
Constitutive NOS Production Is Modulated by Alzheimer’s Disease Pathology Depending on APOE Genotype

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
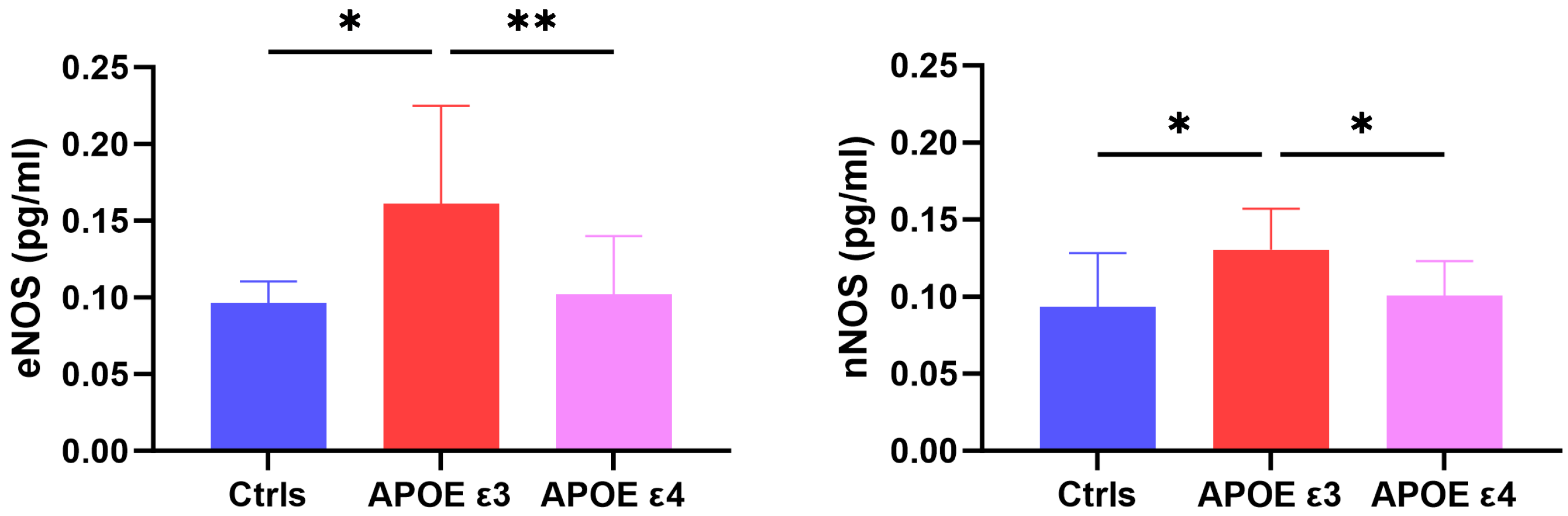
2.1. CSF Levels of eNOS and nNOS across Groups
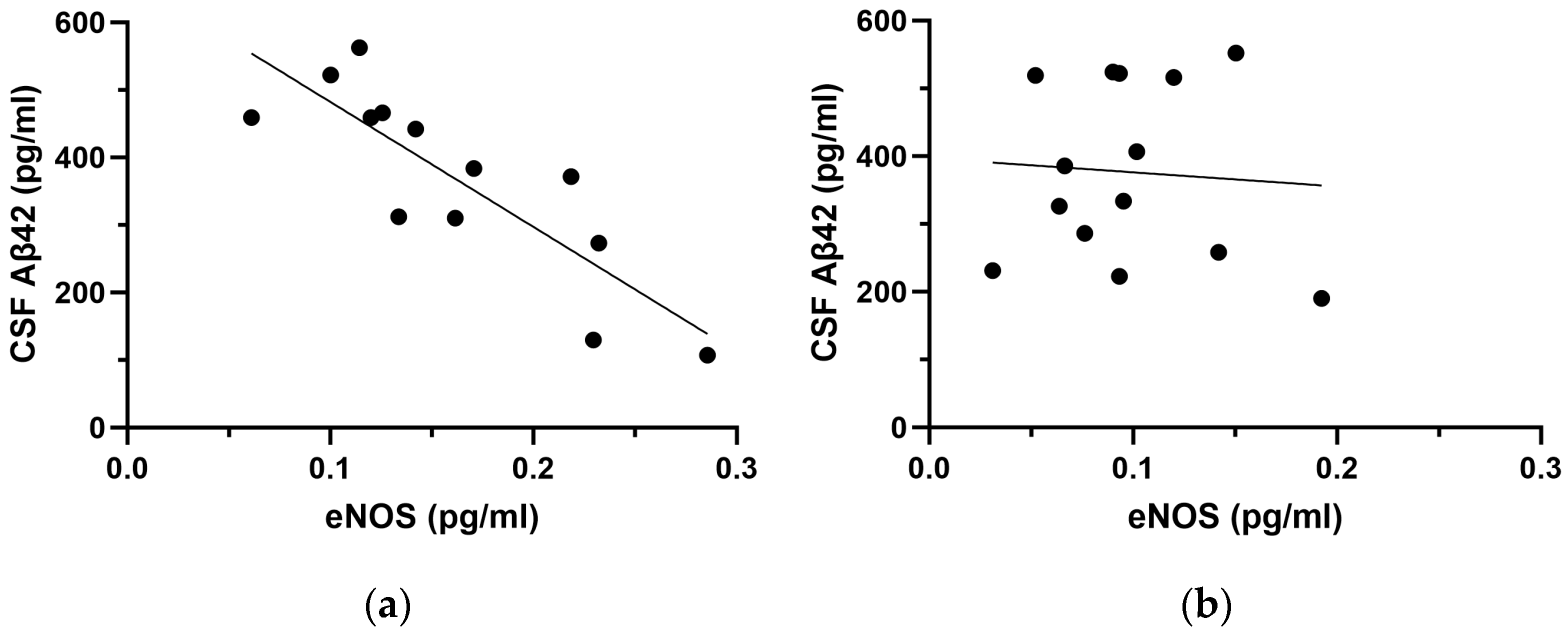
2.2. Correlation Analyses between NOS Species and CSF Aβ42
2.3. Multivariate Regression Analyses in the APOE Subgroups
3. Discussion
4. Materials and Methods
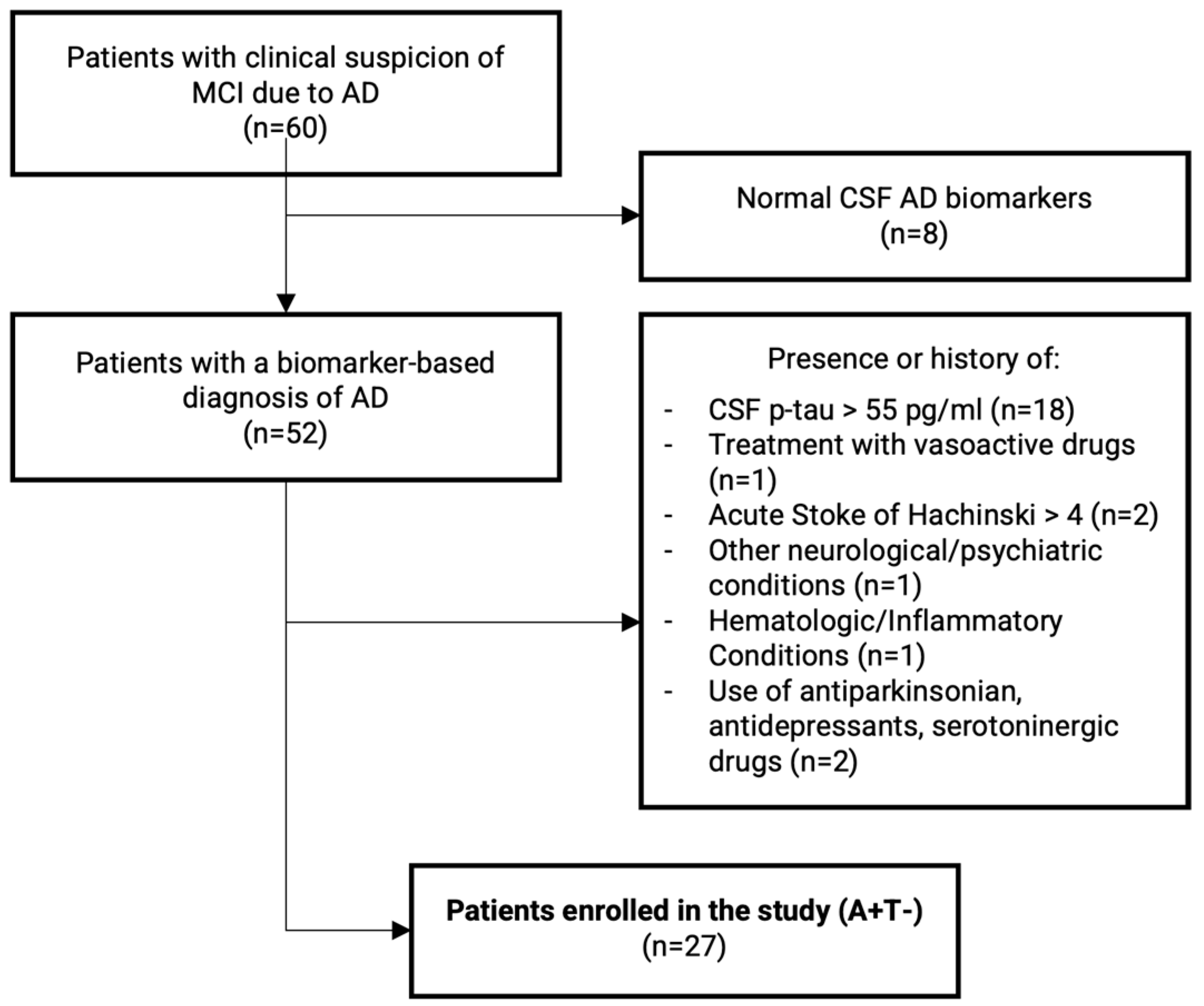
4.1. Subjects’ Enrollment
4.2. CSF Sampling for AD Biomarkers Analysis and APOE Genotype
4.3. NOS Analysis
4.4. Data Management and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Amyloid-β–Induced Neuronal Dysfunction in Alzheimer’s Disease: From Synapses toward Neural Networks. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. Vascular Dysfunction—The Disregarded Partner of Alzheimer’s Disease. Alzheimer’s Dement. 2019, 15, 158–167. [Google Scholar] [CrossRef] [PubMed]
- He, J.T.; Zhao, X.; Xu, L.; Mao, C.Y. Vascular Risk Factors and Alzheimer’s Disease: Blood-Brain Barrier Disruption, Metabolic Syndromes, and Molecular Links. J. Alzheimer’s Dis. 2020, 73, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular Pathways to Neurodegeneration in Alzheimer’s Disease and Other Disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Kapasi, A.; Schneider, J.A. Vascular Contributions to Cognitive Impairment, Clinical Alzheimer’s Disease, and Dementia in Older Persons. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Puig-Pijoan, A.; Jimenez-Balado, J.; Fernández-Lebrero, A.; García-Escobar, G.; Navalpotro-Gómez, I.; Contador, J.; Manero-Borràs, R.M.; Puente-Periz, V.; Suárez, A.; Muñoz, F.J.; et al. Risk of Cognitive Decline Progression Is Associated to Increased Blood-Brain-Barrier Permeability: A Longitudinal Study in a Memory Unit Clinical Cohort. Alzheimer’s Dement. 2023, 20, 538–548. [Google Scholar] [CrossRef]
- Ferrari, A.U.; Radaelli, A.; Mori, I.; Mircoli, L.; Perlini, S.; Meregalli, P.; Fedele, L.; Mancia, G. Nitric Oxide-Dependent Vasodilation and the Regulation of Arterial Blood Pressure. J. Cardiovasc. Pharmacol. 2001, 38 (Suppl. S2), S19–S22. [Google Scholar] [CrossRef] [PubMed]
- Azargoonjahromi, A. Dual Role of Nitric Oxide in Alzheimer’s Disease. Nitric Oxide 2023, 134–135, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Spiers, J.G.; Chen, H.J.C.; Bourgognon, J.M.; Steinert, J.R. Dysregulation of Stress Systems and Nitric Oxide Signaling Underlies Neuronal Dysfunction in Alzheimer’s Disease. Free. Radic. Biol. Med. 2019, 134, 468–483. [Google Scholar] [CrossRef]
- Balez, R.; Ooi, L. Getting to NO Alzheimer’s Disease: Neuroprotection versus Neurotoxicity Mediated by Nitric Oxide. Oxidative Med. Cell. Longev. 2016, 2016, 3806157. [Google Scholar] [CrossRef]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric Oxide: Physiology, Pathophysiology, and Pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar] [PubMed]
- Persichini, T.; Cantoni, O.; Suzuki, H.; Colasanti, M. Cross-Talk Between Constitutive and Inducible NO Synthase: An Update. Antioxid. Redox Signal. 2006, 8, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Katusic, Z.S.; D’Uscio, L.V.; He, T. Emerging Roles of Endothelial Nitric Oxide in Preservation of Cognitive Health. Stroke 2023, 54, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Atochin, D.N.; Huang, P.L. Role of Endothelial Nitric Oxide in Cerebrovascular Regulation. Curr. Pharm. Biotechnol. 2011, 12, 1334. [Google Scholar] [CrossRef]
- Garthwaite, J. Concepts of Neural Nitric Oxide-Mediated Transmission. Eur. J. Neurosci. 2008, 27, 2783–2802. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, G.; Bartus, K.; Malcolm, D.; Goodwin, D.; Kollb-Sielecka, M.; Dooldeniya, C.; Garthwaite, J. Signaling from Blood Vessels to CNS Axons through Nitric Oxide. J. Neurosci. 2006, 26, 7730–7740. [Google Scholar] [CrossRef]
- D’Uscio, L.V.; He, T.; Katusic, Z.S. Expression and Processing of Amyloid Precursor Protein in Vascular Endothelium. Physiology 2017, 32, 20–32. [Google Scholar] [CrossRef]
- Austin, S.A.; Santhanam, A.V.; Katusic, Z.S. Endothelial Nitric Oxide Modulates Expression and Processing of Amyloid Precursor Protein. Circ. Res. 2010, 107, 1498–1502. [Google Scholar] [CrossRef]
- Canepa, E.; Fossati, S. Impact of Tau on Neurovascular Pathology in Alzheimer’s Disease. Front. Neurol. 2021, 11, 573324. [Google Scholar] [CrossRef]
- Austin, S.A.; Katusic, Z.S. Loss of Endothelial Nitric Oxide Synthase Promotes P25 Generation and Tau Phosphorylation in a Murine Model of Alzheimer’s Disease. Circ. Res. 2016, 119, 1128–1134. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a Biological Definition of Alzheimer’s Disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Ojo, J.O.; Reed, J.M.; Crynen, G.; Vallabhaneni, P.; Evans, J.; Shackleton, B.; Eisenbaum, M.; Ringland, C.; Edsell, A.; Mullan, M.; et al. APOE Genotype Dependent Molecular Abnormalities in the Cerebrovasculature of Alzheimer’s Disease and Age-Matched Non-Demented Brains. Mol. Brain 2021, 14, 110. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein e and Alzheimer Disease: Risk, Mechanisms and Therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 Leads to Blood–Brain Barrier Dysfunction Predicting Cognitive Decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, J.W.; Bula, M.; Davila-Velderrain, J.; Akay, L.A.; Zhu, L.; Frank, A.; Victor, M.B.; Bonner, J.M.; Mathys, H.; Lin, Y.T.; et al. Reconstruction of the Human Blood-Brain Barrier in Vitro Reveals a Pathogenic Mechanism of APOE4 in Pericytes. Nat. Med. 2020, 26, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; William Rebeck, G.; Vonsattel, J.P.G.; Gomez-Isla, T.; Hyman, B.T. Apolipoprotein E Ε4 and Cerebral Hemorrhage Associated with Amyloid Angiopathy. Ann. Neurol. 1995, 38, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Thambisetty, M.; Beason-Held, L.; An, Y.; Kraut, M.A.; Resnick, S.M. APOE Epsilon4 Genotype and Longitudinal Changes in Cerebral Blood Flow in Normal Aging. Arch. Neurol. 2010, 67, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Murray, M.E.; Frank, R.D.; Shinohara, M.; DeTure, M.; Yamazaki, Y.; Tachibana, M.; Atagi, Y.; Davis, M.D.; Liu, C.C.; et al. Impact of Sex and APOE4 on Cerebral Amyloid Angiopathy in Alzheimer’s Disease. Acta Neuropathol. 2016, 132, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Hong, H.; Li, K.; Zeng, Q.; Wang, S.; Li, Z.; Fu, Y.; Liu, X.; Hong, L.; Li, J.; et al. Distinct Cerebral Small Vessel Disease Impairment in Early- and Late-Onset Alzheimer’s Disease. Ann. Clin. Transl. Neurol. 2023, 10, 1326–1337. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, X.Y.; Baum, L.; Ng, H.K.; Mok, V.; Wong, K.S. Association Between the Apolipoprotein E Gene Polymorphism and Atherosclerotic Middle Cerebral Artery Stenosis. Neurologist 2018, 23, 47–50. [Google Scholar] [CrossRef]
- Diomedi, M.; Rocco, A.; Bonomi, C.G.; Mascolo, A.P.; De Lucia, V.; Marrama, F.; Sallustio, F.; Koch, G.; Martorana, A. Haemodynamic Impairment along the Alzheimer’s Disease Continuum. Eur. J. Neurol. 2021, 28, 2168–2173. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The Diagnosis of Mild Cognitive Impairment Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Motta, C.; Di Donna, M.G.; Bonomi, C.G.; Assogna, M.; Chiaravalloti, A.; Mercuri, N.B.; Koch, G.; Martorana, A. Different Associations between Amyloid-Βeta 42, Amyloid-Βeta 40, and Amyloid-Βeta 42/40 with Soluble Phosphorylated-Tau and Disease Burden in Alzheimer’s Disease: A Cerebrospinal Fluid and Fluorodeoxyglucose-Positron Emission Tomography Study. Alzheimer’s Res. Ther. 2023, 15, 144. [Google Scholar] [CrossRef] [PubMed]
- Toda, N. Age-Related Changes in Endothelial Function and Blood Flow Regulation. Pharmacol. Ther. 2012, 133, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid b Oligomers Constrict Human Capillaries in Alzheimer’s Disease via Signaling to Pericytes. Science 2019, 365, eaav9518. [Google Scholar] [CrossRef]
- Ulrich, V.; Konaniah, E.S.; Herz, J.; Gerard, R.D.; Jung, E.; Yuhanna, I.S.; Ahmed, M.; Hui, D.Y.; Mineo, C.; Shaul, P.W. Genetic Variants of ApoE and ApoER2 Differentially Modulate Endothelial Function. Proc. Natl. Acad. Sci. USA 2014, 111, 13493–13498. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein e Controls Cerebrovascular Integrity via Cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, S.; Iadecola, C. Revisiting the Neurovascular Unit. Nat. Neurosci. 2021, 24, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Santhanam, A.V.R.; D’Uscio, L.V.; He, T.; Das, P.; Younkin, S.G.; Katusic, Z.S. Uncoupling of Endothelial Nitric Oxide Synthase in Cerebral Vasculature of Tg2576 Mice. J. Neurochem. 2015, 134, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Tropea, M.R.; Gulisano, W.; Vacanti, V.; Arancio, O.; Puzzo, D.; Palmeri, A. Nitric Oxide/CGMP/CREB Pathway and Amyloid-Beta Crosstalk: From Physiology to Alzheimer’s Disease. Free Radic. Biol. Med. 2022, 193, 657–668. [Google Scholar] [CrossRef]
- Tricoire, L.; Vitalis, T. Neuronal Nitric Oxide Synthase Expressing Neurons: A Journey from Birth to Neuronal Circuits. Front. Neural Circuits 2012, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Kourosh-Arami, M.; Hosseini, N.; Mohsenzadegan, M.; Komaki, A.; Joghataei, M.T. Neurophysiologic Implications of Neuronal Nitric Oxide Synthase. Rev. Neurosci. 2020, 31, 617–636. [Google Scholar] [CrossRef] [PubMed]
- Fuentealba, P.; Begum, R.; Capogna, M.; Jinno, S.; Márton, L.F.; Csicsvari, J.; Thomson, A.; Somogyi, P.; Klausberger, T. Ivy Cells: A Population of Nitric-Oxide-Producing, Slow-Spiking GABAergic Neurons and Their Involvement in Hippocampal Network Activity. Neuron 2008, 57, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Zoubovsky, S.P.; Pogorelov, V.M.; Taniguchi, Y.; Kim, S.H.; Yoon, P.; Nwulia, E.; Sawa, A.; Pletnikov, M.V.; Kamiya, A. Working Memory Deficits in Neuronal Nitric Oxide Synthase Knockout Mice: Potential Impairments in Prefrontal Cortex Mediated Cognitive Function. Biochem. Biophys. Res. Commun. 2011, 408, 707–712. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, T.J.; Hawkins, R.D.; Kandel, E.R.; Arancio, O. Tests of the Roles of Two Diffusible Substances in Long-Term Potentiation: Evidence for Nitric Oxide as a Possible Early Retrograde Messenger. Proc. Natl. Acad. Sci. USA 1991, 88, 11285–11289. [Google Scholar] [CrossRef] [PubMed]
- Martorana, A.; Semprini, R.; Koch, G. Clinical Profile of Alzheimer’s Disease Non-Responder Patient. In Advanced Understanding of Neurodegenerative Diseases; InTech: London, UK, 2011. [Google Scholar]
- Strittmatter, W.J.; Weisgraber, K.H.; Huang, D.Y.; Dong, L.M.; Salvesen, G.S.; Pericak-Vance, M.; Schmechel, D.; Saunders, A.M.; Goldgaber, D.; Roses, A.D. Binding of Human Apolipoprotein E to Synthetic Amyloid Beta Peptide: Isoform-Specific Effects and Implications for Late-Onset Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1993, 90, 8098–8102. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.; Newland, P.L.; Zaben, M.; Attard, G.S.; Gray, W.P. Intracellular Nitric Oxide Mediates Neuroproliferative Effect of Neuropeptide y on Postnatal Hippocampal Precursor Cells. J. Biol. Chem. 2012, 287, 20187–20196. [Google Scholar] [CrossRef]
- Carlesimo, G.A.; Caltagirone, C.; Gainotti, G.; Facida, L.; Gallassi, R.; Lorusso, S.; Marfia, G.; Marra, C.; Nocentini, U.; Parnett, L. The Mental Deterioration Battery: Normative Data, Diagnostic Reliability and Qualitative Analyses of Cognitive Impairment. Eur. Neurol. 1996, 36, 378–384. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| HC (n = 8) | APOE ε3 (n = 13) | APOE ε4 (n = 14) | p | |
|---|---|---|---|---|
| Age | 67.25 ± 10.36 | 72.62 ± 7.33 | 70.50 ± 8.14 | 0.400 |
| Sex (%F) | 62.5% | 46.15% | 35.71% | 0.479 |
| Disease Duration | n.a. | 16.2 ± 4.2 | 15.9 ± 5.1 | 0.672 |
| MMSE | 28.62 ± 1.06 | 22.23 ± 2.71 | 21.71 ± 2.43 | <0.001 *** |
| Qalb | 6.97 ± 2.72 | 6.35 ± 2.86 | 7.35 ± 2.52 | 0.619 |
| Aβ42 (pg/mL) | 919.21 ± 329.22 | 369.16 ± 139.63 | 376.74 ± 130.36 | <0.001 *** |
| Aβ40 (pg/mL) | 5417.03 ± 1808.92 | 3917.48 ± 2515.43 | 4519.43 ± 1845.67 | 0.159 |
| p-tau (pg/mL) | 29.81 ± 13.46 | 34.00 ± 18.39 | 35.32 ± 16.97 | 0.767 |
| t-tau (pg/mL) | 205.14 ± 112.18 | 210.17 ± 103.82 | 207.06 ± 101.77 | 0.901 |
| eNOS (pg/mL) | 0.097 ± 0.014 | 0.161 ± 0.064 | 0.098 ± 0.042 | 0.005 ** |
| nNOS (pg/mL) | 0.094 ± 0.036 | 0.131 ± 0.026 | 0.100 ± 0.023 | 0.016 * |
| All Patients (n = 27) | APOE ε3 (n = 13) | APOE ε4 (n = 14) | |||||
|---|---|---|---|---|---|---|---|
| eNOS (pg/mL) | β | p | β | p | β | p | |
| Age | −0.195 | 0.291 | −0.341 | 0.053 | −0.511 | 0.138 | |
| Sex | −0.162 | 0.477 | 0.070 | 0.797 | −0.048 | 0.920 | |
| Qalb | −0.241 | 0.293 | −0.001 | 0.997 | −0.182 | 0.608 | |
| MMSE | 0.384 | 0.050 | 0.251 | 0.295 | −0.050 | 0.914 | |
| Aβ42 | −0.592 | 0.008 ** | −0.698 | 0.003 ** | −0.165 | 0.750 | |
| p−tau | −0.276 | 0.163 | 0.145 | 0.433 | −0.552 | 0.237 | |
| R2 | 0.440 | 0.901 | 0.509 | ||||
| nNOS (pg/mL) | β | p | β | p | β | p | |
| Age | −0.258 | 0.235 | −0.227 | 0.576 | −0.684 | 0.007 ** | |
| Sex | 0.105 | 0.691 | −0.262 | 0.723 | 0.613 | 0.061 | |
| Qalb | −0.377 | 0.163 | −0.805 | 0.386 | −0.088 | 0.668 | |
| MMSE | −0.097 | 0.656 | 0.164 | 0.790 | −0.038 | 0.887 | |
| Aβ42 | 0.006 | 0.978 | 0.120 | 0.766 | −0.170 | 0.575 | |
| p-tau | −0.120 | 0.597 | −0.007 | 0.988 | −0.623 | 0.043 * | |
| R2 | 0.234 | 0.276 | 0.835 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonomi, C.G.; Martorana, A.; Fiorelli, D.; Nuccetelli, M.; Placidi, F.; Mercuri, N.B.; Motta, C. Constitutive NOS Production Is Modulated by Alzheimer’s Disease Pathology Depending on APOE Genotype. Int. J. Mol. Sci. 2024, 25, 3725. https://doi.org/10.3390/ijms25073725
Bonomi CG, Martorana A, Fiorelli D, Nuccetelli M, Placidi F, Mercuri NB, Motta C. Constitutive NOS Production Is Modulated by Alzheimer’s Disease Pathology Depending on APOE Genotype. International Journal of Molecular Sciences. 2024; 25(7):3725. https://doi.org/10.3390/ijms25073725
Chicago/Turabian StyleBonomi, Chiara Giuseppina, Alessandro Martorana, Denise Fiorelli, Marzia Nuccetelli, Fabio Placidi, Nicola Biagio Mercuri, and Caterina Motta. 2024. "Constitutive NOS Production Is Modulated by Alzheimer’s Disease Pathology Depending on APOE Genotype" International Journal of Molecular Sciences 25, no. 7: 3725. https://doi.org/10.3390/ijms25073725
APA StyleBonomi, C. G., Martorana, A., Fiorelli, D., Nuccetelli, M., Placidi, F., Mercuri, N. B., & Motta, C. (2024). Constitutive NOS Production Is Modulated by Alzheimer’s Disease Pathology Depending on APOE Genotype. International Journal of Molecular Sciences, 25(7), 3725. https://doi.org/10.3390/ijms25073725