
Alterations in Proteostasis Mechanisms in Niemann–Pick Type C Disease


 ,
,  ,
,  and
and
Abstract
1. Introduction
1.1. N-Glycosylation, Implication in NPC1 Function
1.2. ERAD Pathway and Chaperone Folding
1.3. Folding by Heat Shock Proteins
2. Autophagy in NPC
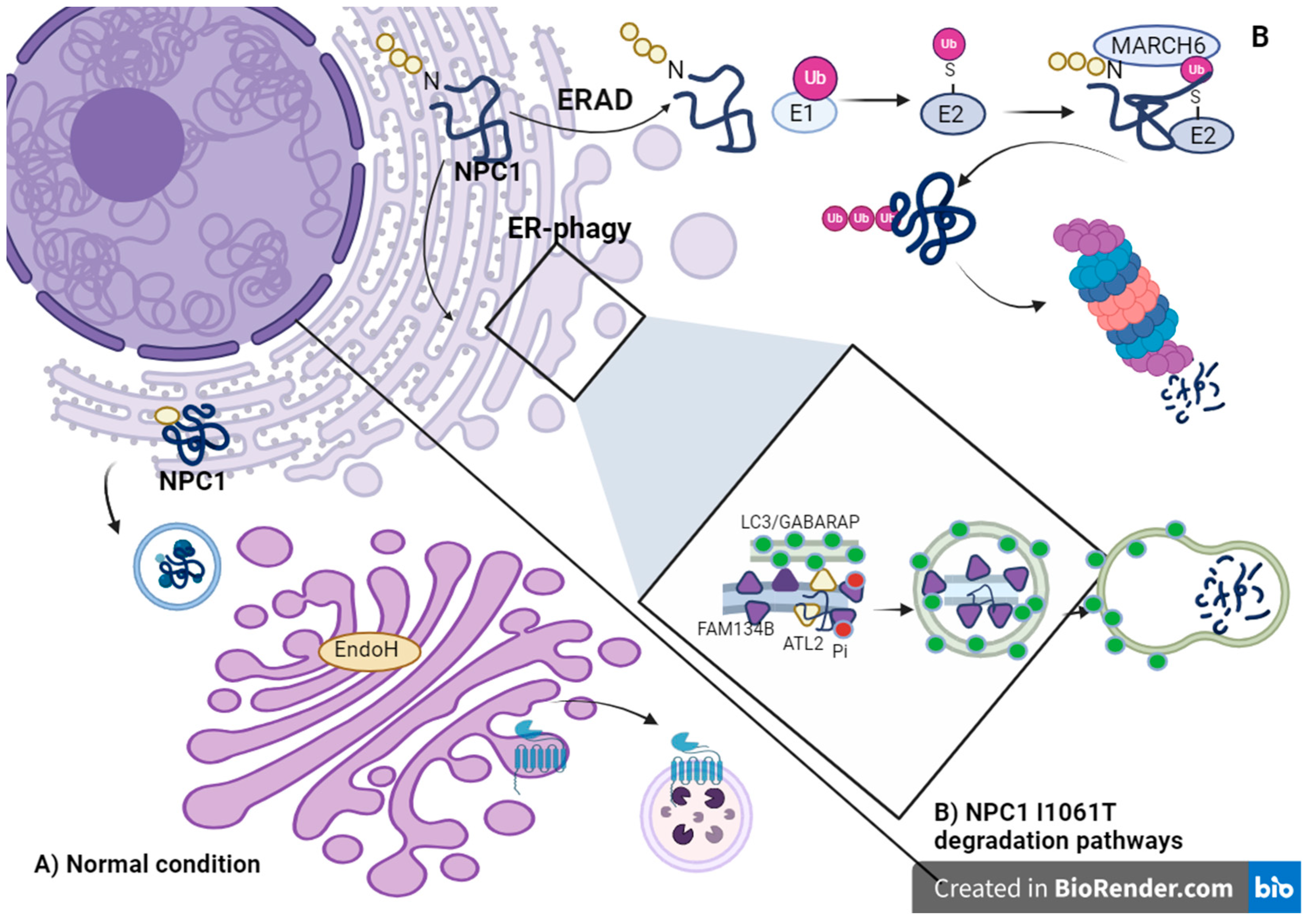
Alterations in ER-Phagy
3. Lysosomal Degradation Pathway
3.1. Alterations in Endocytosis Due to Cholesterol Accumulation in NPC
3.2. Alterations in Lysosomal Glycocalyx
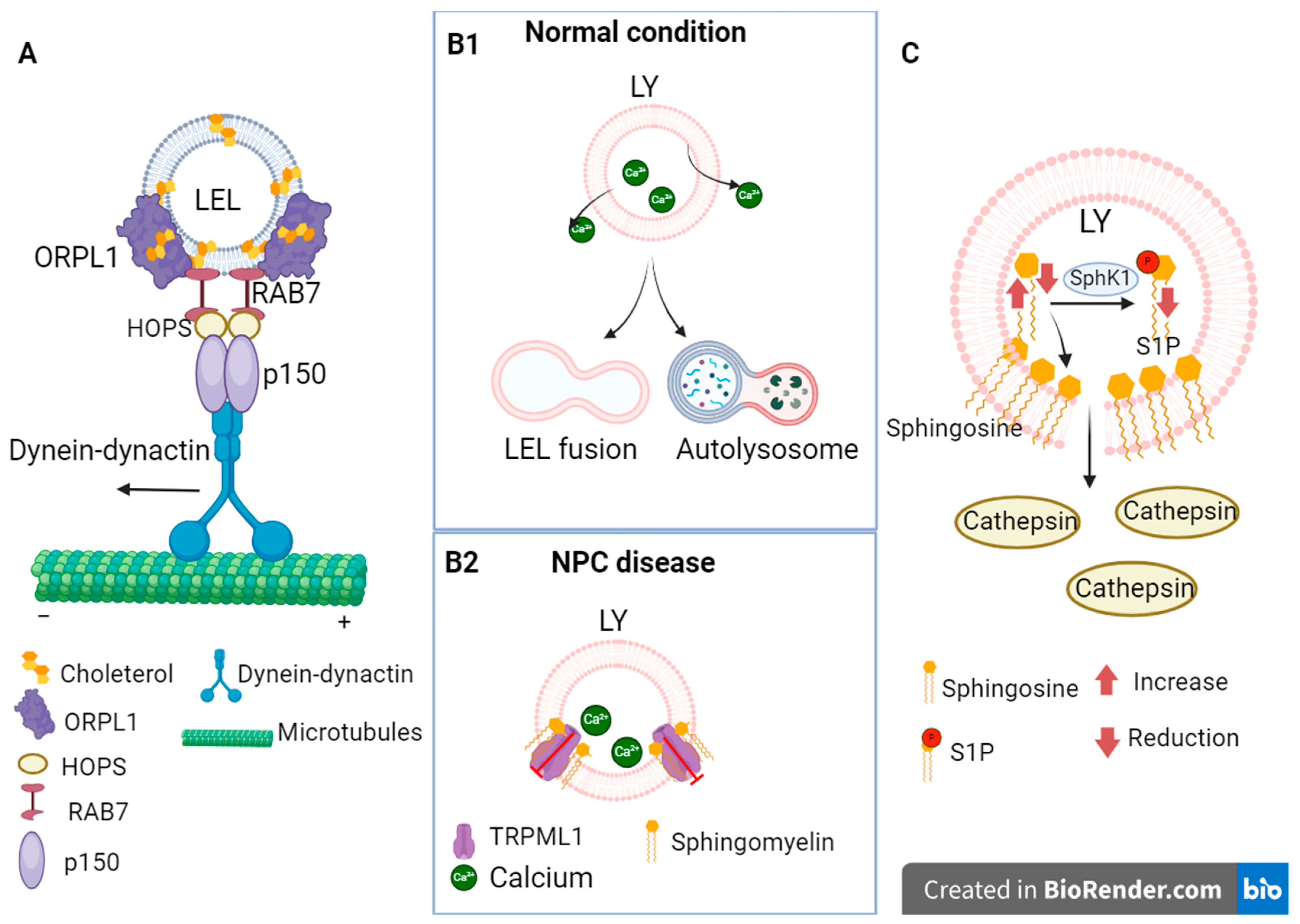
3.3. Dysregulation of Calcium Concentration
4. Ubiquitin–Proteosome Pathway
5. Therapies Based on the Improvement of Proteostasis
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Merrick, W.C.; Pavitt, G.D. Protein Synthesis Initiation in Eukaryotic Cells. Cold Spring Harb. Perspect. Biol. 2018, 10, a033092. [Google Scholar] [CrossRef] [PubMed]
- Watari, H.; Blanchette-Mackie, E.J.; Dwyer, N.K.; Watari, M.; Neufeld, E.B.; Patel, S.; Pentchev, P.G.; Strauss, J.F., 3rd. Mutations in the leucine zipper motif and sterol-sensing domain inactivate the Niemann-Pick C1 glycoprotein. J. Biol. Chem. 1999, 274, 21861–21866. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Paudel, R.R.; Lu, D.; Roy Chowdhury, S.; Monroy, E.Y.; Wang, J. Targeted Protein Degradation via Lysosomes. Biochemistry 2023, 62, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.L.; Ciechanover, A. Targeting proteins for destruction by the ubiquitin system: Implications for human pathobiology. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 73–96. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Cao, Z.; Zheng, P.; Vitolo, O.V.; Liu, S.; Staniszewski, A.; Moolman, D.; Zhang, H.; Shelanski, M.; Arancio, O. Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell 2006, 126, 775–788. [Google Scholar] [CrossRef]
- Leigh, P.N.; Whitwell, H.; Garofalo, O.; Buller, J.; Swash, M.; Martin, J.E.; Gallo, J.M.; Weller, R.O.; Anderton, B.H. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain A J. Neurol. 1991, 114 Pt 2, 775–788. [Google Scholar] [CrossRef]
- Poliquin, S.; Kang, J.Q. Disruption of the Ubiquitin-Proteasome System and Elevated Endoplasmic Reticulum Stress in Epilepsy. Biomedicines 2022, 10, 647. [Google Scholar] [CrossRef]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of Ubiquitin-Proteasome System in Neurodegenerative Diseases. Front. Aging Neurosci. 2016, 8, 303. [Google Scholar] [CrossRef]
- Chiba, Y.; Komori, H.; Takei, S.; Hasegawa-Ishii, S.; Kawamura, N.; Adachi, K.; Nanba, E.; Hosokawa, M.; Enokido, Y.; Kouchi, Z.; et al. Niemann-Pick disease type C1 predominantly involving the frontotemporal region, with cortical and brainstem Lewy bodies: An autopsy case. Neuropathol. Off. J. Jpn. Soc. Neuropathol. 2014, 34, 49–57. [Google Scholar] [CrossRef]
- Liu, E.A.; Mori, E.; Hamasaki, F.; Lieberman, A.P. TDP-43 proteinopathy occurs independently of autophagic substrate accumulation and underlies nuclear defects in Niemann-Pick C disease. Neuropathol. Appl. Neurobiol. 2021, 47, 1019–1032. [Google Scholar] [CrossRef]
- Love, S.; Bridges, L.R.; Case, C.P. Neurofibrillary tangles in Niemann-Pick disease type C. Brain A J. Neurol. 1995, 118 Pt 1, 119–129. [Google Scholar] [CrossRef]
- Saito, Y.; Suzuki, K.; Hulette, C.M.; Murayama, S. Aberrant phosphorylation of alpha-synuclein in human Niemann-Pick type C1 disease. J. Neuropathol. Exp. Neurol. 2004, 63, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.; Sillence, D.J. Niemann-Pick type C disease: Cellular pathology and pharmacotherapy. J. Neurochem. 2020, 153, 674–692. [Google Scholar] [CrossRef] [PubMed]
- Bräuer, A.U.; Kuhla, A.; Holzmann, C.; Wree, A.; Witt, M. Current Challenges in Understanding the Cellular and Molecular Mechanisms in Niemann-Pick Disease Type C1. Int. J. Mol. Sci. 2019, 20, 4392. [Google Scholar] [CrossRef] [PubMed]
- Shammas, H.; Kuech, E.M.; Rizk, S.; Das, A.M.; Naim, H.Y. Different Niemann-Pick C1 Genotypes Generate Protein Phenotypes that Vary in their Intracellular Processing, Trafficking and Localization. Sci. Rep. 2019, 9, 5292. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.L.; Schache, K.J.; Azaria, R.D.; Kuiper, E.Q.; Erwood, S.; Ivakine, E.A.; Farhat, N.Y.; Porter, F.D.; Pathmasiri, K.C.; Cologna, S.M.; et al. Species-specific differences in NPC1 protein trafficking govern therapeutic response in Niemann-Pick type C disease. JCI Insight 2022, 7, e160308. [Google Scholar] [CrossRef]
- Chikh, K.; Vey, S.; Simonot, C.; Vanier, M.T.; Millat, G. Niemann-Pick type C disease: Importance of N-glycosylation sites for function and cellular location of the NPC2 protein. Mol. Genet. Metab. 2004, 83, 220–230. [Google Scholar] [CrossRef]
- Gelsthorpe, M.E.; Baumann, N.; Millard, E.; Gale, S.E.; Langmade, S.J.; Schaffer, J.E.; Ory, D.S. Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J. Biol. Chem. 2008, 283, 8229–8236. [Google Scholar] [CrossRef] [PubMed]
- Völkner, C.; Pantoom, S.; Liedtke, M.; Lukas, J.; Hermann, A.; Frech, M.J. Assessment of FDA-Approved Drugs as a Therapeutic Approach for Niemann-Pick Disease Type C1 Using Patient-Specific iPSC-Based Model Systems. Cells 2022, 11, 319. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Chung, C.; Shen, D.; Xu, H.; Lieberman, A.P. Ryanodine receptor antagonists adapt NPC1 proteostasis to ameliorate lipid storage in Niemann-Pick type C disease fibroblasts. Hum. Mol. Genet. 2012, 21, 3205–3214. [Google Scholar] [CrossRef] [PubMed]
- Grabbe, C.; Husnjak, K.; Dikic, I. The spatial and temporal organization of ubiquitin networks. Nat. Rev. Mol. Cell Biol. 2011, 12, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T.; Gray, J.; Priestman, D.A.; Wallom, K.L.; Atkins, J.; Olsen, O.D.; Klein, A.; Drndarski, S.; Petersen, N.H.; Ingemann, L.; et al. Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci. Transl. Med. 2016, 8, 355ra118. [Google Scholar] [CrossRef] [PubMed]
- Mengel, E.; Patterson, M.C.; Da Riol, R.M.; Del Toro, M.; Deodato, F.; Gautschi, M.; Grunewald, S.; Grønborg, S.; Harmatz, P.; Héron, B.; et al. Efficacy and safety of arimoclomol in Niemann-Pick disease type C: Results from a double-blind, randomised, placebo-controlled, multinational phase 2/3 trial of a novel treatment. J. Inherit. Metab. Dis. 2021, 44, 1463–1480. [Google Scholar] [CrossRef] [PubMed]
- Pipalia, N.H.; Saad, S.Z.; Subramanian, K.; Cross, A.; Al-Motawa, A.; Garg, K.; Blagg, B.S.J.; Neckers, L.; Helquist, P.; Wiest, O.; et al. HSP90 inhibitors reduce cholesterol storage in Niemann-Pick type C1 mutant fibroblasts. J. Lipid Res. 2021, 62, 100114. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Scott, S.M.; Sun, S.; Zhao, P.; Hutt, D.M.; Shao, H.; Gestwicki, J.E.; Balch, W.E. Individualized management of genetic diversity in Niemann-Pick C1 through modulation of the Hsp70 chaperone system. Hum. Mol. Genet. 2020, 29, 1–19. [Google Scholar] [CrossRef]
- Lebrand, C.; Corti, M.; Goodson, H.; Cosson, P.; Cavalli, V.; Mayran, N.; Fauré, J.; Gruenberg, J. Late endosome motility depends on lipids via the small GTPase Rab7. EMBO J. 2002, 21, 1289–1300. [Google Scholar] [CrossRef]
- Rocha, N.; Kuijl, C.; van der Kant, R.; Janssen, L.; Houben, D.; Janssen, H.; Zwart, W.; Neefjes, J. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J. Cell Biol. 2009, 185, 1209–1225. [Google Scholar] [CrossRef]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Van der Kant, R.; Fish, A.; Janssen, L.; Janssen, H.; Krom, S.; Ho, N.; Brummelkamp, T.; Carette, J.; Rocha, N.; Neefjes, J. Late endosomal transport and tethering are coupled processes controlled by RILP and the cholesterol sensor ORP1L. J. Cell Sci. 2013, 126 Pt 15, 3462–3474. [Google Scholar] [CrossRef]
- Carreira, A.C.; Pokorna, S.; Ventura, A.E.; Walker, M.W.; Futerman, A.H.; Lloyd-Evans, E.; de Almeida, R.F.M.; Silva, L.C. Biophysical impact of sphingosine and other abnormal lipid accumulation in Niemann-Pick disease type C cell models. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158944. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Fiorio Pla, A.; Lim, D.; Lodola, F.; Gerbino, A. Intracellular Ca2+ signalling: Unexpected new roles for the usual suspect. Front. Physiol. 2023, 14, 1210085. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Gao, Q.; Yang, M.; Zhang, X.; Yu, L.; Lawas, M.; Li, X.; Bryant-Genevier, M.; Southall, N.T.; Marugan, J.; et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc. Natl. Acad. Sci. USA 2015, 112, E1373–E1381. [Google Scholar] [CrossRef] [PubMed]
- Kosicek, M.; Gudelj, I.; Horvatic, A.; Jovic, T.; Vuckovic, F.; Lauc, G.; Hecimovic, S. N-glycome of the Lysosomal Glycocalyx is Altered in Niemann-Pick Type C Disease (NPC) Model Cells. Mol. Cell. Proteom. 2018, 17, 631–642. [Google Scholar] [CrossRef]
- Schultz, M.L.; Krus, K.L.; Kaushik, S.; Dang, D.; Chopra, R.; Qi, L.; Shakkottai, V.G.; Cuervo, A.M.; Lieberman, A.P. Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat. Commun. 2018, 9, 3671. [Google Scholar] [CrossRef] [PubMed]
- Willenborg, M.; Schmidt, C.K.; Braun, P.; Landgrebe, J.; von Figura, K.; Saftig, P.; Eskelinen, E.L. Mannose 6-phosphate receptors, Niemann-Pick C2 protein, and lysosomal cholesterol accumulation. J. Lipid Res. 2005, 46, 2559–2569. [Google Scholar] [CrossRef]
- Brodsky, J.L. Cleaning up: ER-associated degradation to the rescue. Cell 2012, 151, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Shioi, R.; Karaki, F.; Yoshioka, H.; Noguchi-Yachide, T.; Ishikawa, M.; Dodo, K.; Hashimoto, Y.; Sodeoka, M.; Ohgane, K. Image-based screen capturing misfolding status of Niemann-Pick type C1 identifies potential candidates for chaperone drugs. PLoS ONE 2020, 15, e0243746. [Google Scholar] [CrossRef]
- Mohamed, F.E.; Al-Gazali, L.; Al-Jasmi, F.; Ali, B.R. Pharmaceutical Chaperones and Proteostasis Regulators in the Therapy of Lysosomal Storage Disorders: Current Perspective and Future Promises. Front. Pharmacol. 2017, 8, 448. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD®): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Ingemann, L.; Kirkegaard, T. Lysosomal storage diseases and the heat shock response: Convergences and therapeutic opportunities. J. Lipid Res. 2014, 55, 2198–2210. [Google Scholar] [CrossRef] [PubMed]
- Jäättelä, M. Heat shock proteins as cellular lifeguards. Ann. Med. 1999, 31, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nature reviews. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- Hargitai, J.; Lewis, H.; Boros, I.; Rácz, T.; Fiser, A.; Kurucz, I.; Benjamin, I.; Vígh, L.; Pénzes, Z.; Csermely, P.; et al. Bimoclomol, a heat shock protein co-inducer, acts by the prolonged activation of heat shock factor-1. Biochem. Biophys. Res. Commun. 2003, 307, 689–695. [Google Scholar] [CrossRef]
- Gray, J.; Fernández-Suárez, M.E.; Falah, M.; Smith, D.; Smith, C.; Kaya, E.; Palmer, A.M.; Fog, C.K.; Kirkegaard, T.; Platt, F.M. Heat shock protein amplification improves cerebellar myelination in the Npc1nih mouse model. EBioMedicine 2022, 86, 104374. [Google Scholar] [CrossRef]
- Gonçalves, C.C.; Sharon, I.; Schmeing, T.M.; Ramos CH, I.; Young, J.C. The chaperone HSPB1 prepares protein aggregates for resolubilization by HSP70. Sci. Rep. 2021, 11, 17139. [Google Scholar] [CrossRef]
- Baughman, H.E.R.; Clouser, A.F.; Klevit, R.E.; Nath, A. HspB1 and Hsc70 chaperones engage distinct tau species and have different inhibitory effects on amyloid formation. J. Biol. Chem. 2018, 293, 2687–2700. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Elrick, M.J.; Dell’Orco, J.M.; Qin, Z.S.; Kalyana-Sundaram, S.; Chinnaiyan, A.M.; Shakkottai, V.G.; Lieberman, A.P. Heat Shock Protein Beta-1 Modifies Anterior to Posterior Purkinje Cell Vulnerability in a Mouse Model of Niemann-Pick Type C Disease. PLoS Genet. 2016, 12, e1006042. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, C.D.; Kunkel, R.; Lieberman, A.P. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum. Mol. Genet. 2007, 16, 1495–1503. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H. Regulation of Autophagy by mTOR Signaling Pathway. Adv. Exp. Med. Biol. 2019, 1206, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.A.; Sykes, A.M.; Mellick, G.D. ER-phagy in neurodegeneration. J. Neurosci. Res. 2023, 101, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, K.; Blessinger, S.; Bailey, N.T.; Scaife, R.; Liu, G.; Khambu, B. Therapeutic regulation of autophagy in hepatic metabolism. Yao Xue Xue Bao 2022, 12, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.C.; Milenkovic, L.; Beier, S.M.; Manuel, H.; Buchanan, J.; Scott, M.P. Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet. 2005, 1, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.D.; Qin, Z.H. Beclin 1, Bcl-2 and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 109–126. [Google Scholar] [CrossRef]
- Sarkar, S.; Carroll, B.; Buganim, Y.; Maetzel, D.; Ng, A.H.; Cassady, J.P.; Cohen, M.A.; Chakraborty, S.; Wang, H.; Spooner, E.; et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep. 2013, 5, 1302–1315. [Google Scholar] [CrossRef]
- Bernales, S.; Schuck, S.; Walter, P. ER-phagy: Selective autophagy of the endoplasmic reticulum. Autophagy 2007, 3, 285–287. [Google Scholar] [CrossRef]
- Lipatova, Z.; Gyurkovska, V.; Zhao, S.F.; Segev, N. Characterization of constitutive ER-phagy of excess membrane proteins. PLoS Genet. 2020, 16, e1009255. [Google Scholar] [CrossRef]
- Reggiori, F.; Molinari, M. ER-phagy: Mechanisms, regulation, and diseases connected to the lysosomal clearance of the endoplasmic reticulum. Physiol. Rev. 2022, 102, 1393–1448. [Google Scholar] [CrossRef]
- Forrester, A.; De Leonibus, C.; Grumati, P.; Fasana, E.; Piemontese, M.; Staiano, L.; Fregno, I.; Raimondi, A.; Marazza, A.; Bruno, G.; et al. A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J. 2019, 38, e99847. [Google Scholar] [CrossRef] [PubMed]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Falcão de Campos, C.; Vidailhet, M.; Toutain, A.; de Becdelièvre, A.; Funalot, B.; Bonello-Palot, N.; Stojkovic, T. Hereditary sensory autonomic neuropathy type II: Report of two novel mutations in the FAM134B gene. J. Peripher. Nerv. Syst. 2019, 24, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Kurth, I. Hereditary Sensory and Autonomic Neuropathy Type II. In GeneReviews®; Adam, M.P., Fedman, J., Mirzaa, G.M., Pagon, R.A., Wallace, E.E., Bean, L.J., Gripp, W.K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2010. [Google Scholar]
- Kurth, I.; Pamminger, T.; Hennings, J.C.; Soehendra, D.; Huebner, A.K.; Rotthier, A.; Baets, J.; Senderek, J.; Topaloglu, H.; Farrell, S.A.; et al. Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat. Genet. 2009, 41, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; He, W.; Wang, K.; Han, H.; Xiao, T.; Chen, X.; Zhou, B.; Tan, J.; Xia, K.; Tang, B.; et al. Identification of rare RTN3 variants in Alzheimer’s disease in Han Chinese. Hum. Genet. 2018, 137, 141–150. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Zoncu, R. The Lysosome as a Regulatory Hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253. [Google Scholar] [CrossRef]
- Yang, C.; Wang, X. Lysosome biogenesis: Regulation and functions. J. Cell Biol. 2021, 220, e202102001. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nature reviews. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.B.; Wastney, M.; Patel, S.; Suresh, S.; Cooney, A.M.; Dwyer, N.K.; Roff, C.F.; Ohno, K.; Morris, J.A.; Carstea, E.D.; et al. The Niemann-Pick C1 protein resides in a vesicular compartment linked to retrograde transport of multiple lysosomal cargo. J. Biol. Chem. 1999, 274, 9627–9635. [Google Scholar] [CrossRef] [PubMed]
- Bananis, E.; Nath, S.; Gordon, K.; Satir, P.; Stockert, R.J.; Murray, J.W.; Wolkoff, A.W. Microtubule-dependent movement of late endocytic vesicles in vitro: Requirements for Dynein and Kinesin. Mol. Biol. Cell 2004, 15, 3688–3697. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, J.; Low, P.S.; Cheng, J.X. Cholesterol level regulates endosome motility via Rab proteins. Biophys. J. 2008, 94, 1508–1520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Dwyer, N.K.; Love, D.C.; Cooney, A.; Comly, M.; Neufeld, E.; Pentchev, P.G.; Blanchette-Mackie, E.J.; Hanover, J.A. Cessation of rapid late endosomal tubulovesicular trafficking in Niemann-Pick type C1 disease. Proc. Natl. Acad. Sci. USA 2001, 98, 4466–4471. [Google Scholar] [CrossRef] [PubMed]
- Cabeza, C.; Figueroa, A.; Lazo, O.M.; Galleguillos, C.; Pissani, C.; Klein, A.; Gonzalez-Billault, C.; Inestrosa, N.C.; Alvarez, A.R.; Zanlungo, S.; et al. Cholinergic abnormalities, endosomal alterations and up-regulation of nerve growth factor signaling in Niemann-Pick type C disease. Mol. Neurodegener. 2012, 7, 11. [Google Scholar] [CrossRef]
- Mbua, N.E.; Flanagan-Steet, H.; Johnson, S.; Wolfert, M.A.; Boons, G.J.; Steet, R. Abnormal accumulation and recycling of glycoproteins visualized in Niemann-Pick type C cells using the chemical reporter strategy. Proc. Natl. Acad. Sci. USA 2013, 110, 10207–10212. [Google Scholar] [CrossRef]
- Jahn, R.; Scheller, R.H. SNAREs—Engines for membrane fusion. Nature reviews. Mol. Cell Biol. 2006, 7, 631–643. [Google Scholar] [CrossRef]
- Enrich, C.; Rentero, C.; Hierro, A.; Grewal, T. Role of cholesterol in SNARE-mediated trafficking on intracellular membranes. J. Cell Sci. 2015, 128, 1071–1081. [Google Scholar] [CrossRef]
- Cougnoux, A.; Pergande, M.R.; Serna-Perez, F.; Cologna, S.M. Investigation of 2-Hydroxypropyl-β-Cyclodextrin Treatment in a Neuronal-Like Cell Model of Niemann-Pick Type C Using Quantitative Proteomics. J. Am. Soc. Mass Spectrom. 2023, 34, 668–675. [Google Scholar] [CrossRef]
- Megías-Vericat, J.E.; García-Robles, A.; Company-Albir, M.J.; Fernández-Megía, M.J.; Pérez-Miralles, F.C.; López-Briz, E.; Casanova, B.; Poveda, J.L. Early experience with compassionate use of 2 hydroxypropyl-beta-cyclodextrin for Niemann-Pick type C disease: Review of initial published cases. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2017, 38, 727–743. [Google Scholar] [CrossRef] [PubMed]
- Calias, P. 2-Hydroxypropyl-β-cyclodextrins and the Blood-Brain Barrier: Considerations for Niemann-Pick Disease Type C1. Curr. Pharm. Des. 2017, 23, 6231–6238. [Google Scholar] [CrossRef]
- Goldin, E.; Roff, C.F.; Miller, S.P.; Rodriguez-Lafrasse, C.; Vanier, M.T.; Brady, R.O.; Pentchev, P.G. Type C Niemann-Pick disease: A murine model of the lysosomal cholesterol lipidosis accumulates sphingosine and sphinganine in liver. Biochim. Biophys. Acta 1992, 1127, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Cuvillier, O. Sphingosine in apoptosis signaling. Biochim. Biophys. Acta 2002, 1585, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, J. Sphingosine and ceramide signalling in apoptosis. IUBMB Life 2006, 58, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.; Palladino EN, D.; Weigel, C.; Maceyka, M.; Gräler, M.H.; Senkal, C.E.; Enriz, R.D.; Marvanova, P.; Jampilek, J.; Lima, S.; et al. Targeting defective sphingosine kinase 1 in Niemann-Pick type C disease with an activator mitigates cholesterol accumulation. J. Biol. Chem. 2020, 295, 9121–9133. [Google Scholar] [CrossRef]
- Cawley, N.X.; Giddens, S.; Farhat, N.M.; Luke, R.A.; Scott KE, J.; Mohamed, H.O.; Dang Do, A.; Berry-Kravis, E.; Cologna, S.M.; Liu, F.; et al. Elevated cerebrospinal fluid ubiquitin C-terminal hydrolase-L1 levels correlate with phenotypic severity and therapeutic response in Niemann-Pick disease, type C1. Mol. Genet. Metab. 2023, 140, 107656, Advance online publication. [Google Scholar] [CrossRef]
- Cawley, N.X.; Lyons, A.T.; Abebe, D.; Luke, R.; Yerger, J.; Telese, R.; Wassif, C.A.; Bailey-Wilson, J.E.; Porter, F.D. Complex N-Linked Glycosylation: A Potential Modifier of Niemann-Pick Disease, Type C1 Pathology. Int. J. Mol. Sci. 2022, 23, 5082. [Google Scholar] [CrossRef]
- Medina, D.L. Lysosomal calcium and autophagy. Int. Rev. Cell Mol. Biol. 2021, 362, 141–170. [Google Scholar] [CrossRef]
- Pozzan, T.; Rizzuto, R.; Volpe, P.; Meldolesi, J. Molecular and cellular physiology of intracellular calcium stores. Physiol. Rev. 1994, 74, 595–636. [Google Scholar] [CrossRef]
- Bojarski, L.; Herms, J.; Kuznicki, J. Calcium dysregulation in Alzheimer’s disease. Neurochem. Int. 2008, 52, 621–633. [Google Scholar] [CrossRef]
- Liu, E.A.; Lieberman, A.P. The intersection of lysosomal and endoplasmic reticulum calcium with autophagy defects in lysosomal diseases. Neurosci. Lett. 2019, 697, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Evans, E.; Platt, F.M. Lysosomal Ca (2+) homeostasis: Role in pathogenesis of lysosomal storage diseases. Cell Calcium 2011, 50, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Wang, X.; Li, X.; Zhang, X.; Yao, Z.; Dibble, S.; Dong, X.P.; Yu, T.; Lieberman, A.P.; Showalter, H.D.; et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012, 3, 731. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Lesimple, A.; Denis, M.; Vincent, J.; Larsen, A.; Mamer, O.; Krimbou, L.; Genest, J.; Marcil, M. Increased sphingomyelin content impairs HDL biogenesis and maturation in human Niemann-Pick disease type B. J. Lipid Res. 2006, 47, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Levran, O.; Desnick, R.J.; Schuchman, E.H. Niemann-Pick disease: A frequent missense mutation in the acid sphingomyelinase gene of Ashkenazi Jewish type A and B patients. Proc. Natl. Acad. Sci. USA 1991, 88, 3748–3752. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, Y.; Hao, X.; Lin, W.; Su, W.; Chiang, E.; Baudry, M.; Bi, X. LAMTOR1 inhibition of TRPML1-dependent lysosomal calcium release regulates dendritic lysosome trafficking and hippocampal neuronal function. EMBO J. 2022, 41, e108119. [Google Scholar] [CrossRef]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Cascella, R.; Fani, G.; Capitini, C.; Rusmini, P.; Poletti, A.; Cecchi, C.; Chiti, F. Quantitative assessment of the degradation of aggregated TDP-43 mediated by the ubiquitin proteasome system and macroautophagy. FASEBJ. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 5609–5624. [Google Scholar] [CrossRef]
- Lee, M.J.; Lee, J.H.; Rubinsztein, D.C. Tau degradation: The ubiquitin-proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 2013, 105, 49–59. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. α-Synuclein and protein degradation systems: A reciprocal relationship. Mol. Neurobiol. 2013, 47, 537–551. [Google Scholar] [CrossRef]
- Mi, Z.; Graham, S.H. Role of UCHL1 in the pathogenesis of neurodegenerative diseases and brain injury. Ageing Res. Rev. 2023, 86, 101856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Cai, F.; Zhang, S.; Zhang, S.; Song, W. Overexpression of ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) delays Alzheimer’s progression in vivo. Sci. Rep. 2014, 4, 7298. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, S.; Bembi, B.; Rosso, N.; Filocamo, M.; Dardis, A. Treatment of Human Fibroblasts Carrying NPC1 Missense Mutations with MG132 Leads to an Improvement of Intracellular Cholesterol Trafficking. JIMD Rep. 2012, 2, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Macías-Vidal, J.; Girós, M.; Guerrero, M.; Gascón, P.; Serratosa, J.; Bachs, O.; Coll, M.J. The proteasome inhibitor bortezomib reduced cholesterol accumulation in fibroblasts from Niemann-Pick type C patients carrying missense mutations. FEBS J. 2014, 281, 4450–4466. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, K.; Hutt, D.M.; Scott, S.M.; Gupta, V.; Mao, S.; Balch, W.E. Correction of Niemann-Pick type C1 trafficking and activity with the histone deacetylase inhibitor valproic acid. J. Biol. Chem. 2020, 295, 8017–8035. [Google Scholar] [CrossRef] [PubMed]
- Madison, B.B. Srebp2: A master regulator of sterol and fatty acid synthesis. J. Lipid Res. 2016, 57, 333–335. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.R.; St Louis, K.J.; Tradewell, M.L.; Gentil, B.J.; Minotti, S.; Jaffer, Z.M.; Chen, R.; Rubenstein, A.E.; Durham, H.D. A novel small molecule HSP90 inhibitor, NXD30001, differentially induces heat shock proteins in nervous tissue in culture and in vivo. Cell Stress Chaperones 2014, 19, 421–435. [Google Scholar] [CrossRef]
- Tan, C.R.C.; Abdul-Majeed, S.; Cael, B.; Barta, S.K. Clinical Pharmacokinetics and Pharmacodynamics of Bortezomib. Clin. Pharmacokinet. 2019, 58, 157–168. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Mechanism of Proteostasis in Which It Is Involved | Therapy or Therapeutic Target | Mechanism of Action | Effects | Considerations | Ref. |
|---|---|---|---|---|---|
| Synthesis | Valproic acid (VPA) and chloroquine | VPA reduces HDAC7 expression and increases NPC1 acetylation, and chloroquine neutralizes lysosomal hyperacidity. | Promotes folding and transport of NPC1-I1061T to LE/Ly, reduces cholesterol accumulation in NPC1-I1061T fibroblasts. | Valproic acid can cross the blood–brain barrier and can be administered orally. Treatment with VPA can alter the pH of the lysosome, so that combined treatment with chloroquine stabilizes the pH and has better effects. | [108] |
| Glycosylation | Itraconazole | Inhibits protein glycosylation; highly glycosylated proteins coat the lysosomal inner membrane, so the inhibition of glycosylation may cause increased permeability of the lysosomal glycocalyx, which increases the release of cholesterol into the lysosomes. | At high concentrations (10 μM), it significantly reduces cholesterol accumulation. | It has a slight effect on reducing cholesterol accumulation at low concentrations, possibly because it also inhibits the glycosylation of the NPC1 protein, which affects its function. | [39] |
| Folding | Abiraterone acetate | Interacts with NTD of NPC1 to mediate its folding. | Reduction in cholesterol concentration; upregulation and co-localization in lysosomes of NPC1 resistant to EndoH in three different mutations NPC1 E612D/P543Rfs*, I1061T, NPC1 Y394H/Y394H; these results show it as an effective treatment. | It is a drug approved by the FDA and the EMA for prostate cancer; its active metabolite is abiraterone; in animal models, it has been observed that it crosses the blood–brain barrier. | [21] |
| DHBP | Inhibits ryanodine receptors and increases calcium levels in the ER; promotes the folding of NPC1 by calcium-dependent chaperones. | Increases levels of EndoH-resistant NPC1 I1061T, promotes the trafficking of NPC1 I1061T to lysosomes, and regulates the accumulation of cholesterol and sphingolipids. | With this treatment, it was observed that only the trafficking of a small fraction of NPC1 I1061T improved, but the reduction in cholesterol was significant. | [22] | |
| Calnexine | Mediates NPC1 I1061T folding. | Enhances trafficking of NPC1 I1061T to lysosomes and regulates the accumulation of cholesterol and sphingolipids. | Despite only increasing the transport of a low level of NPC1 I1061T NPC1, the reduction in cholesterol was significant. | [22] | |
| Arimoclomol | Co-induces heat shock proteins by stabilizing their interaction between heat shock factor (HSF-1) and HSES, which mediate the transcription of HSPs. | The treatment had significant effects on reducing the progression of NPC; increased HSP70 expression and reduced lipid accumulation. | Safety studies in patients with CPN show that the most common adverse event was vomiting (23.5%), and six of the patients presented increased serum creatinine. | [24,25] | |
| Recombinant human heat shock protein 70 (rhHSP70) | Amplification of HSP70 and therefore enhancement of sphingolipid-degrading enzymes. | Normalizes the oligodendrocyte population and rescues it from cerebellar atrophy; improved myelination in cerebellum; reduced GSL accumulation and improved NPC phenotype in a murine model. | In the study, treatment with rhHSP70 showed an improvement in myelination in the cerebellum; however, it is not known if the treatment is effective for increasing Npc1 levels and transport or if it is able to reduce cholesterol accumulation. | [46] | |
| Recombinant HSP70 | Enhance NPC1 folding and enhancement of sphingolipid-degrading enzymes. | Reduced GM1 ganglioside accumulation; improved the motor phenotype in a murine model. | This treatment is not effective in knockout models, only in nonsense mutations. | [24] | |
| JG98 inhibitor | Disrupts the interaction between Hsp70 and BAG-1 and -3, which mediate the degradation of Hsp70 by the ubiquitin–proteasome system or the lysosomal autophagic pathway. | Correction in the trafficking of 58 NPC1 variants and reduction in the activation of SREBP-2, a transcription factor that regulates the expression of genes that participate in the synthesis of fatty acids. | This treatment showed significant responses even in variants with null or reduced activity of NPC1 transport and function; however, one of the disadvantages is that it is a fluorescent molecule, so that monitoring of cholesterol accumulation by filipin staining is not feasible. | [27,109] | |
| AUY922 | Inhibition of HSP90, which when inactive mediates the release of HSF1 factor and promotes the expression of HSPs. | Overexpression of HSP70 and HSP40, increased transport of NPC1 from ER to LE/Ly and decreased cholesterol concentration. | The treatment with this drug for NPC has only been performed in vitro, but in the treatment of solid tumors it has been observed that the main adverse event is fatigue and decreased appetite. | [26,110] | |
| Overexpression of hspb1 | Phosphorylated Hspb1 provides a neuroprotective effect by having anti-apoptotic functions. | In mice, there was delayed motor impairment and decreased Purkinje cell loss. In cells, promotes neuronal survival. | Hspb1 as a therapeutic target was only proven to be effective in reducing Purkinje cell loss in a murine model, but it was not evaluated whether there was a reduction in the accumulation of cholesterol or other fatty acids, so its effectiveness in this aspect cannot be determined. | [49] | |
| Lysosomal pathway | Overexpression of TRPML1 receptor | Increasing its expression improves lysosomal calcium release, which is favorable for the fusion of LE and Ly and Ly with autophagosomes. | Enhances lysosomal trafficking and rescues it from cholesterol storage. | This treatment offers another approach that does not act directly on NPC1 or NPC2 proteins but improves lysosomal trafficking; also decreases cholesterol concentrations. | [96] |
| Degradation | Bortezomib treatment | Inhibits the 26s proteasome by inactivation of the chymotrypsin-like site in the proteolytic nucleus. | Partially increases NPC1 levels and reduces cholesterol content. | This treatment is not completely effective because it does not increase the co-localization of NPC1 to lysosomes despite showing a reduction in cholesterol accumulation. | [107,111] |
| MG132 | Inhibits the 26s proteasome | Increased NPC1 protein expression and co-localization within the endolysosomal compartment, reduction of GM1 accumulation, reduced cholesterol concentrations. | According to the study, it is not a safe treatment; at doses higher than 500 nM, there is reduction in cell viability; this treatment was shown to be effective in increasing NPC1 protein levels in fibroblast cell lines of six mutant variants. | [106] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Servín Muñoz, I.V.; Ortuño-Sahagún, D.; Griñán-Ferré, C.; Pallàs, M.; González-Castillo, C. Alterations in Proteostasis Mechanisms in Niemann–Pick Type C Disease. Int. J. Mol. Sci. 2024, 25, 3806. https://doi.org/10.3390/ijms25073806
Servín Muñoz IV, Ortuño-Sahagún D, Griñán-Ferré C, Pallàs M, González-Castillo C. Alterations in Proteostasis Mechanisms in Niemann–Pick Type C Disease. International Journal of Molecular Sciences. 2024; 25(7):3806. https://doi.org/10.3390/ijms25073806
Chicago/Turabian StyleServín Muñoz, Iris Valeria, Daniel Ortuño-Sahagún, Christian Griñán-Ferré, Mercè Pallàs, and Celia González-Castillo. 2024. "Alterations in Proteostasis Mechanisms in Niemann–Pick Type C Disease" International Journal of Molecular Sciences 25, no. 7: 3806. https://doi.org/10.3390/ijms25073806
APA StyleServín Muñoz, I. V., Ortuño-Sahagún, D., Griñán-Ferré, C., Pallàs, M., & González-Castillo, C. (2024). Alterations in Proteostasis Mechanisms in Niemann–Pick Type C Disease. International Journal of Molecular Sciences, 25(7), 3806. https://doi.org/10.3390/ijms25073806