 1 fatal neurotoxicity event |
| Siddiqi T. [94] | I/II | Median of five prior therapies; 80% of patients were double-refractory; 100% BTKi, 80% venetoclax, 86% chemoimmunotherapy; 6% SCT, 25% PI3Ki | Lisocabtagene maraleucel (CD4+ CD8+ CAR-T cells) | 48%/18% | 17.87 months | Most common AEs: 85% CRS, 67% anemia, 62% neutropenia, 50% thrombocytopenia; 5 fatal events, 1 related to treatment due to hemophagocytic lymphohistiocytosis | |
| CAR-NK | Liu E. et al. [95] | I/II | Median of four prior therapies; 18% of patients were double-refractory; 45% BTKi, 18% venetoclax; 36% autoSCT | anti-CD19 CAR-NK | 73%/64% | NR | Most common AEs of grade ≥3: 91% neutropenia, 91% lymphopenia, 18% anemia |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zygmunciak, P.; Robak, T.; Puła, B. Treatment of Double-Refractory Chronic Lymphocytic Leukemia—An Unmet Clinical Need. Int. J. Mol. Sci. 2024, 25, 1589. https://doi.org/10.3390/ijms25031589
Zygmunciak P, Robak T, Puła B. Treatment of Double-Refractory Chronic Lymphocytic Leukemia—An Unmet Clinical Need. International Journal of Molecular Sciences. 2024; 25(3):1589. https://doi.org/10.3390/ijms25031589
Chicago/Turabian StyleZygmunciak, Przemysław, Tadeusz Robak, and Bartosz Puła. 2024. "Treatment of Double-Refractory Chronic Lymphocytic Leukemia—An Unmet Clinical Need" International Journal of Molecular Sciences 25, no. 3: 1589. https://doi.org/10.3390/ijms25031589
APA StyleZygmunciak, P., Robak, T., & Puła, B. (2024). Treatment of Double-Refractory Chronic Lymphocytic Leukemia—An Unmet Clinical Need. International Journal of Molecular Sciences, 25(3), 1589. https://doi.org/10.3390/ijms25031589