
Targeting Interleukin-13 Receptor α2 and EphA2 in Aggressive Breast Cancer Subtypes with Special References to Chimeric Antigen Receptor T-Cell Therapy


Abstract
1. Introduction
2. Structure and Function of IL-13Rα2 and EphA2
3. IL-13Rα2 and EphA2 in Her-2-Enriched and TNBC Pathology
4. Overcoming Therapeutic Hurdles: The Quest to Tame Aggressive Her-2-Positive and TNBC Breast Cancers
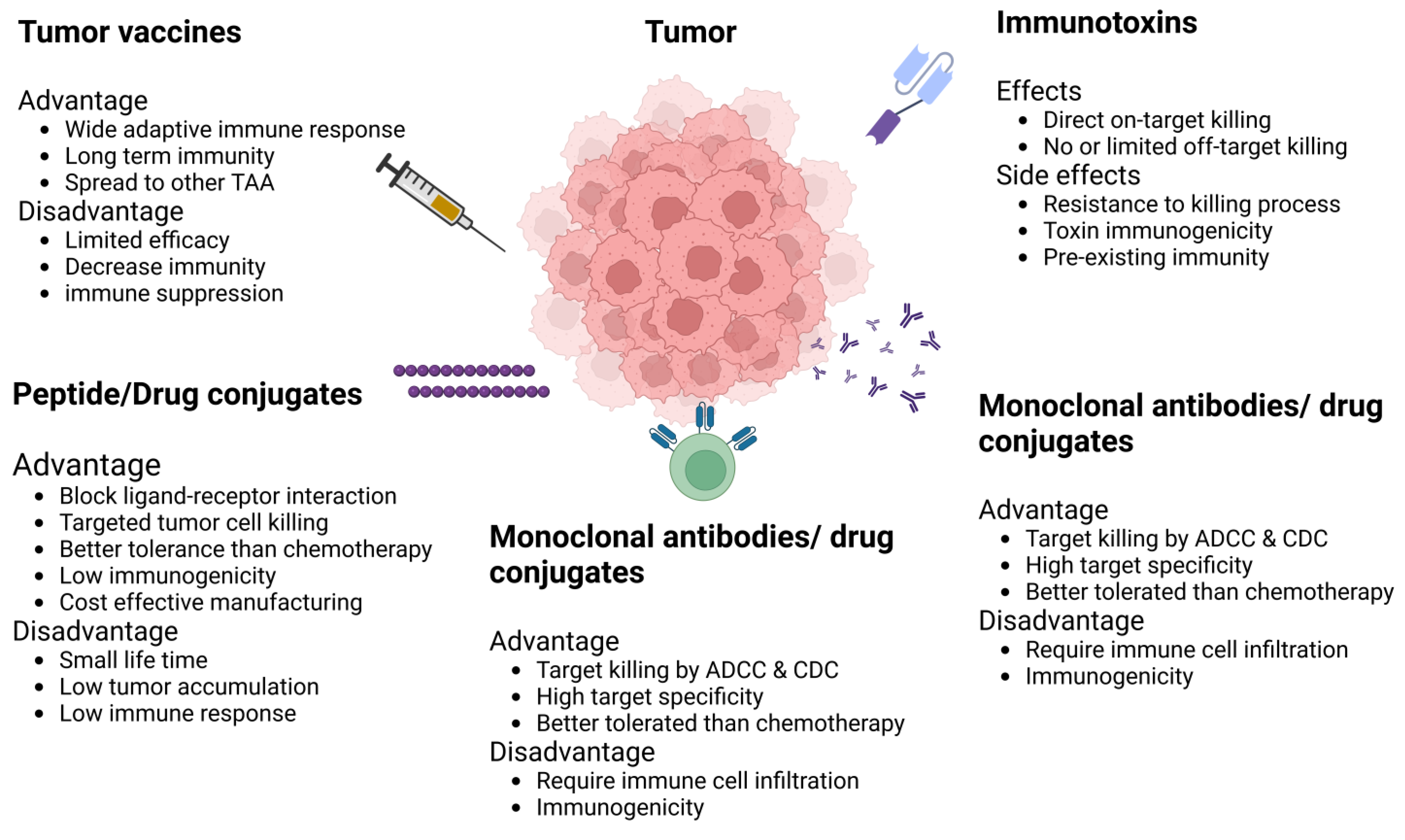
5. Immunotherapy for BCA
6. IL-13R Alpha-2 and EphA2 as Therapeutic Targets
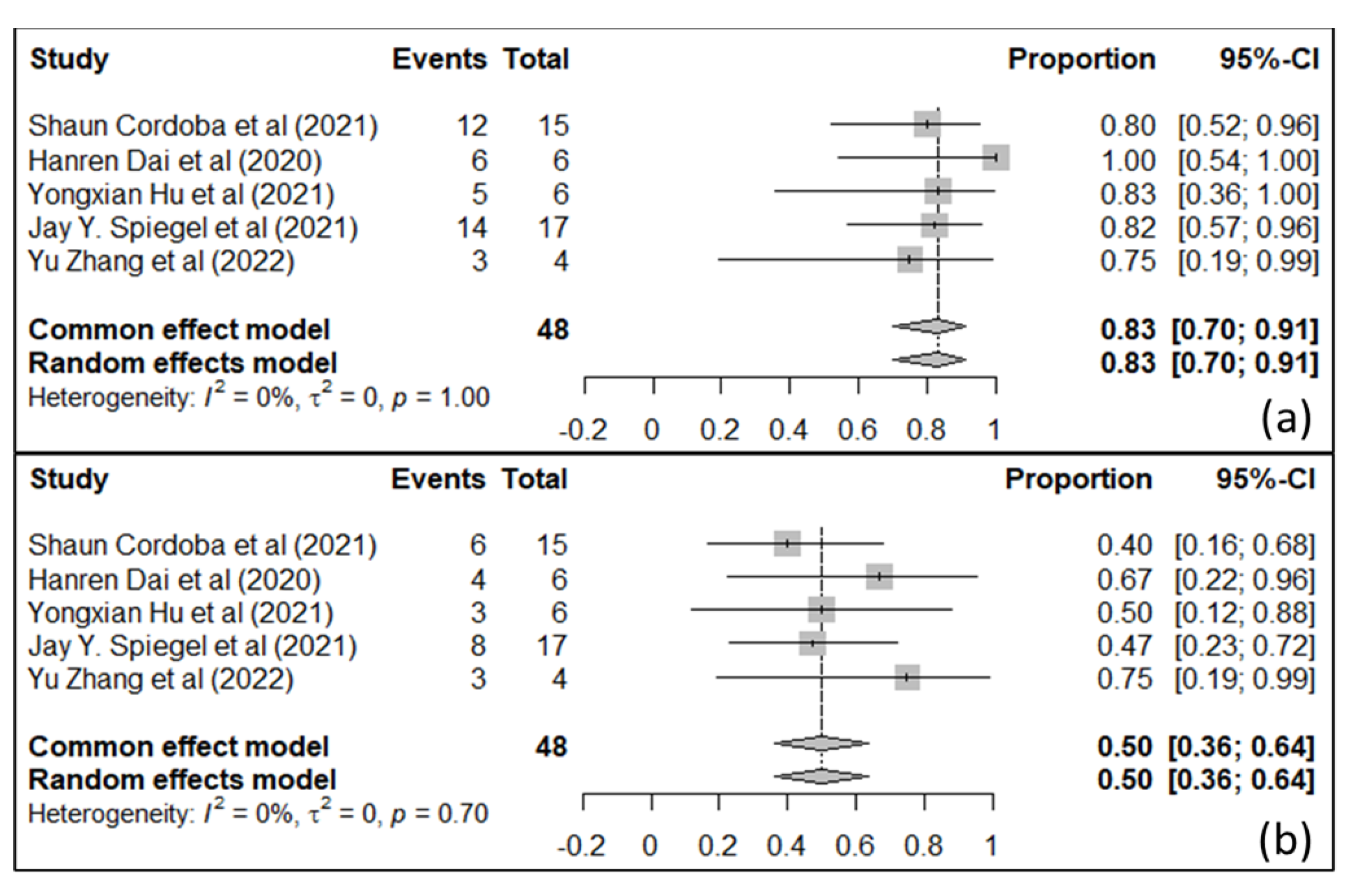
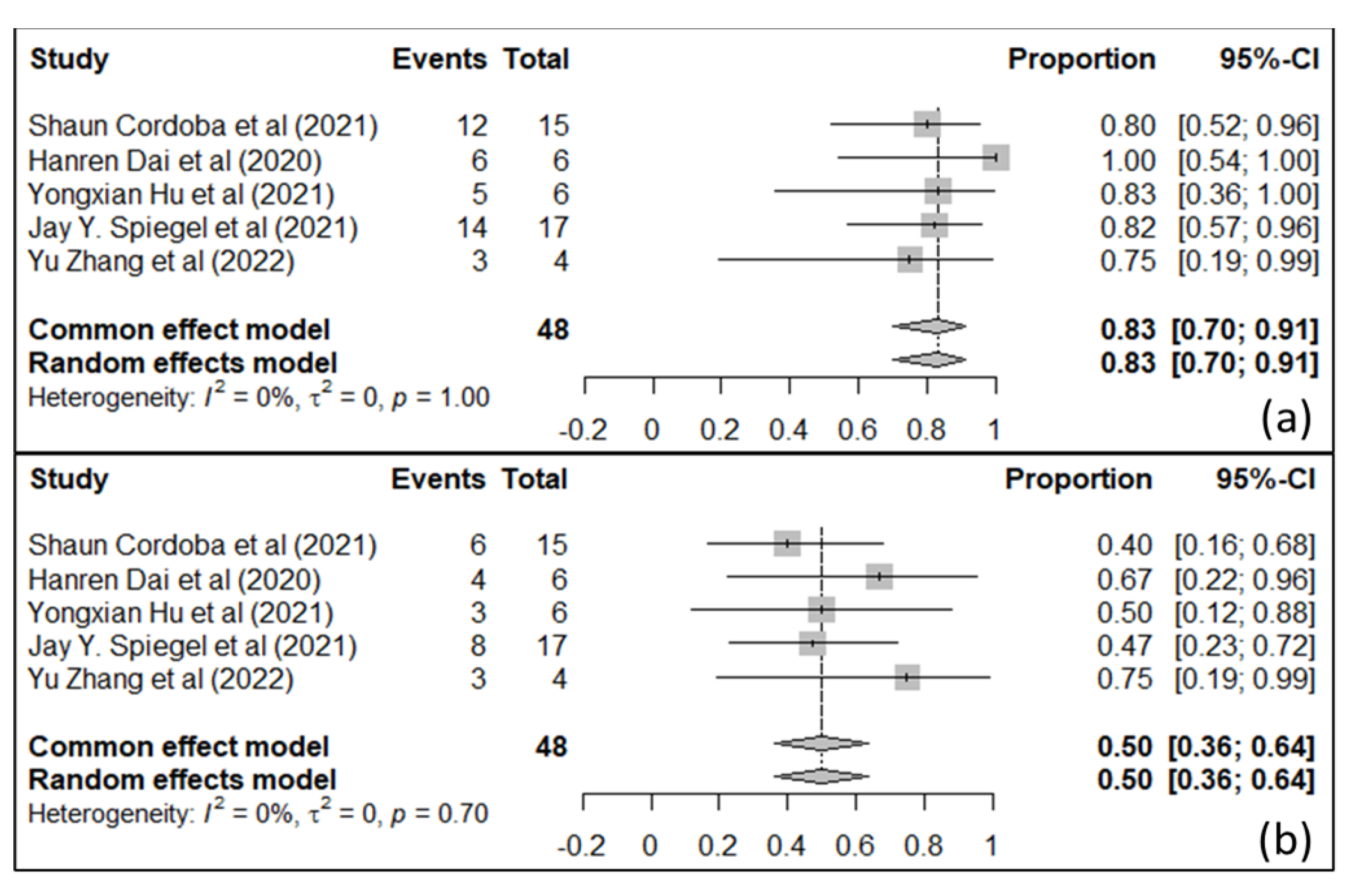
7. IL-13Rα-2 and EphA2 as CAR T-Cell Targets
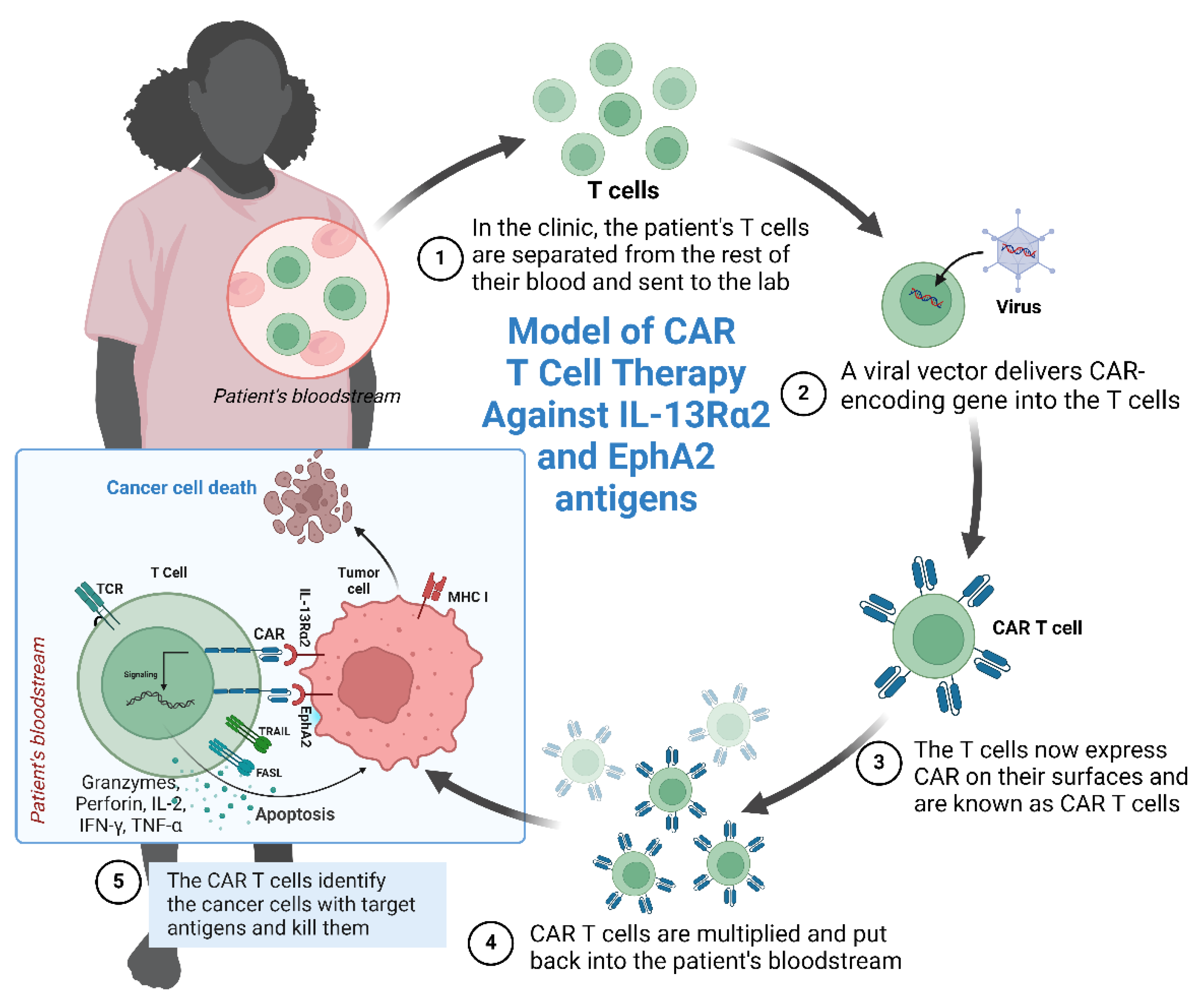
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Lainetti, P.F.; Leis-Filho, A.F.; Laufer-Amorim, R.; Battazza, A.; Fonseca-Alves, C.E. Mechanisms of Resistance to Chemotherapy in Breast Cancer and Possible Targets in Drug Delivery Systems. Pharmaceutics 2020, 12, 1193. [Google Scholar] [CrossRef]
- Marquez-Ortiz, R.A.; Contreras-Zarate, M.J.; Tesic, V.; Alvarez-Eraso, K.L.F.; Kwak, G.; Littrell, Z.; Costello, J.C.; Sreekanth, V.; Ormond, D.R.; Karam, S.D.; et al. IL13Ralpha2 Promotes Proliferation and Outgrowth of Breast Cancer Brain Metastases. Clin. Cancer Res. 2021, 27, 6209–6221. [Google Scholar] [CrossRef]
- Zhou, L.; Lu, X.; Zhang, B.; Shi, Y.; Li, Z. EphA2 as a new target for breast cancer and its potential clinical application. Int. J. Clin. Exp. Pathol. 2021, 14, 484–492. [Google Scholar]
- Chavent, M.; Seiradake, E.; Jones, E.Y.; Sansom, M.S. Structures of the EphA2 Receptor at the Membrane: Role of Lipid Interactions. Structure 2016, 24, 337–347. [Google Scholar] [CrossRef]
- Singh, D.R.; Kanvinde, P.; King, C.; Pasquale, E.B.; Hristova, K. The EphA2 receptor is activated through induction of distinct, ligand-dependent oligomeric structures. Commun. Biol. 2018, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Son, A.I.; Zhou, R. Roles of EphA2 in Development and Disease. Genes 2013, 4, 334–357. [Google Scholar] [CrossRef]
- Wilson, K.; Shiuan, E.; Brantley-Sieders, D.M. Oncogenic functions and therapeutic targeting of EphA2 in cancer. Oncogene 2021, 40, 2483–2495. [Google Scholar] [CrossRef] [PubMed]
- Vaught, D.B.; Merkel, A.R.; Lynch, C.C.; Edwards, J.; Tantawy, M.N.; Hilliard, T.; Wang, S.; Peterson, T.; Johnson, R.W.; Sterling, J.A.; et al. EphA2 Is a Clinically Relevant Target for Breast Cancer Bone Metastatic Disease. JBMR Plus 2021, 5, e10465. [Google Scholar] [CrossRef]
- Zhao, P.; Jiang, D.; Huang, Y.; Chen, C. EphA2: A promising therapeutic target in breast cancer. J. Genet. Genom. 2021, 48, 261–267. [Google Scholar] [CrossRef]
- Kumar, A.; Bellayr, I.H.; Singh, H.S.; Puri, R.K. IL-13Ralpha2 gene expression is a biomarker of adverse outcome in patients with adrenocortical carcinoma. PLoS ONE 2021, 16, e0246632. [Google Scholar] [CrossRef]
- Liang, R.; Wu, C.; Liu, S.; Zhao, W. Targeting interleukin-13 receptor alpha2 (IL-13Ralpha2) for glioblastoma therapy with surface functionalized nanocarriers. Drug Deliv. 2022, 29, 1620–1630. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, L.; Xu, W. IL-13Ralpha2 mediates PNR-induced migration and metastasis in ERalpha-negative breast cancer. Oncogene 2015, 34, 1596–1607. [Google Scholar] [CrossRef] [PubMed]
- Popovic, B.; Breed, J.; Rees, D.G.; Gardener, M.J.; Vinall, L.M.; Kemp, B.; Spooner, J.; Keen, J.; Minter, R.; Uddin, F.; et al. Structural Characterisation Reveals Mechanism of IL-13-Neutralising Monoclonal Antibody Tralokinumab as Inhibition of Binding to IL-13Ralpha1 and IL-13Ralpha2. J. Mol. Biol. 2017, 429, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, R.; Suzuki, A.; Leland, P.; Joshi, B.H.; Puri, R.K. Identification of a novel role of IL-13Ralpha2 in human Glioblastoma multiforme: Interleukin-13 mediates signal transduction through AP-1 pathway. J. Transl. Med. 2018, 16, 369. [Google Scholar] [CrossRef] [PubMed]
- Jannoo, R.; Xia, Z.; Row, P.E.; Kanamarlapudi, V. Targeting of the Interleukin-13 Receptor (IL-13R)alpha2 Expressing Prostate Cancer by a Novel Hybrid Lytic Peptide. Biomolecules 2023, 13, 356. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Wu, X.J.; Zhang, J.J.; He, C.S. IL-13 receptor alpha2 is a negative prognostic factor in human lung cancer and stimulates lung cancer growth in mice. Oncotarget 2015, 6, 32902–32913. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Joshi, B.H.; Puri, R.K. IL-13 regulates cancer invasion and metastasis through IL-13Ralpha2 via ERK/AP-1 pathway in mouse model of human ovarian cancer. Int. J. Cancer 2012, 131, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Joshi, B.H.; Takahashi, S.; Takasaki, Y.; Suzuki, A.; Ito, K.; Ochiai, K.; Tomishima, K.; Ishii, S.; Puri, R.K.; et al. IL-13Ralpha2 Is a Biomarker of Diagnosis and Therapeutic Response in Human Pancreatic Cancer. Diagnostics 2021, 11, 1140. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Liu, H.; Zhang, H.; He, H.; Li, H.; Shen, Z.; Qin, J.; Qin, X.; Xu, J.; Sun, Y. Interleukin-13 receptor alpha2 is associated with poor prognosis in patients with gastric cancer after gastrectomy. Oncotarget 2016, 7, 49281–49288. [Google Scholar] [CrossRef]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef] [PubMed]
- Sahu, M.; Suryawanshi, H. Immunotherapy: The future of cancer treatment. J. Oral Maxillofac. Pathol. 2021, 25, 371. [Google Scholar] [CrossRef]
- Jogalekar, M.P.; Rajendran, R.L.; Khan, F.; Dmello, C.; Gangadaran, P.; Ahn, B.C. CAR T-Cell-Based gene therapy for cancers: New perspectives, challenges, and clinical developments. Front. Immunol. 2022, 13, 925985. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Xiao, Y.; Wang, W.; Tang, Y.Y.; Xiao, Z.; Su, M. Targeting EphA2 in cancer. J. Hematol. Oncol. 2020, 13, 114. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.B.; Brantley-Sieders, D.M.; Hwang, Y.; Ham, A.J.; Chen, J. Identification and functional analysis of phosphorylated tyrosine residues within EphA2 receptor tyrosine kinase. J. Biol. Chem. 2008, 283, 16017–16026. [Google Scholar] [CrossRef] [PubMed]
- Jannoo, R.; Kanamarlapudi, V. Interleukin-13 Receptor Subunit Alpha-2 (IL-13Rα2). In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Springer: New York, NY, USA, 2017; pp. 1–7. [Google Scholar] [CrossRef]
- Jaen, M.; Martin-Regalado, A.; Bartolome, R.A.; Robles, J.; Casal, J.I. Interleukin 13 receptor alpha 2 (IL13Ralpha2): Expression, signaling pathways and therapeutic applications in cancer. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188802. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, R.A.; Garcia-Palmero, I.; Torres, S.; Lopez-Lucendo, M.; Balyasnikova, I.V.; Casal, J.I. IL13 Receptor alpha2 Signaling Requires a Scaffold Protein, FAM120A, to Activate the FAK and PI3K Pathways in Colon Cancer Metastasis. Cancer Res. 2015, 75, 2434–2444. [Google Scholar] [CrossRef] [PubMed]
- Papageorgis, P.; Ozturk, S.; Lambert, A.W.; Neophytou, C.M.; Tzatsos, A.; Wong, C.K.; Thiagalingam, S.; Constantinou, A.I. Targeting IL13Ralpha2 activates STAT6-TP63 pathway to suppress breast cancer lung metastasis. Breast Cancer Res. 2015, 17, 98. [Google Scholar] [CrossRef]
- Youngblood, V.M.; Kim, L.C.; Edwards, D.N.; Hwang, Y.; Santapuram, P.R.; Stirdivant, S.M.; Lu, P.; Ye, F.; Brantley-Sieders, D.M.; Chen, J. The Ephrin-A1/EPHA2 Signaling Axis Regulates Glutamine Metabolism in HER2-Positive Breast Cancer. Cancer Res. 2016, 76, 1825–1836. [Google Scholar] [CrossRef]
- Fox, B.P.; Kandpal, R.P. Invasiveness of breast carcinoma cells and transcript profile: Eph receptors and ephrin ligands as molecular markers of potential diagnostic and prognostic application. Biochem. Biophys. Res. Commun. 2004, 318, 882–892. [Google Scholar] [CrossRef]
- Brantley-Sieders, D.M.; Zhuang, G.; Hicks, D.; Fang, W.B.; Hwang, Y.; Cates, J.M.; Coffman, K.; Jackson, D.; Bruckheimer, E.; Muraoka-Cook, R.S.; et al. The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J. Clin. Investig. 2008, 118, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Torres-Adorno, A.M.; Vitrac, H.; Qi, Y.; Tan, L.; Levental, K.R.; Fan, Y.Y.; Yang, P.; Chapkin, R.S.; Eckhardt, B.L.; Ueno, N.T. Eicosapentaenoic acid in combination with EPHA2 inhibition shows efficacy in preclinical models of triple-negative breast cancer by disrupting cellular cholesterol efflux. Oncogene 2019, 38, 2135–2150. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Hwang, Y.; Youngblood, V.M.; Cook, R.S.; Balko, J.M.; Chen, J.; Brantley-Sieders, D.M. Targeting EphA2 impairs cell cycle progression and growth of basal-like/triple-negative breast cancers. Oncogene 2017, 36, 5620–5630. [Google Scholar] [CrossRef] [PubMed]
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef]
- Demont, Y.; Corbet, C.; Page, A.; Ataman-Onal, Y.; Choquet-Kastylevsky, G.; Fliniaux, I.; Le Bourhis, X.; Toillon, R.A.; Bradshaw, R.A.; Hondermarck, H. Pro-nerve growth factor induces autocrine stimulation of breast cancer cell invasion through tropomyosin-related kinase A (TrkA) and sortilin protein. J. Biol. Chem. 2012, 287, 1923–1931. [Google Scholar] [CrossRef] [PubMed]
- Leveque, R.; Corbet, C.; Aubert, L.; Guilbert, M.; Lagadec, C.; Adriaenssens, E.; Duval, J.; Finetti, P.; Birnbaum, D.; Magne, N.; et al. ProNGF increases breast tumor aggressiveness through functional association of TrkA with EphA2. Cancer Lett. 2019, 449, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, G.; Brantley-Sieders, D.M.; Vaught, D.; Yu, J.; Xie, L.; Wells, S.; Jackson, D.; Muraoka-Cook, R.; Arteaga, C.; Chen, J. Elevation of receptor tyrosine kinase EphA2 mediates resistance to trastuzumab therapy. Cancer Res. 2010, 70, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Sun, J.; Huang, X.; Zhang, X.; Liu, X.; Liu, R.; Du, G.; Gan, W.; Yang, C.; Tang, Y.; et al. Targeting the KLF5-EphA2 axis can restrain cancer stemness and overcome chemoresistance in basal-like breast cancer. Int. J. Biol. Sci. 2023, 19, 1861–1874. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Choi, J.E.; Bae, Y.K. Interleukin-13 receptor alpha 2 expression in tumor cells is associated with reduced disease-free survival in patients with luminal subtype invasive breast cancer. Tumour Biol. 2018, 40, 1010428318783657. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Onitilo, A.A.; Engel, J.M.; Greenlee, R.T.; Mukesh, B.N. Breast cancer subtypes based on ER/PR and Her2 expression: Comparison of clinicopathologic features and survival. Clin. Med. Res. 2009, 7, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.K.; Yang, H.J.; Lee, M.S.; Liu, D.W.; Chen, L.C.; Chew, C.H.; Lin, C.H.; Lee, C.H.; Li, S.C.; Hong, C.L.; et al. Molecular subtypes of breast cancer predicting clinical benefits of radiotherapy after breast-conserving surgery: A propensity-score-matched cohort study. Breast Cancer Res. 2023, 25, 149. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Cronin, K.A.; Kurian, A.W.; Andridge, R. Differences in Breast Cancer Survival by Molecular Subtypes in the United States. Cancer Epidemiol. Biomark. Prev. 2018, 27, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Tong, C.Y.; Cheng, C. A framework to predict the applicability of Oncotype DX, MammaPrint, and E2F4 gene signatures for improving breast cancer prognostic prediction. Sci. Rep. 2022, 12, 2211. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Liu, Y.H.; Martin, T.A.; Jiang, W.G. The Era of Multigene Panels Comes? The Clinical Utility of Oncotype DX and MammaPrint. World J. Oncol. 2017, 8, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Trastuzumab for early-stage, HER2-positive breast cancer: A meta-analysis of 13 864 women in seven randomised trials. Lancet Oncol. 2021, 22, 1139–1150. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Shastry, M.; Hamilton, E. Targeting HER2-positive breast cancer: Advances and future directions. Nat. Rev. Drug Discov. 2023, 22, 101–126. [Google Scholar] [CrossRef]
- Dar, H.; Johansson, A.; Nordenskjold, A.; Iftimi, A.; Yau, C.; Perez-Tenorio, G.; Benz, C.; Nordenskjold, B.; Stal, O.; Esserman, L.J.; et al. Assessment of 25-Year Survival of Women with Estrogen Receptor-Positive/ERBB2-Negative Breast Cancer Treated with and without Tamoxifen Therapy: A Secondary Analysis of Data from the Stockholm Tamoxifen Randomized Clinical Trial. JAMA Netw. Open 2021, 4, e2114904. [Google Scholar] [CrossRef]
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Aromatase inhibitors versus tamoxifen in premenopausal women with oestrogen receptor-positive early-stage breast cancer treated with ovarian suppression: A patient-level meta-analysis of 7030 women from four randomised trials. Lancet Oncol. 2022, 23, 382–392. [Google Scholar] [CrossRef]
- Obidiro, O.; Battogtokh, G.; Akala, E.O. Triple Negative Breast Cancer Treatment Options and Limitations: Future Outlook. Pharmaceutics 2023, 15, 1796. [Google Scholar] [CrossRef]
- Acheampong, T.; Kehm, R.D.; Terry, M.B.; Argov, E.L.; Tehranifar, P. Incidence Trends of Breast Cancer Molecular Subtypes by Age and Race/Ethnicity in the US From 2010 to 2016. JAMA Netw. Open 2020, 3, e2013226. [Google Scholar] [CrossRef]
- Pandit, P.; Patil, R.; Palwe, V.; Gandhe, S.; Patil, R.; Nagarkar, R. Prevalence of Molecular Subtypes of Breast Cancer: A Single Institutional Experience of 2062 Patients. Eur. J. Breast Health 2020, 16, 39–43. [Google Scholar] [CrossRef]
- Tapia, M.; Hernando, C.; Martinez, M.T.; Burgues, O.; Tebar-Sanchez, C.; Lameirinhas, A.; Agreda-Roca, A.; Torres-Ruiz, S.; Garrido-Cano, I.; Lluch, A.; et al. Clinical Impact of New Treatment Strategies for HER2-Positive Metastatic Breast Cancer Patients with Resistance to Classical Anti-HER Therapies. Cancers 2023, 15, 4522. [Google Scholar] [CrossRef] [PubMed]
- Irie, H.; Kawabata, R.; Fujioka, Y.; Nakagawa, F.; Itadani, H.; Nagase, H.; Ito, K.; Uchida, J.; Ohkubo, S.; Matsuo, K. Acquired resistance to trastuzumab/pertuzumab or to T-DM1 in vivo can be overcome by HER2 kinase inhibition with TAS0728. Cancer Sci. 2020, 111, 2123–2131. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Han, Y.Q.; Li, Q.; Mo, H.N.; Li, Y.Q.; Guan, X.W.; Chen, Y.M.; Lin, S.Y.; Xu, B.H.; Li, Q.; et al. A real world study on the relationship between drug resistance of targeted therapy and prognosis of HER-2-positive advanced breast cancer. Zhonghua Zhong Liu Za Zhi 2022, 44, 360–363. [Google Scholar] [CrossRef]
- Kancha, R.K.; von Bubnoff, N.; Bartosch, N.; Peschel, C.; Engh, R.A.; Duyster, J. Differential sensitivity of ERBB2 kinase domain mutations towards lapatinib. PLoS ONE 2011, 6, e26760. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Hodis, E.; Jacobus, S.; Supko, J.G.; Stewart, M.; Choueiri, T.K.; Gandhi, L.; Cleary, J.M.; et al. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov. 2014, 4, 546–553. [Google Scholar] [CrossRef]
- Derakhshani, A.; Rezaei, Z.; Safarpour, H.; Sabri, M.; Mir, A.; Sanati, M.A.; Vahidian, F.; Gholamiyan Moghadam, A.; Aghadoukht, A.; Hajiasgharzadeh, K.; et al. Overcoming trastuzumab resistance in HER2-positive breast cancer using combination therapy. J. Cell. Physiol. 2020, 235, 3142–3156. [Google Scholar] [CrossRef]
- Loibl, S.; Majewski, I.; Guarneri, V.; Nekljudova, V.; Holmes, E.; Bria, E.; Denkert, C.; Schem, C.; Sotiriou, C.; Loi, S.; et al. PIK3CA mutations are associated with reduced pathological complete response rates in primary HER2-positive breast cancer: Pooled analysis of 967 patients from five prospective trials investigating lapatinib and trastuzumab. Ann. Oncol. 2016, 27, 1519–1525. [Google Scholar] [CrossRef]
- Swain, S.M.; Miles, D.; Kim, S.B.; Im, Y.H.; Im, S.A.; Semiglazov, V.; Ciruelos, E.; Schneeweiss, A.; Loi, S.; Monturus, E.; et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): End-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Filho, O.M.; Viale, G.; Stein, S.; Trippa, L.; Yardley, D.A.; Mayer, I.A.; Abramson, V.G.; Arteaga, C.L.; Spring, L.M.; Waks, A.G.; et al. Impact of HER2 Heterogeneity on Treatment Response of Early-Stage HER2-Positive Breast Cancer: Phase II Neoadjuvant Clinical Trial of T-DM1 Combined with Pertuzumab. Cancer Discov. 2021, 11, 2474–2487. [Google Scholar] [CrossRef]
- Zheng, G.; Guo, Z.; Li, W.; Xi, W.; Zuo, B.; Zhang, R.; Wen, W.; Yang, A.G.; Jia, L. Interaction between HLA-G and NK cell receptor KIR2DL4 orchestrates HER2-positive breast cancer resistance to trastuzumab. Signal Transduct. Target Ther. 2021, 6, 236. [Google Scholar] [CrossRef]
- Boyle, P. Triple-negative breast cancer: Epidemiological considerations and recommendations. Ann. Oncol. 2012, 23 (Suppl. S6), vi7–vi12. [Google Scholar] [CrossRef]
- Borri, F.; Granaglia, A. Pathology of triple negative breast cancer. Semin. Cancer Biol. 2021, 72, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, J.; Peng, L.; Sahin, A.A.; Huo, L.; Ward, K.C.; O’Regan, R.; Torres, M.A.; Meisel, J.L. Triple-negative breast cancer has worse overall survival and cause-specific survival than non-triple-negative breast cancer. Breast Cancer Res. Treat. 2017, 161, 279–287. [Google Scholar] [CrossRef]
- Tzikas, A.K.; Nemes, S.; Linderholm, B.K. A comparison between young and old patients with triple-negative breast cancer: Biology, survival and metastatic patterns. Breast Cancer Res. Treat. 2020, 182, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xin, T.; Huang, D.Y.; Shen, W.X.; Li, L.; Lv, Y.J.; Jin, Y.H.; Song, X.W.; Teng, C.; Jiang, Q.Y. Prognosis in very young women with triple-negative breast cancer: Retrospective study of 216 cases. Med. Oncol. 2014, 31, 222. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wu, J.; Zhang, Z.; Tang, Y.; Li, X.; Liu, S.; Cao, S.; Li, X. Association Between BRCA Status and Triple-Negative Breast Cancer: A Meta-Analysis. Front. Pharmacol. 2018, 9, 909. [Google Scholar] [CrossRef]
- So, J.Y.; Ohm, J.; Lipkowitz, S.; Yang, L. Triple negative breast cancer (TNBC): Non-genetic tumor heterogeneity and immune microenvironment: Emerging treatment options. Pharmacol. Ther. 2022, 237, 108253. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Kim, S.K.; Cho, S.W. The Evasion Mechanisms of Cancer Immunity and Drug Intervention in the Tumor Microenvironment. Front. Pharmacol. 2022, 13, 868695. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tie, Y.; Tang, F.; Wei, Y.Q.; Wei, X.W. Immunosuppressive cells in cancer: Mechanisms and potential therapeutic targets. J. Hematol. Oncol. 2022, 15, 61. [Google Scholar] [CrossRef]
- Pilard, C.; Ancion, M.; Delvenne, P.; Jerusalem, G.; Hubert, P.; Herfs, M. Cancer immunotherapy: It’s time to better predict patients’ response. Br. J. Cancer 2021, 125, 927–938. [Google Scholar] [CrossRef]
- Gatti-Mays, M.E.; Balko, J.M.; Gameiro, S.R.; Bear, H.D.; Prabhakaran, S.; Fukui, J.; Disis, M.L.; Nanda, R.; Gulley, J.L.; Kalinsky, K.; et al. If we build it they will come: Targeting the immune response to breast cancer. NPJ Breast Cancer 2019, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Stanton, S.E.; Adams, S.; Disis, M.L. Variation in the Incidence and Magnitude of Tumor-Infiltrating Lymphocytes in Breast Cancer Subtypes: A Systematic Review. JAMA Oncol. 2016, 2, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Loibl, S.; Noske, A.; Roller, M.; Muller, B.M.; Komor, M.; Budczies, J.; Darb-Esfahani, S.; Kronenwett, R.; Hanusch, C.; et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol. 2010, 28, 105–113. [Google Scholar] [CrossRef]
- Denkert, C.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Cortes, J.; Rugo, H.S.; Cescon, D.W.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Perez-Garcia, J.; Iwata, H.; et al. Pembrolizumab plus Chemotherapy in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2022, 387, 217–226. [Google Scholar] [CrossRef]
- Emens, L.A.; Adams, S.; Barrios, C.H.; Dieras, V.; Iwata, H.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Winer, E.P.; Patel, S.; et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann. Oncol. 2021, 32, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Yazdanifar, M.; Zhou, R.; Mukherjee, P. Emerging immunotherapeutics in adenocarcinomas: A focus on CAR-T cells. Curr. Trends Immunol. 2016, 17, 95–115. [Google Scholar] [PubMed]
- Tong, X.; Dong, C.; Liang, S. Mucin1 as a potential molecule for cancer immunotherapy and targeted therapy. J. Cancer 2024, 15, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.D.; Dillon, L.M.; Zhou, R.; Moore, L.J.; Livasy, C.; El-Khoury, J.M.; Puri, R.; Mukherjee, P. A tumor specific antibody to aid breast cancer screening in women with dense breast tissue. Genes Cancer 2017, 8, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Wallstabe, L.; Mades, A.; Frenz, S.; Einsele, H.; Rader, C.; Hudecek, M. CAR T cells targeting alpha(v)beta(3) integrin are effective against advanced cancer in preclinical models. Adv. Cell Gene Ther. 2018, 1, e11. [Google Scholar] [CrossRef] [PubMed]
- Corti, C.; Venetis, K.; Sajjadi, E.; Zattoni, L.; Curigliano, G.; Fusco, N. CAR-T cell therapy for triple-negative breast cancer and other solid tumors: Preclinical and clinical progress. Expert Opin. Investig. Drugs 2022, 31, 593–605. [Google Scholar] [CrossRef]
- Nasiri, F.; Kazemi, M.; Mirarefin, S.M.J.; Mahboubi Kancha, M.; Ahmadi Najafabadi, M.; Salem, F.; Dashti Shokoohi, S.; Evazi Bakhshi, S.; Safarzadeh Kozani, P.; Safarzadeh Kozani, P. CAR-T cell therapy in triple-negative breast cancer: Hunting the invisible devil. Front. Immunol. 2022, 13, 1018786. [Google Scholar] [CrossRef]
- Bajgain, P.; Tawinwung, S.; D’Elia, L.; Sukumaran, S.; Watanabe, N.; Hoyos, V.; Lulla, P.; Brenner, M.K.; Leen, A.M.; Vera, J.F. CAR T cell therapy for breast cancer: Harnessing the tumor milieu to drive T cell activation. J. Immunother. Cancer 2018, 6, 34. [Google Scholar] [CrossRef]
- Minn, A.J.; Gupta, G.P.; Siegel, P.M.; Bos, P.D.; Shu, W.; Giri, D.D.; Viale, A.; Olshen, A.B.; Gerald, W.L.; Massague, J. Genes that mediate breast cancer metastasis to lung. Nature 2005, 436, 518–524. [Google Scholar] [CrossRef]
- Kawakami, K.; Kawakami, M.; Snoy, P.J.; Husain, S.R.; Puri, R.K. In vivo overexpression of IL-13 receptor alpha2 chain inhibits tumorigenicity of human breast and pancreatic tumors in immunodeficient mice. J. Exp. Med. 2001, 194, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Han, X.; Zhu, Y.; Zhang, H.; Tian, R.; Wang, Z.; Cui, Y.; Wang, Z.; Niu, R.; Zhang, F. Drug-resistant cancer cell-derived exosomal EphA2 promotes breast cancer metastasis via the EphA2-Ephrin A1 reverse signaling. Cell Death Dis. 2021, 12, 414. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; He, B.; Yuan, L.; Dai, W.; Zhang, H.; Wang, X.; Wang, J.; Zhang, X.; Zhang, Q. Dual targeting for metastatic breast cancer and tumor neovasculature by EphA2-mediated nanocarriers. Int. J. Pharm. 2015, 493, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, E.; Amani, J.; Bidmeshki Pour, A.; Mahmoodzadeh Hosseini, H. A new scfv-based recombinant immunotoxin against EPHA2-overexpressing breast cancer cells; High in vitro anti-cancer potency. Eur. J. Pharmacol. 2020, 870, 172912. [Google Scholar] [CrossRef] [PubMed]
- Fox, B.P.; Kandpal, R.P. A paradigm shift in EPH receptor interaction: Biological relevance of EPHB6 interaction with EPHA2 and EPHB2 in breast carcinoma cell lines. Cancer Genom. Proteom. 2011, 8, 185–193. [Google Scholar]
- Kiewlich, D.; Zhang, J.; Gross, C.; Xia, W.; Larsen, B.; Cobb, R.R.; Biroc, S.; Gu, J.M.; Sato, T.; Light, D.R.; et al. Anti-EphA2 antibodies decrease EphA2 protein levels in murine CT26 colorectal and human MDA-231 breast tumors but do not inhibit tumor growth. Neoplasia 2006, 8, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Nikas, I.; Giaginis, C.; Petrouska, K.; Alexandrou, P.; Michail, A.; Sarantis, P.; Tsourouflis, G.; Danas, E.; Pergaris, A.; Politis, P.K.; et al. EPHA2, EPHA4, and EPHA7 Expression in Triple-Negative Breast Cancer. Diagnostics 2022, 12, 366. [Google Scholar] [CrossRef]
- Psilopatis, I.; Souferi-Chronopoulou, E.; Vrettou, K.; Troungos, C.; Theocharis, S. EPH/Ephrin-Targeting Treatment in Breast Cancer: A New Chapter in Breast Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 15275. [Google Scholar] [CrossRef] [PubMed]
- Carles-Kinch, K.; Kilpatrick, K.E.; Stewart, J.C.; Kinch, M.S. Antibody targeting of the EphA2 tyrosine kinase inhibits malignant cell behavior. Cancer Res. 2002, 62, 2840–2847. [Google Scholar]
- Bruckheimer, E.M.; Fazenbaker, C.A.; Gallagher, S.; Mulgrew, K.; Fuhrmann, S.; Coffman, K.T.; Walsh, W.; Ready, S.; Cook, K.; Damschroder, M.; et al. Antibody-dependent cell-mediated cytotoxicity effector-enhanced EphA2 agonist monoclonal antibody demonstrates potent activity against human tumors. Neoplasia 2009, 11, 509–517. [Google Scholar] [CrossRef]
- Gokmen-Polar, Y.; Toroni, R.A.; Hocevar, B.A.; Badve, S.; Zhao, Q.; Shen, C.; Bruckheimer, E.; Kinch, M.S.; Miller, K.D. Dual targeting of EphA2 and ER restores tamoxifen sensitivity in ER/EphA2-positive breast cancer. Breast Cancer Res. Treat. 2011, 127, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, J.; Ma, X.; Yang, Y.; Sun, W.; Jin, W.; Wang, L.; He, Y.; Yang, F.; Yi, Z.; et al. Inhibition of HDACs-EphA2 Signaling Axis with WW437 Demonstrates Promising Preclinical Antitumor Activity in Breast Cancer. EBioMedicine 2018, 31, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tao, Z.; Zhang, Q.; Wan, S.; Zhang, F.; Zhang, Y.; Wu, G.; Wang, J. YSA-conjugated mesoporous silica nanoparticles effectively target EphA2-overexpressing breast cancer cells. Cancer Chemother. Pharmacol. 2018, 81, 687–695. [Google Scholar] [CrossRef]
- Salem, A.F.; Wang, S.; Billet, S.; Chen, J.F.; Udompholkul, P.; Gambini, L.; Baggio, C.; Tseng, H.R.; Posadas, E.M.; Bhowmick, N.A.; et al. Reduction of Circulating Cancer Cells and Metastases in Breast-Cancer Models by a Potent EphA2-Agonistic Peptide-Drug Conjugate. J. Med. Chem. 2018, 61, 2052–2061. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, W.S.; Dugast, A.S.; Suchy, J.J.; Grabow, S.; Fulton, R.B.; Sampson, J.F.; Luus, L.; Santiago, M.; Koshkaryev, A.; Sun, G.; et al. Synergy between EphA2-ILs-DTXp, a Novel EphA2-Targeted Nanoliposomal Taxane, and PD-1 Inhibitors in Preclinical Tumor Models. Mol. Cancer Ther. 2020, 19, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, H.; Terabe, M.; Berzofsky, J.A.; Husain, S.R.; Puri, R.K. A novel combination immunotherapy for cancer by IL-13Ralpha2-targeted DNA vaccine and immunotoxin in murine tumor models. J. Immunol. 2011, 187, 4935–4946. [Google Scholar] [CrossRef] [PubMed]
- Knudson, K.M.; Hwang, S.; McCann, M.S.; Joshi, B.H.; Husain, S.R.; Puri, R.K. Recent Advances in IL-13Ralpha2-Directed Cancer Immunotherapy. Front. Immunol. 2022, 13, 878365. [Google Scholar] [CrossRef]
- Stern, L.A.; Gholamin, S.; Moraga, I.; Yang, X.; Saravanakumar, S.; Cohen, J.R.; Starr, R.; Aguilar, B.; Salvary, V.; Hibbard, J.C.; et al. Engineered IL13 variants direct specificity of IL13Ralpha2-targeted CAR T cell therapy. Proc. Natl. Acad. Sci. USA 2022, 119, e2112006119. [Google Scholar] [CrossRef]
- Pandya, H.; Gibo, D.M.; Garg, S.; Kridel, S.; Debinski, W. An interleukin 13 receptor alpha 2-specific peptide homes to human Glioblastoma multiforme xenografts. Neuro-Oncol. 2012, 14, 6–18. [Google Scholar] [CrossRef]
- Bartolome, R.A.; Jaen, M.; Casal, J.I. An IL13Ralpha2 peptide exhibits therapeutic activity against metastatic colorectal cancer. Br. J. Cancer 2018, 119, 940–949. [Google Scholar] [CrossRef]
- Balyasnikova, I.V.; Wainwright, D.A.; Solomaha, E.; Lee, G.; Han, Y.; Thaci, B.; Lesniak, M.S. Characterization and immunotherapeutic implications for a novel antibody targeting interleukin (IL)-13 receptor alpha2. J. Biol. Chem. 2012, 287, 30215–30227. [Google Scholar] [CrossRef]
- Phuphanich, S.; Wheeler, C.J.; Rudnick, J.D.; Mazer, M.; Wang, H.; Nuno, M.A.; Richardson, J.E.; Fan, X.; Ji, J.; Chu, R.M.; et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol. Immunother. 2013, 62, 125–135. [Google Scholar] [CrossRef]
- Okada, H.; Kalinski, P.; Ueda, R.; Hoji, A.; Kohanbash, G.; Donegan, T.E.; Mintz, A.H.; Engh, J.A.; Bartlett, D.L.; Brown, C.K.; et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with alpha-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J. Clin. Oncol. 2011, 29, 330–336. [Google Scholar] [CrossRef]
- Pant, A.; Lim, M. CAR-T Therapy in GBM: Current Challenges and Avenues for Improvement. Cancers 2023, 15, 1249. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.F.; Orange, J.S.; Sumazin, P.; Man, T.K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef]
- Zhang, A.Q.; Hostetler, A.; Chen, L.E.; Mukkamala, V.; Abraham, W.; Padilla, L.T.; Wolff, A.N.; Maiorino, L.; Backlund, C.M.; Aung, A.; et al. Universal redirection of CAR T cells against solid tumours via membrane-inserted ligands for the CAR. Nat. Biomed. Eng. 2023, 7, 1113–1128. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef]
- Yan, T.; Zhu, L.; Chen, J. Current advances and challenges in CAR T-Cell therapy for solid tumors: Tumor-associated antigens and the tumor microenvironment. Exp. Hematol. Oncol. 2023, 12, 14. [Google Scholar] [CrossRef]
- Feng, K.C.; Guo, Y.L.; Liu, Y.; Dai, H.R.; Wang, Y.; Lv, H.Y.; Huang, J.H.; Yang, Q.M.; Han, W.D. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol. 2017, 10, 4. [Google Scholar] [CrossRef]
- Wei, X.; Lai, Y.; Li, J.; Qin, L.; Xu, Y.; Zhao, R.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology 2017, 6, e1284722. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Wang, G.; Westwood, J.A.; Pachynski, R.K.; Tiffany, H.L.; Marincola, F.M.; Wang, E.; Young, H.A.; Murphy, P.M.; Hwu, P. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum. Gene Ther. 2002, 13, 1971–1980. [Google Scholar] [CrossRef]
- Wang, W.; Ma, Y.; Li, J.; Shi, H.S.; Wang, L.Q.; Guo, F.C.; Zhang, J.; Li, D.; Mo, B.H.; Wen, F.; et al. Specificity redirection by CAR with human VEGFR-1 affinity endows T lymphocytes with tumor-killing ability and anti-angiogenic potency. Gene Ther. 2013, 20, 970–978. [Google Scholar] [CrossRef]
- Jaspers, J.E.; Brentjens, R.J. Development of CAR T cells designed to improve antitumor efficacy and safety. Pharmacol. Ther. 2017, 178, 83–91. [Google Scholar] [CrossRef]
- Sun, S.; Hao, H.; Yang, G.; Zhang, Y.; Fu, Y. Immunotherapy with CAR-Modified T Cells: Toxicities and Overcoming Strategies. J. Immunol. Res. 2018, 2018, 2386187. [Google Scholar] [CrossRef]
- Flugel, C.L.; Majzner, R.G.; Krenciute, G.; Dotti, G.; Riddell, S.R.; Wagner, D.L.; Abou-El-Enein, M. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat. Rev. Clin. Oncol. 2023, 20, 49–62. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, Z.; Dang, Q.; Xu, H.; Lv, J.; Li, H.; Han, X. Immunosuppression in tumor immune microenvironment and its optimization from CAR-T cell therapy. Theranostics 2022, 12, 6273–6290. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Hong, R.; Zhao, H.; Wei, G.; Wu, W.; Xu, H.; Cui, J.; Zhang, Y.; Chang, A.H.; et al. A retrospective comparison of CD19 single and CD19/CD22 bispecific targeted chimeric antigen receptor T cell therapy in patients with relapsed/refractory acute lymphoblastic leukemia. Blood Cancer J. 2020, 10, 105. [Google Scholar] [CrossRef]
- Wang, N.; Hu, X.; Cao, W.; Li, C.; Xiao, Y.; Cao, Y.; Gu, C.; Zhang, S.; Chen, L.; Cheng, J.; et al. Efficacy and safety of CAR19/22 T-cell cocktail therapy in patients with refractory/relapsed B-cell malignancies. Blood 2020, 135, 17–27. [Google Scholar] [CrossRef]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: A phase 1 trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef]
- Dai, H.; Wu, Z.; Jia, H.; Tong, C.; Guo, Y.; Ti, D.; Han, X.; Liu, Y.; Zhang, W.; Wang, C.; et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J. Hematol. Oncol. 2020, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Becerril-Rico, J.; Delgado-Montes, Y.A.; Ortiz-Sanchez, E. Differences in efficacy and safety among CAR-Ts anti-CD19/CD22, anti-CD19, and anti-CD22, in adult patients with relapse/refractory B-cell acute lymphoblastic leukemia: A meta-analysis and systematic review. Leuk. Lymphoma 2023, 64, 1822–1831. [Google Scholar] [CrossRef]
- Huang, M.; Deng, J.; Gao, L.; Zhou, J. Innovative strategies to advance CAR T cell therapy for solid tumors. Am. J. Cancer Res. 2020, 10, 1979–1992. [Google Scholar]
- Ramello, M.C.; Haura, E.B.; Abate-Daga, D. CAR-T cells and combination therapies: What’s next in the immunotherapy revolution? Pharmacol. Res. 2018, 129, 194–203. [Google Scholar] [CrossRef]
- Cordoba, S.; Onuoha, S.; Thomas, S.; Pignataro, D.S.; Hough, R.; Ghorashian, S.; Vora, A.; Bonney, D.; Veys, P.; Rao, K.; et al. CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: A phase 1 trial. Nat Med. 2021, 10, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhou, Y.; Zhang, M.; Ge, W.; Li, Y.; Yang, L.; Wei, G.; Han, L.; Wang, H.; Yu, S.; et al. CRISPR/Cas9-Engineered Universal CD19/CD22 Dual-Targeted CAR-T Cell Therapy for Relapsed/Refractory B-cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2021, 27, 2764–2772. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, S.; Wang, Y.; Lu, Y.; Xu, Y.; Rao, Q.; Wang, H.; Xing, H.; Tian, Z.; Tang, K.; et al. A novel and efficient CD22 CAR-T therapy induced a robust antitumor effect in relapsed/refractory leukemia patients when combined with CD19 CAR-T treatment as a sequential therapy. Exp. Hematol Oncol. 2022, 11, 15. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Thanh Nhu, N.; Chen, C.L.; Lin, C.F. Effectiveness and safety of CD22 and CD19 dual-targeting chimeric antigen receptor T-cell therapy in patients with relapsed or refractory B-cell malignancies: A meta-analysis. Cancer Med. 2023, 12, 18767–18785. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, K.; Du, H.; Xu, Y.; Shou, P.; Zhou, X.; Fuca, G.; Landoni, E.; Sun, C.; Chen, Y.; Savoldo, B.; et al. Dual Targeting CAR-T Cells with Optimal Costimulation and Metabolic Fitness enhance Antitumor Activity and Prevent Escape in Solid Tumors. Nat. Cancer 2021, 2, 904–918. [Google Scholar] [CrossRef]
- Simon, S.; Riddell, S.R. Dual Targeting with CAR T Cells to Limit Antigen Escape in Multiple Myeloma. Blood Cancer Discov. 2020, 1, 130–133. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Compound | Agent Type | Phase | Trial Identifier | Status |
|---|---|---|---|---|
| DS-8895a | Monoclonal antibody, immunotherapy | 1 | NCT02252211 | Completed |
| DOPC-liposomal EphA2 siRNA | Silencing RNA/RNAi, nanotechnology | 1 | NCT01591356 | Recruiting |
| Dasatinib + Zoledronic Acid | Small-molecule inhibitor | 1/2 | NCT00566618 | Completed |
| Dasatinib | Small-molecule inhibitor | 2 | CA180059 | Completed |
| Dasatinib | Small-molecule inhibitor | 2 | NCT02720185 | Recruiting |
| Various agents, including Dasatinib and Ponatinib | Precision medicine | 1b | NCT03878524 | Recruiting |
| Various agents, including Dasatinib | Precision medicine | 2 | NCT02465060 | Recruiting |
| BT5528 | Small-molecule inhibitor | 1/2 | NCT04180371 | Recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kashyap, D.; Salman, H. Targeting Interleukin-13 Receptor α2 and EphA2 in Aggressive Breast Cancer Subtypes with Special References to Chimeric Antigen Receptor T-Cell Therapy. Int. J. Mol. Sci. 2024, 25, 3780. https://doi.org/10.3390/ijms25073780
Kashyap D, Salman H. Targeting Interleukin-13 Receptor α2 and EphA2 in Aggressive Breast Cancer Subtypes with Special References to Chimeric Antigen Receptor T-Cell Therapy. International Journal of Molecular Sciences. 2024; 25(7):3780. https://doi.org/10.3390/ijms25073780
Chicago/Turabian StyleKashyap, Dharambir, and Huda Salman. 2024. "Targeting Interleukin-13 Receptor α2 and EphA2 in Aggressive Breast Cancer Subtypes with Special References to Chimeric Antigen Receptor T-Cell Therapy" International Journal of Molecular Sciences 25, no. 7: 3780. https://doi.org/10.3390/ijms25073780
APA StyleKashyap, D., & Salman, H. (2024). Targeting Interleukin-13 Receptor α2 and EphA2 in Aggressive Breast Cancer Subtypes with Special References to Chimeric Antigen Receptor T-Cell Therapy. International Journal of Molecular Sciences, 25(7), 3780. https://doi.org/10.3390/ijms25073780