
Aflibercept Off-Target Effects in Diabetic Macular Edema: An In Silico Modeling Approach

 ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. DME Interactome Generation and IAI-Treated DME General Model Evaluation
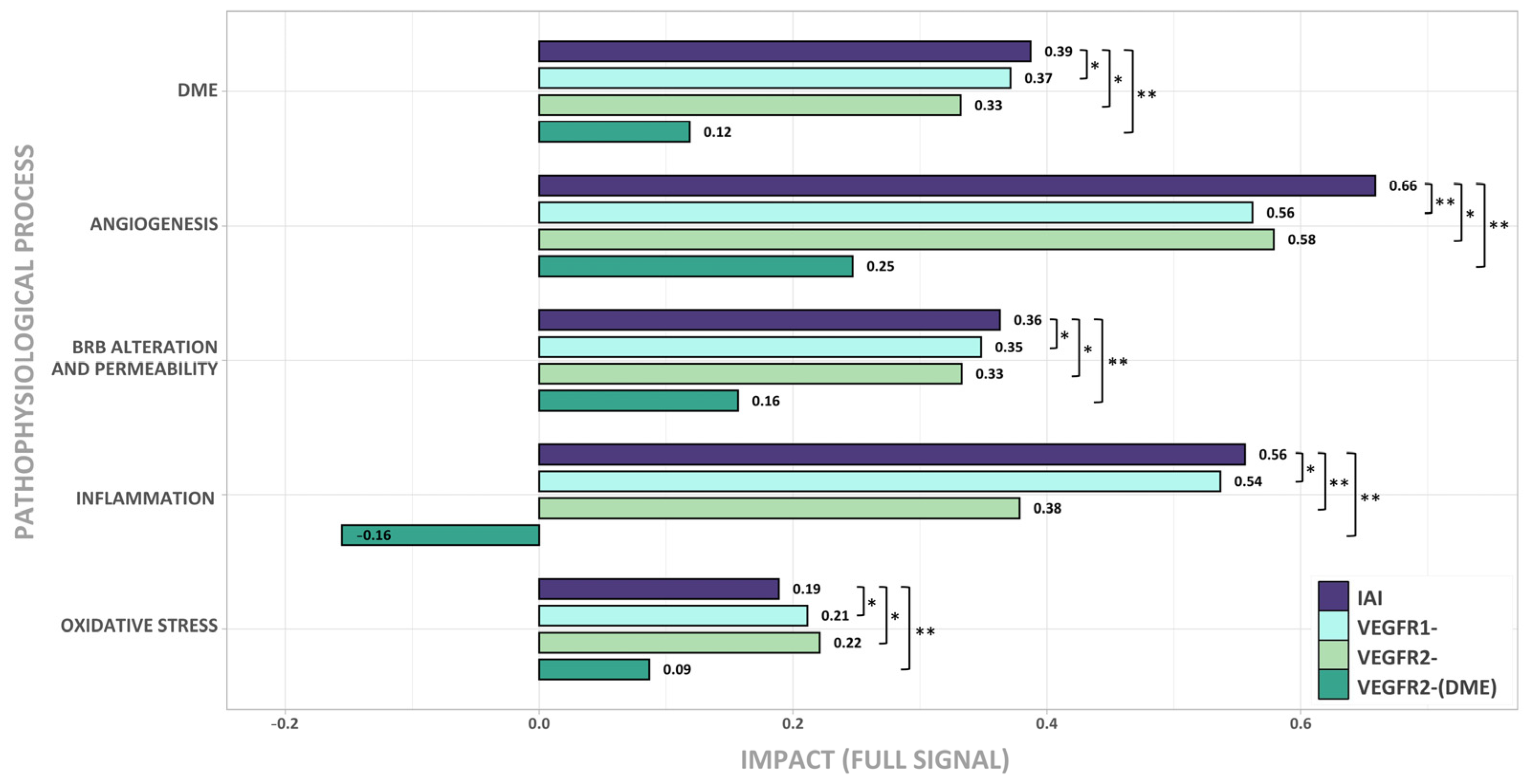
2.2. Effect of IAI, VEGFR1, and VEGFR2 Signaling upon DME
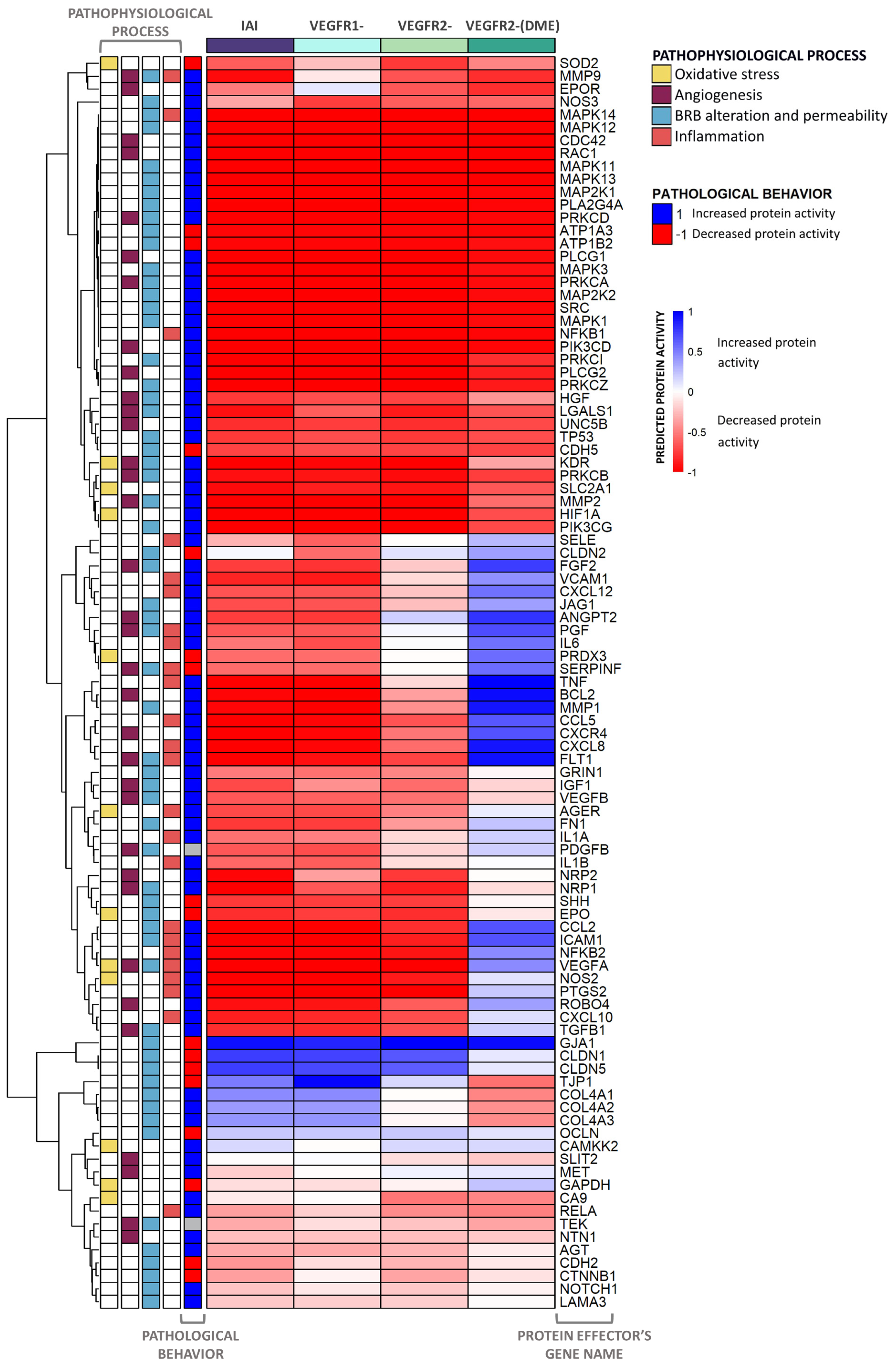
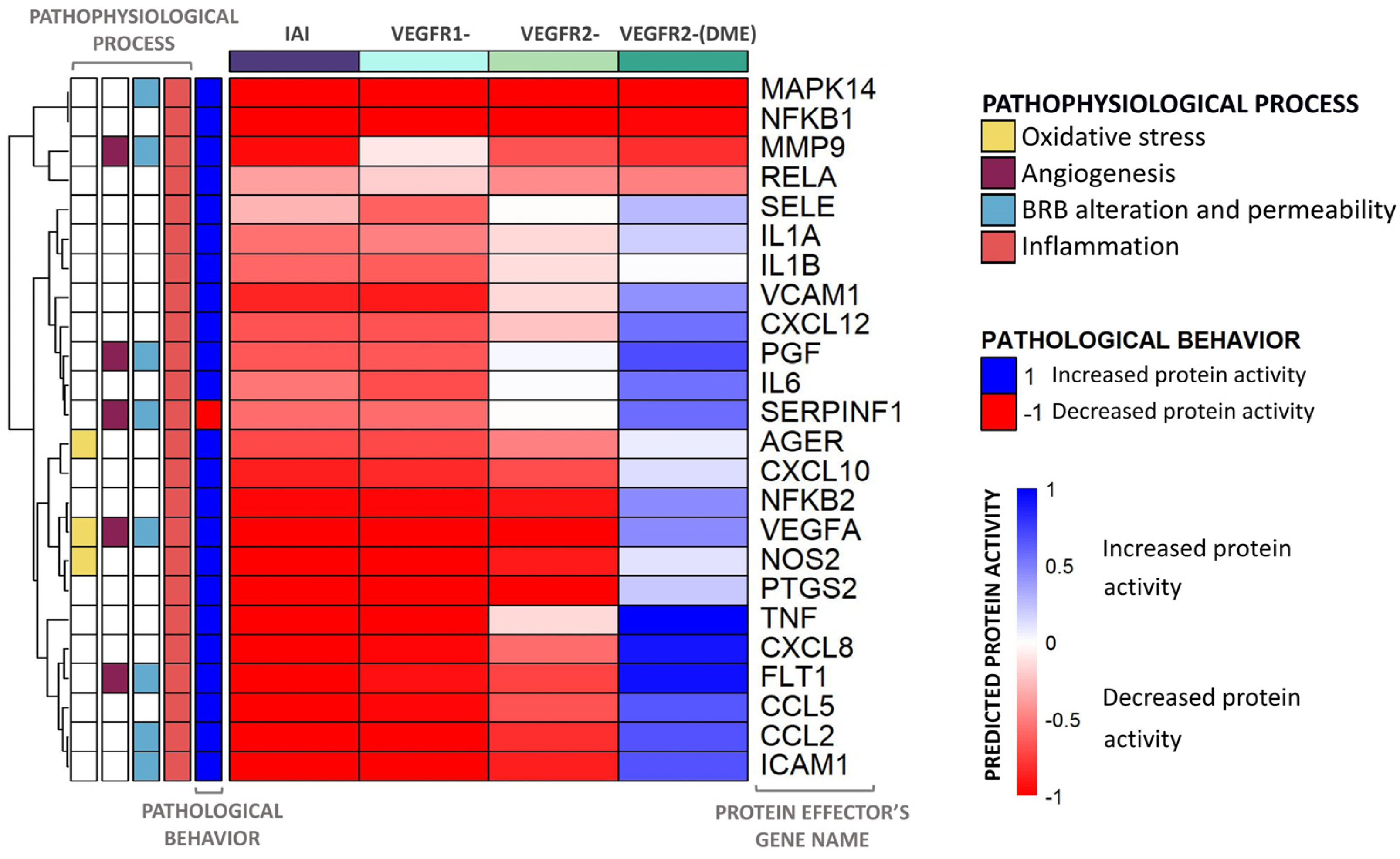
2.3. Molecular Differences of IAI vs. VEGFR2-Specific Inhibition over DME
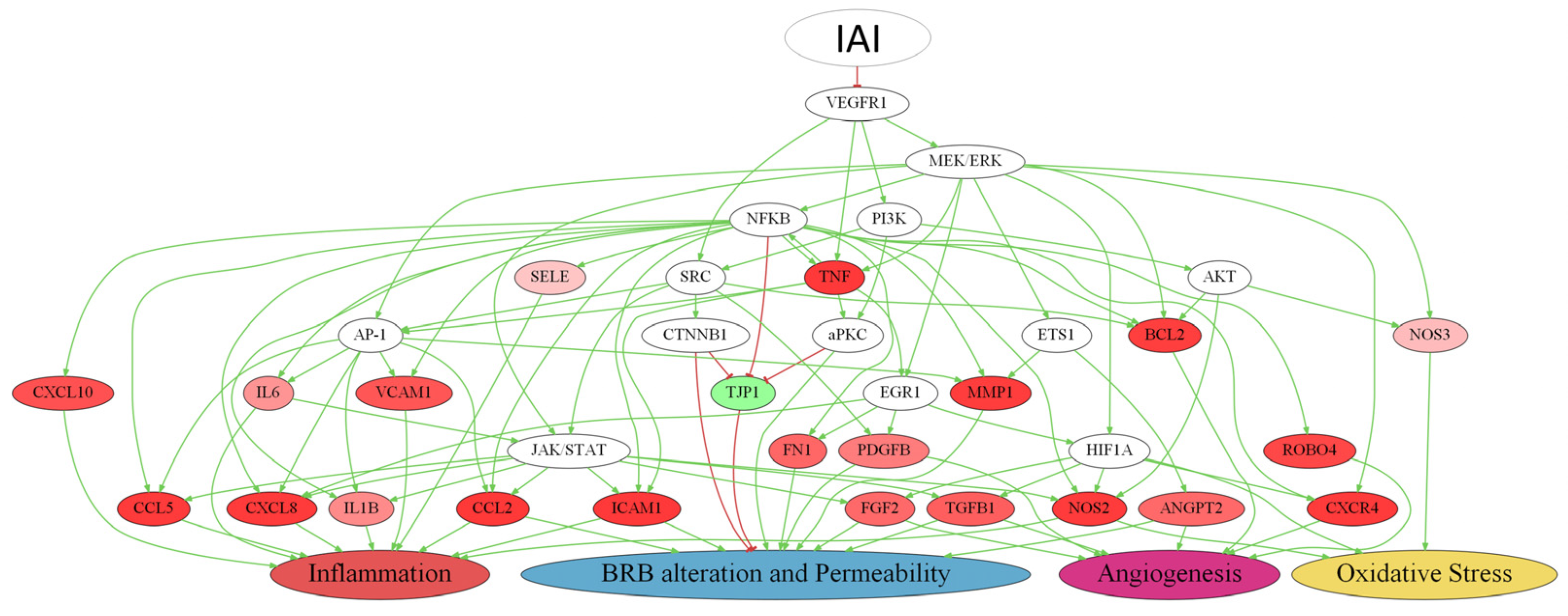
2.4. Detailed IAI-Specific In Silico Molecular Mechanism
3. Discussion
Study Limitations
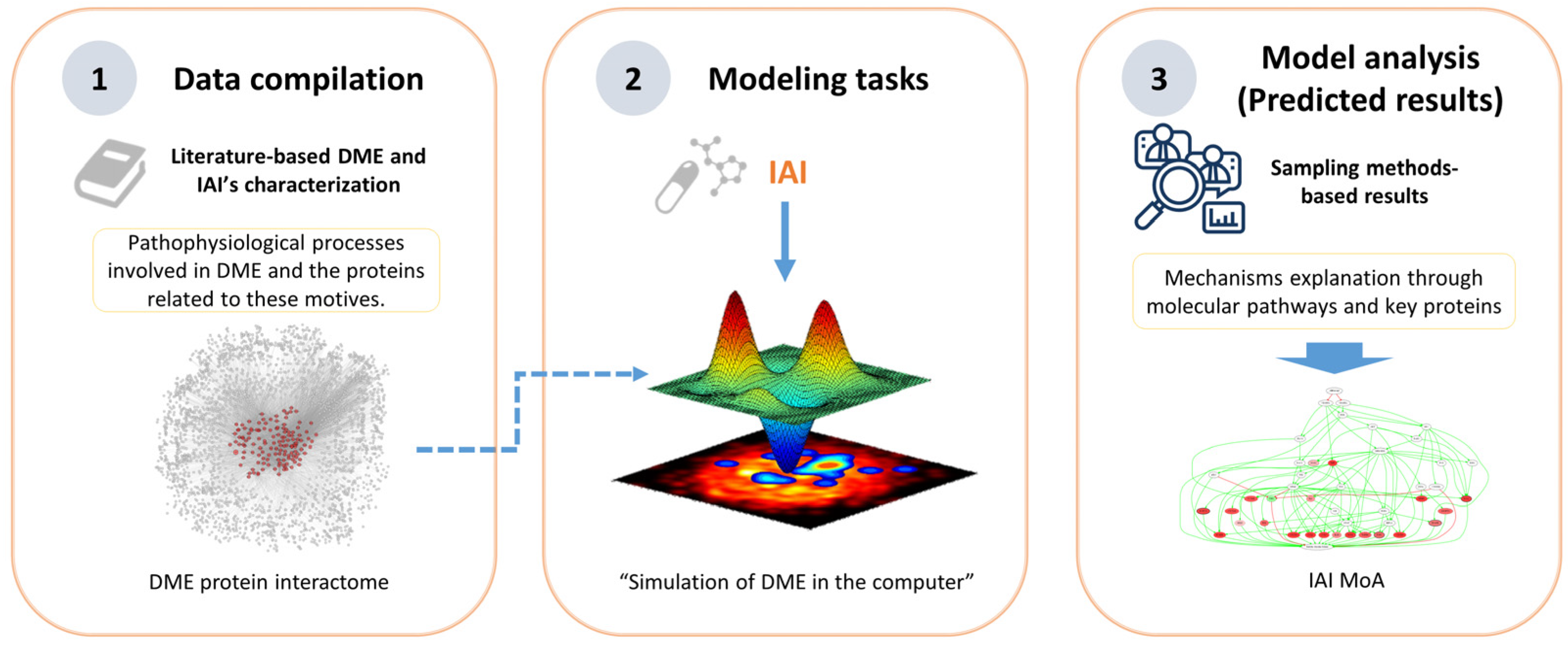
4. Materials and Methods
4.1. Drug and Disease Characterization
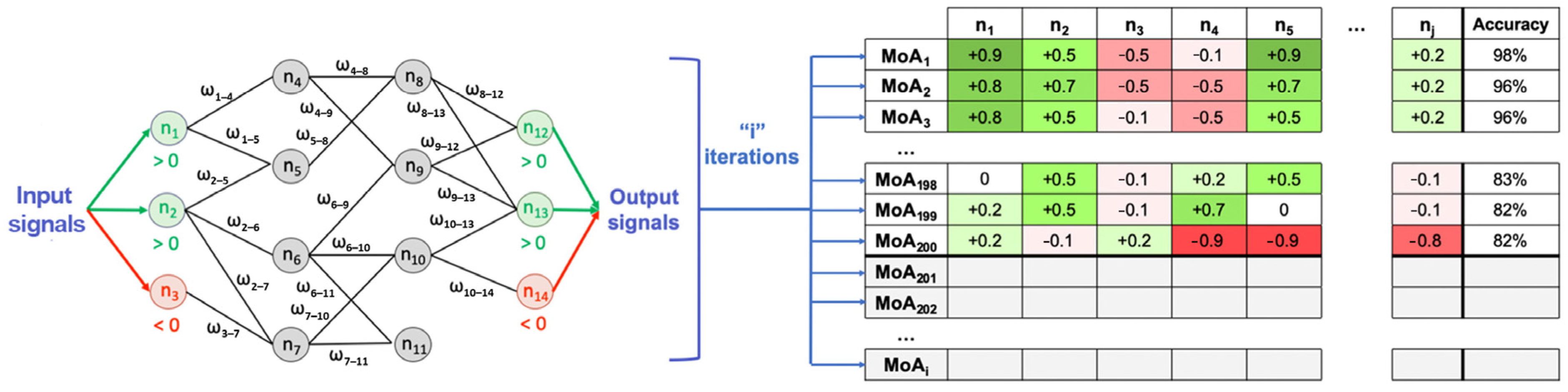
4.2. Modeling: Therapeutic Performance Mapping System and Sampling Methods
4.3. Analysis
4.3.1. Enrichment Analysis
4.3.2. Evaluation of Predicted Protein Activity per VEGFR: Heatmaps
4.3.3. Multi-Target Mechanism of Action and Representation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Acan, D.; Calan, M.; Er, D.; Arkan, T.; Kocak, N.; Bayraktar, F.; Kaynak, S. The Prevalence and Systemic Risk Factors of Diabetic Macular Edema: A Cross-Sectional Study from Turkey. BMC Ophthalmol. 2018, 18, 91. [Google Scholar] [CrossRef] [PubMed]
- Silvia, R.C.; Vijayalakshmi, R. Detection of Non-Proliferative Diabetic Retinopathy in Fundus Images of the Human Retina. In Proceedings of the 2013 International Conference on Information Communication and Embedded Systems (ICICES), Chennai, Tamilnadu, 21–22 February 2013; IEEE: Chennai, Tamilnadu, 2013; pp. 978–983. [Google Scholar]
- Zhang, X.; Zeng, H.; Bao, S.; Wang, N.; Gillies, M.C. Diabetic Macular Edema: New Concepts in Patho-Physiology and Treatment. Cell Biosci. 2014, 4, 27. [Google Scholar] [CrossRef]
- Kleinman, M.E.; Baffi, J.Z.; Ambati, J. The Multifactorial Nature of Retinal Vascular Disease. Ophthalmologica 2010, 224, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Al-Kharashi, A.S. Role of Oxidative Stress, Inflammation, Hypoxia and Angiogenesis in the Development of Diabetic Retinopathy. Saudi J. Ophthalmol. Off. J. Saudi Ophthalmol. Soc. 2018, 32, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Penn, J.S.; Madan, A.; Caldwell, R.B.; Bartoli, M.; Caldwell, R.W.; Hartnett, M.E. Vascular Endothelial Growth Factor in Eye Disease. Prog. Retin. Eye Res. 2008, 27, 331–371. [Google Scholar] [CrossRef]
- Nguyen, Q.D.; De Falco, S.; Behar-Cohen, F.; Lam, W.-C.; Li, X.; Reichhart, N.; Ricci, F.; Pluim, J.; Li, W.W. Placental Growth Factor and Its Potential Role in Diabetic Retinopathy and Other Ocular Neovascular Diseases. Acta Ophthalmol. 2018, 96, e1–e9. [Google Scholar] [CrossRef]
- Khan, M.; Aziz, A.A.; Shafi, N.A.; Abbas, T.; Khanani, A.M. Targeting Angiopoietin in Retinal Vascular Diseases: A Literature Review and Summary of Clinical Trials Involving Faricimab. Cells 2020, 9, 1869. [Google Scholar] [CrossRef]
- Rahimi, N. VEGFR-1 and VEGFR-2: Two Non-Identical Twins with a Unique Physiognomy. Front. Biosci. 2006, 11, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Kanda, A.; Noda, K.; Saito, W.; Ishida, S. Aflibercept Traps Galectin-1, an Angiogenic Factor Associated with Diabetic Retinopathy. Sci. Rep. 2015, 5, 17946. [Google Scholar] [CrossRef]
- Hsieh, S.H.; Ying, N.W.; Wu, M.H.; Chiang, W.F.; Hsu, C.L.; Wong, T.Y.; Jin, Y.T.; Hong, T.M.; Chen, Y.L. Galectin-1, a Novel Ligand of Neuropilin-1, Activates VEGFR-2 Signaling and Modulates the Migration of Vascular Endothelial Cells. Oncogene 2008, 27, 3746–3753. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Uemura, A.; Fruttiger, M.; D’Amore, P.A.; De Falco, S.; Joussen, A.M.; Sennlaub, F.; Brunck, L.R.; Johnson, K.T.; Lambrou, G.N.; Rittenhouse, K.D.; et al. VEGFR1 Signaling in Retinal Angiogenesis and Microinflammation. Prog. Retin. Eye Res. 2021, 84, 100954. [Google Scholar] [CrossRef]
- Figueira, J.; Henriques, J.; Carneiro, Â.; Marques-Neves, C.; Flores, R.; Castro-Sousa, J.P.; Meireles, A.; Gomes, N.; Nascimento, J.; Amaro, M.; et al. Guidelines for the Management of Center-Involving Diabetic Macular Edema: Treatment Options and Patient Monitorization. Clin. Ophthalmol. 2021, 15, 3221–3230. [Google Scholar] [CrossRef]
- Fogli, S.; Del Re, M.; Rofi, E.; Posarelli, C.; Figus, M.; Danesi, R. Clinical Pharmacology of Intravitreal Anti-VEGF Drugs. Eye 2018, 32, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Anguita, R.; Tasiopoulou, A.; Shahid, S.; Roth, J.; Sim, S.Y.; Patel, P.J. A Review of Aflibercept Treatment for Macular Disease. Ophthalmol. Ther. 2021, 10, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Liberski, S.; Wichrowska, M.; Kocięcki, J. Aflibercept versus Faricimab in the Treatment of Neovascular Age-Related Macular Degeneration and Diabetic Macular Edema: A Review. Int. J. Mol. Sci. 2022, 23, 9424. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Fan, R.; Fan, X.; Shao, Y.; Li, X. Progress and Challenges of Anti-VEGF Agents and Their Sustained-Release Strategies for Retinal Angiogenesis. Drug Des. Dev. Ther. 2022, 16, 3241–3262. [Google Scholar] [CrossRef] [PubMed]
- Do, D.V.; Schmidt-Erfurth, U.; Gonzalez, V.H.; Gordon, C.M.; Tolentino, M.; Berliner, A.J.; Vitti, R.; Rückert, R.; Sandbrink, R.; Stein, D.; et al. The DA VINCI Study: Phase 2 Primary Results of VEGF Trap-Eye in Patients with Diabetic Macular Edema. Ophthalmology 2011, 118, 1819–1826. [Google Scholar] [CrossRef]
- Wells, J.A.; Glassman, A.R.; Ayala, A.R.; Jampol, L.M.; Bressler, N.M.; Bressler, S.B.; Brucker, A.J.; Ferris, F.L.; Hampton, G.R.; Jhaveri, C.; et al. Aflibercept, Bevacizumab, or Ranibizumab for Diabetic Macular Edema: Two-Year Results from a Comparative Effectiveness Randomized Clinical Trial. Ophthalmology 2016, 123, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Heier, J.S.; Korobelnik, J.-F.; Brown, D.M.; Schmidt-Erfurth, U.; Do, D.V.; Midena, E.; Boyer, D.S.; Terasaki, H.; Kaiser, P.K.; Marcus, D.M.; et al. Intravitreal Aflibercept for Diabetic Macular Edema: 148-Week Results from the VISTA and VIVID Studies. Ophthalmology 2016, 123, 2376–2385. [Google Scholar] [CrossRef]
- Chauhan, M.Z.; Rather, P.A.; Samarah, S.M.; Elhusseiny, A.M.; Sallam, A.B. Current and Novel Therapeutic Approaches for Treatment of Diabetic Macular Edema. Cells 2022, 11, 1950. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.D.; Cave, J.W.; Cho, S. Aflibercept, a VEGF (Vascular Endothelial Growth Factor)-Trap, Reduces Vascular Permeability and Stroke-Induced Brain Swelling in Obese Mice. Stroke 2021, 52, 2637–2648. [Google Scholar] [CrossRef]
- Lazzara, F.; Fidilio, A.; Platania, C.B.M.; Giurdanella, G.; Salomone, S.; Leggio, G.M.; Tarallo, V.; Cicatiello, V.; De Falco, S.; Eandi, C.M.; et al. Aflibercept Regulates Retinal Inflammation Elicited by High Glucose via the PlGF/ERK Pathway. Biochem. Pharmacol. 2019, 168, 341–351. [Google Scholar] [CrossRef]
- Ji, Z.; Yan, K.; Li, W.; Hu, H.; Zhu, X. Mathematical and Computational Modeling in Complex Biological Systems. BioMed Res. Int. 2017, 2017, 5958321. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Oliveira, R.H.M.; Zhao, C.; Popel, A.S. Systems Biology of Angiogenesis Signaling: Computational Models and Omics. WIREs Mech. Dis. 2022, 14, e1550. [Google Scholar] [CrossRef] [PubMed]
- Segú-Vergés, C.; Gómez, J.; Terradas-Montana, P.; Artigas, L.; Smeets, S.; Ferrer, M.; Savic, S. Unveiling Chronic Spontaneous Urticaria Pathophysiology through Systems Biology. J. Allergy Clin. Immunol. 2023, 151, 1005–1014. [Google Scholar] [CrossRef]
- Lorén, V.; Garcia-Jaraquemada, A.; Naves, J.E.; Carmona, X.; Mañosa, M.; Aransay, A.M.; Lavin, J.L.; Sánchez, I.; Cabré, E.; Manyé, J.; et al. ANP32E, a Protein Involved in Steroid-Refractoriness in Ulcerative Colitis, Identified by a Systems Biology Approach. J. Crohn’s Colitis 2019, 13, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Jorba, G.; Aguirre-Plans, J.; Junet, V.; Segu-Verges, C.; Ruiz, J.L.; Pujol, A.; Fernandez-Fuentes, N.; Mas, J.M.; Oliva, B. In-Silico Simulated Prototype-Patients Using TPMS Technology to Study a Potential Adverse Effect of Sacubitril and Valsartan. PLoS ONE 2020, 15, e0228926. [Google Scholar] [CrossRef]
- Bayes-Genis, A.; Iborra-Egea, O.; Spitaleri, G.; Domingo, M.; Revuelta-López, E.; Codina, P.; Cediel, G.; Santiago-Vacas, E.; Cserkóová, A.; Pascual-Figal, D.; et al. Decoding Empagliflozin’s Molecular Mechanism of Action in Heart Failure with Preserved Ejection Fraction Using Artificial Intelligence. Sci. Rep. 2021, 11, 12025. [Google Scholar] [CrossRef]
- Segu-Verges, C.; Coma, M.; Kessel, C.; Smeets, S.; Foell, D.; Aldea, A. Application of Systems Biology-Based in Silico Tools to Optimize Treatment Strategy Identification in Still’s Disease. Arthritis Res. Ther. 2021, 23, 126. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Martin, J.; Ruan, Q.; Rafique, A.; Rosconi, M.P.; Shi, E.; Pyles, E.A.; Yancopoulos, G.D.; Stahl, N.; Wiegand, S.J. Binding and Neutralization of Vascular Endothelial Growth Factor (VEGF) and Related Ligands by VEGF Trap, Ranibizumab and Bevacizumab. Angiogenesis 2012, 15, 171–185. [Google Scholar] [CrossRef]
- Holash, J.; Davis, S.; Papadopoulos, N.; Croll, S.D.; Ho, L.; Russell, M.; Boland, P.; Leidich, R.; Hylton, D.; Burova, E.; et al. VEGF-Trap: A VEGF Blocker with Potent Antitumor Effects. Proc. Natl. Acad. Sci. USA 2002, 99, 11393–11398. [Google Scholar] [CrossRef]
- Mesquita, J.; Castro-de-Sousa, J.P.; Vaz-Pereira, S.; Neves, A.; Passarinha, L.A.; Tomaz, C.T. Evaluation of the Growth Factors VEGF-a and VEGF-B in the Vitreous and Serum of Patients with Macular and Retinal Vascular Diseases. Growth Factors 2018, 36, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Lennikov, A.; Saddala, M.S.; Gozal, D.; Grab, D.J.; Khalyfa, A.; Fan, L. Placental Growth Factor Negatively Regulates Retinal Endothelial Cell Barrier Function through Suppression of Glucose-6-phosphate Dehydrogenase and Antioxidant Defense Systems. FASEB J. 2019, 33, 13695–13709. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Huang, H.; Shen, J.; Zacchigna, S.; Zentilin, L.; Giacca, M.; Vinores, S.A. Vascular Endothelial Growth Factor-B Gene Transfer Exacerbates Retinal and Choroidal Neovascularization and Vasopermeability without Promoting Inflammation. Mol. Vis. 2011, 17, 492–507. [Google Scholar] [PubMed]
- Hase, K.; Kanda, A.; Noda, K.; Ishida, S. Increased Plasma Galectin-1 Correlates with Advanced Glycation End Products and Interleukin-1β in Patients with Proliferative Diabetic Retinopathy. Int. J. Ophthalmol. 2019, 12, 692–694. [Google Scholar] [CrossRef]
- Odorisio, T.; Schietroma, C.; Zaccaria, M.L.; Cianfarani, F.; Tiveron, C.; Tatangelo, L.; Failla, C.M.; Zambruno, G. Mice Overexpressing Placenta Growth Factor Exhibit Increased Vascularization and Vessel Permeability. J. Cell Sci. 2002, 115, 2559–2567. [Google Scholar] [CrossRef]
- Rakic, J.-M.; Lambert, V.; Devy, L.; Luttun, A.; Carmeliet, P.; Claes, C.; Nguyen, L.; Foidart, J.-M.; Noe¨l, A.; Munaut, C. Placental Growth Factor, a Member of the VEGF Family, Contributes to the Development of Choroidal Neovascularization. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3186. [Google Scholar] [CrossRef]
- Huang, H.; Shen, J.; Vinores, S.A. Blockade of VEGFR1 and 2 Suppresses Pathological Angiogenesis and Vascular Leakage in the Eye. PLoS ONE 2011, 6, e21411. [Google Scholar] [CrossRef]
- Hiratsuka, S.; Minowa, O.; Kuno, J.; Noda, T.; Shibuya, M. Flt-1 Lacking the Tyrosine Kinase Domain Is Sufficient for Normal Development and Angiogenesis in Mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9349–9354. [Google Scholar] [CrossRef]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; De Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Beck, H.; et al. Synergism between Vascular Endothelial Growth Factor and Placental Growth Factor Contributes to Angiogenesis and Plasma Extravasation in Pathological Conditions. Nat. Med. 2001, 7, 575–583. [Google Scholar] [CrossRef]
- Crespo-Garcia, S.; Corkhill, C.; Roubeix, C.; Davids, A.-M.; Kociok, N.; Strauss, O.; Joussen, A.M.; Reichhart, N. Inhibition of Placenta Growth Factor Reduces Subretinal Mononuclear Phagocyte Accumulation in Choroidal Neovascularization. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4997. [Google Scholar] [CrossRef]
- Fischer, C.; Mazzone, M.; Jonckx, B.; Carmeliet, P. FLT1 and Its Ligands VEGFB and PlGF: Drug Targets for Anti-Angiogenic Therapy? Nat. Rev. Cancer 2008, 8, 942–956. [Google Scholar] [CrossRef] [PubMed]
- Ziche, M.; Maglione, D.; Ribatti, D.; Morbidelli, L.; Lago, C.; Battisti, M.; Paoletti, I.; Barra, A.; Tucci, M.; Parise, G.; et al. Placenta Growth Factor-1 Is Chemotactic, Mitogenic, and Angiogenic. Lab. Investig. 1997, 76, 517–531. [Google Scholar] [PubMed]
- Feliers, D.; Chen, X.; Akis, N.; Choudhury, G.G.; Madaio, M.; Kasinath, B.S. VEGF Regulation of Endothelial Nitric Oxide Synthase in Glomerular Endothelial Cells. Kidney Int. 2005, 68, 1648–1659. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, J.; Zhang, C.; Zhang, J.; Gu, L.; Luo, D.; Qiu, Q. Diabetic Macular Edema: Current Understanding, Molecular Mechanisms and Therapeutic Implications. Cells 2022, 11, 3362. [Google Scholar] [CrossRef] [PubMed]
- Vujosevic, S.; Micera, A.; Bini, S.; Berton, M.; Esposito, G.; Midena, E. Proteome Analysis of Retinal Glia Cells-Related Inflammatory Cytokines in the Aqueous Humour of Diabetic Patients. Acta Ophthalmol. 2016, 94, 56–64. [Google Scholar] [CrossRef]
- Funatsu, H.; Yamashita, H.; Sakata, K.; Noma, H.; Mimura, T.; Suzuki, M.; Eguchi, S.; Hori, S. Vitreous Levels of Vascular Endothelial Growth Factor and Intercellular Adhesion Molecule 1 Are Related to Diabetic Macular Edema. Ophthalmology 2005, 112, 806–816. [Google Scholar] [CrossRef]
- Funatsu, H.; Noma, H.; Mimura, T.; Eguchi, S.; Hori, S. Association of Vitreous Inflammatory Factors with Diabetic Macular Edema. Ophthalmology 2009, 116, 73–79. [Google Scholar] [CrossRef]
- Owen, L.A.; Hartnett, M.E. Soluble Mediators of Diabetic Macular Edema: The Diagnostic Role of Aqueous VEGF and Cytokine Levels in Diabetic Macular Edema. Curr. Diabetes Rep. 2013, 13, 476–480. [Google Scholar] [CrossRef]
- Minaker, S.A.; Mason, R.H.; Lahaie Luna, G.; Farahvash, A.; Garg, A.; Bhambra, N.; Bapat, P.; Muni, R.H. Changes in Aqueous and Vitreous Inflammatory Cytokine Levels in Diabetic Macular Oedema: A Systematic Review and Meta-analysis. Acta Ophthalmol. 2022, 100, e53–e70. [Google Scholar] [CrossRef]
- Carpi-Santos, R.; De Melo Reis, R.A.; Gomes, F.C.A.; Calaza, K.C. Contribution of Müller Cells in the Diabetic Retinopathy Development: Focus on Oxidative Stress and Inflammation. Antioxidants 2022, 11, 617. [Google Scholar] [CrossRef]
- Olvera-Montaño, C.; Castellanos-González, J.A.; Navarro-Partida, J.; Cardona-Muñoz, E.G.; López-Contreras, A.K.; Roman-Pintos, L.M.; Robles-Rivera, R.-R.; Rodríguez-Carrizalez, A.D. Oxidative Stress as the Main Target in Diabetic Retinopathy Pathophysiology. J. Diabetes Res. 2019, 2019, 8562408. [Google Scholar] [CrossRef]
- Suryavanshi, S.V.; Kulkarni, Y.A. NF-κβ: A Potential Target in the Management of Vascular Complications of Diabetes. Front. Pharmacol. 2017, 8, 798. [Google Scholar] [CrossRef]
- Morris, K.R.; Lutz, R.D.; Choi, H.S.; Kamitani, T.; Chmura, K.; Chan, E.D. Role of the NF-κB Signaling Pathway and κB Cis-Regulatory Elements on the IRF-1 and iNOS Promoter Regions in Mycobacterial Lipoarabinomannan Induction of Nitric Oxide. Infect. Immun. 2003, 71, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, e17023. [Google Scholar] [CrossRef] [PubMed]
- Yuuki, T.; Kanda, T.; Kimura, Y.; Kotajima, N.; Tamura, J.; Kobayashi, I.; Kishi, S. Inflammatory Cytokines in Vitreous Fluid and Serum of Patients with Diabetic Vitreoretinopathy. J. Diabetes Its Complicat. 2001, 15, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF Family Cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Peach, C.; Mignone, V.; Arruda, M.; Alcobia, D.; Hill, S.; Kilpatrick, L.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef]
- Ahmad, S.; Hewett, P.W.; Wang, P.; Al-Ani, B.; Cudmore, M.; Fujisawa, T.; Haigh, J.J.; Le Noble, F.; Wang, L.; Mukhopadhyay, D.; et al. Direct Evidence for Endothelial Vascular Endothelial Growth Factor Receptor-1 Function in Nitric Oxide–Mediated Angiogenesis. Circ. Res. 2006, 99, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Vinores, S.A. Breakdown of the Blood–Retinal Barrier. In Encyclopedia of the Eye; Elsevier: Amsterdam, The Netherlands, 2010; pp. 216–222. ISBN 978-0-12-374203-2. [Google Scholar]
- Shibuya, M. Differential Roles of Vascular Endothelial Growth Factor Receptor-1 and Receptor-2 in Angiogenesis. J. Biochem. Mol. Biol. 2006, 39, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.A.; Samaranayake, G.J.; Browning, A.C.; Hopkinson, A.; Amoaku, W.M. Comparison of Choroidal and Retinal Endothelial Cells: Characteristics and Response to VEGF Isoforms and Anti-VEGF Treatments. Exp. Eye Res. 2011, 93, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Xue, Y.; Hedlund, E.-M.; Zhong, Z.; Tritsaris, K.; Tondelli, B.; Lucchini, F.; Zhu, Z.; Dissing, S.; Cao, Y. VEGFR1–Mediated Pericyte Ablation Links VEGF and PlGF to Cancer-Associated Retinopathy. Proc. Natl. Acad. Sci. USA 2010, 107, 856–861. [Google Scholar] [CrossRef]
- Stitt, A.W.; Simpson, D.A.C.; Boocock, C.; Gardiner, T.A.; Murphy, G.M.; Archer, D.B. Expression of Vascular Endothelial Growth Factor (VEGF) and Its Receptors Is Regulated in Eyes with Intra-Ocular Tumours. J. Pathol. 1998, 186, 306–312. [Google Scholar] [CrossRef]
- Luo, L.; Uehara, H.; Zhang, X.; Das, S.K.; Olsen, T.; Holt, D.; Simonis, J.M.; Jackman, K.; Singh, N.; Miya, T.R.; et al. Photoreceptor Avascular Privilege Is Shielded by Soluble VEGF Receptor-1. eLife 2013, 2, e00324. [Google Scholar] [CrossRef]
- Maglione, D.; Guerriero, V.; Viglietto, G.; Delli-Bovi, P.; Persico, M.G. Isolation of a Human Placenta cDNA Coding for a Protein Related to the Vascular Permeability Factor. Proc. Natl. Acad. Sci. USA 1991, 88, 9267–9271. [Google Scholar] [CrossRef]
- De Falco, S.; Gigante, B.; Persico, M.G. Structure and Function of Placental Growth Factor. Trends Cardiovasc. Med. 2002, 12, 241–246. [Google Scholar] [CrossRef]
- Clauss, M.; Weich, H.; Breier, G.; Knies, U.; Röckl, W.; Waltenberger, J.; Risau, W. The Vascular Endothelial Growth Factor Receptor Flt-1 Mediates Biological Activities. J. Biol. Chem. 1996, 271, 17629–17634. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; Glassman, A.R.; Ayala, A.R.; Jampol, L.M.; Aiello, L.P.; Antoszyk, A.N.; Arnold-Bush, B.; Baker, C.W.; Bressler, N.M.; Browning, D.J.; et al. Aflibercept, Bevacizumab, or Ranibizumab for Diabetic Macular Edema. N. Engl. J. Med. 2015, 372, 1193–1203. [Google Scholar] [CrossRef]
- Bressler, N.M.; Beaulieu, W.T.; Glassman, A.R.; Blinder, K.J.; Bressler, S.B.; Jampol, L.M.; Melia, M.; Wells, J.A.; for the Diabetic Retinopathy Clinical Research Network. Persistent Macular Thickening Following Intravitreous Aflibercept, Bevacizumab, or Ranibizumab for Central-Involved Diabetic Macular Edema with Vision Impairment: A Secondary Analysis of a Randomized Clinical Trial. JAMA Ophthalmol. 2018, 136, 257. [Google Scholar] [CrossRef]
- Cai, S.; Bressler, N.M. Aflibercept, Bevacizumab or Ranibizumab for Diabetic Macular Oedema: Recent Clinically Relevant Findings from DRCR.Net Protocol T. Curr. Opin. Ophthalmol. 2017, 28, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Jampol, L.M.; Glassman, A.R.; Bressler, N.M.; Wells, J.A.; Ayala, A.R.; for the Diabetic Retinopathy Clinical Research Network. Anti–Vascular Endothelial Growth Factor Comparative Effectiveness Trial for Diabetic Macular Edema: Additional Efficacy Post Hoc Analyses of a Randomized Clinical Trial. JAMA Ophthalmol. 2016, 134, 1429. [Google Scholar] [CrossRef]
- Heier, J.S.; Bressler, N.M.; Avery, R.L.; Bakri, S.J.; Boyer, D.S.; Brown, D.M.; Dugel, P.U.; Freund, K.B.; Glassman, A.R.; Kim, J.E.; et al. Comparison of Aflibercept, Bevacizumab, and Ranibizumab for Treatment of Diabetic Macular Edema: Extrapolation of Data to Clinical Practice. JAMA Ophthalmol. 2016, 134, 95. [Google Scholar] [CrossRef]
- Wang, X.; Bove, A.M.; Simone, G.; Ma, B. Molecular Bases of VEGFR-2-Mediated Physiological Function and Pathological Role. Front. Cell Dev. Biol. 2020, 8, 599281. [Google Scholar] [CrossRef] [PubMed]
- Ledebur, H.C.; Parks, T.P. Transcriptional Regulation of the Intercellular Adhesion Molecule-1 Gene by Inflammatory Cytokines in Human Endothelial Cells. J. Biol. Chem. 1995, 270, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S. THE NF-κB AND IκB PROTEINS: New Discoveries and Insights. Annu. Rev. Immunol. 1996, 14, 649–681. [Google Scholar] [CrossRef]
- Marumo, T.; Schini-Kerth, V.B.; Fisslthaler, B.; Busse, R. Platelet-Derived Growth Factor–Stimulated Superoxide Anion Production Modulates Activation of Transcription Factor NF-κB and Expression of Monocyte Chemoattractant Protein 1 in Human Aortic Smooth Muscle Cells. Circulation 1997, 9, 2361–2367. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Erfurth, U.; Garcia-Arumi, J.; Bandello, F.; Berg, K.; Chakravarthy, U.; Gerendas, B.S.; Jonas, J.; Larsen, M.; Tadayoni, R.; Loewenstein, A. Guidelines for the Management of Diabetic Macular Edema by the European Society of Retina Specialists (EURETINA). Ophthalmologica 2017, 237, 185–222. [Google Scholar] [CrossRef]
- Lozano, M.L.; Segú-Vergés, C.; Coma, M.; Álvarez-Roman, M.T.; González-Porras, J.R.; Gutiérrez, L.; Valcárcel, D.; Butta, N. Elucidating the Mechanism of Action of the Attributed Immunomodulatory Role of Eltrombopag in Primary Immune Thrombocytopenia: An In Silico Approach. Int. J. Mol. Sci. 2021, 22, 6907. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Casares, J.R.; Quintero, J.; Jorba, G.; Junet, V.; Martínez, V.; Pozo-Rubio, T.; Oliva, B.; Daura, X.; Mas, J.M.; Montoto, C. Methods to Develop an in Silico Clinical Trial: Computational Head-to-Head Comparison of Lisdexamfetamine and Methylphenidate. Front. Psychiatry 2021, 12, 741170. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Casares, J.R.; Quintero, J.; Segú-Vergés, C.; Rodríguez Monterde, P.; Pozo-Rubio, T.; Coma, M.; Montoto, C. In Silico Clinical Trial Evaluating Lisdexamfetamine’s and Methylphenidate’s Mechanism of Action Computational Models in an Attention-Deficit/Hyperactivity Disorder Virtual Patients’ Population. Front. Psychiatry 2023, 14, 939650. [Google Scholar] [CrossRef]
- Dinah, C.; Ghulakhszian, A.; Sim, S.Y.; Minocha, A.; Nokhostin, S.; Posner, E.; Cheong-Lee, R.; George, S. Aflibercept for Treatment-Naïve Diabetic Macula Oedema in a Multi-Ethnic Population: Real-World Outcomes from North West London. PLoS ONE 2021, 16, e0246626. [Google Scholar] [CrossRef] [PubMed]
- Hein, M.; Vukmirovic, A.; Constable, I.J.; Raja, V.; Athwal, A.; Freund, K.B.; Balaratnasingam, C. Angiographic Biomarkers Are Significant Predictors of Treatment Response to Intravitreal Aflibercept in Diabetic Macular Edema. Sci. Rep. 2023, 13, 8128. [Google Scholar] [CrossRef]
- Han, Y.E.; Jo, J.; Kim, Y.J.; Lee, J. Factors Affecting Intensive Aflibercept Treatment Response in Diabetic Macular Edema: A Real-World Study. J. Diabetes Res. 2023, 2023, 1485059. [Google Scholar] [CrossRef]
- Coto-Segura, P.; Segú-Vergés, C.; Martorell, A.; Moreno-Ramírez, D.; Jorba, G.; Junet, V.; Guerri, F.; Daura, X.; Oliva, B.; Cara, C.; et al. A Quantitative Systems Pharmacology Model for Certolizumab Pegol Treatment in Moderate-to-Severe Psoriasis. Front. Immunol. 2023, 14, 1212981. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural Apoptosis in the Retina during Experimental and Human Diabetes. Early Onset and Effect of Insulin. J. Clin. Investig. 1998, 102, 783–791. [Google Scholar] [CrossRef]
- Barber, A.J.; Van Guilder, H.D.; Gastinger, M.J. The Neuronal Influence on Retinal Vascular Pathology. In Retinal Vascular Disease; Springer: Heidelberg, Germany, 2007; pp. 108–120. [Google Scholar]
- Starace, V.; Battista, M.; Brambati, M.; Cavalleri, M.; Bertuzzi, F.; Amato, A.; Lattanzio, R.; Bandello, F.; Cicinelli, M.V. The Role of Inflammation and Neurodegeneration in Diabetic Macular Edema. Ther. Adv. Ophtalmol. 2021, 13, 1–17. [Google Scholar] [CrossRef]
- PubMed Database. Available online: https://www.ncbi.nlm.nih.gov/pubmed (accessed on 23 December 2020).
- European Medicines Agency Website. Available online: https://www.ema.europa.eu/en (accessed on 23 December 2020).
- US Food and Drug Administration Website. Available online: https://www.fda.gov/ (accessed on 23 December 2020).
- Drugbank Website. Available online: http://www.drugbank.ca/ (accessed on 23 December 2020).
- Stitch Website. Available online: http://stitch.embl.de/ (accessed on 23 December 2020).
- Supertarget Website. Available online: http://insilico.charite.de/supertarget/ (accessed on 23 December 2020).
- Perera, S.; Artigas, L.; Mulet, R.; Mas, J.M.; Sardón, T. Systems Biology Applied to Non-Alcoholic Fatty Liver Disease (NAFLD): Treatment Selection Based on the Mechanism of Action of Nutraceuticals. Nutrafoods 2014, 13, 61–68. [Google Scholar] [CrossRef]
- Moncunill, G.; Scholzen, A.; Mpina, M.; Nhabomba, A.; Hounkpatin, A.B.; Osaba, L.; Valls, R.; Campo, J.J.; Sanz, H.; Jairoce, C.; et al. Antigen-Stimulated PBMC Transcriptional Protective Signatures for Malaria Immunization. Sci. Transl. Med. 2020, 12, eaay8924. [Google Scholar] [CrossRef]
- Artigas, L.; Coma, M.; Matos-Filipe, P.; Aguirre-Plans, J.; Farrés, J.; Valls, R.; Fernandez-Fuentes, N.; De La Haba-Rodriguez, J.; Olvera, A.; Barbera, J.; et al. In-Silico Drug Repurposing Study Predicts the Combination of Pirfenidone and Melatonin as a Promising Candidate Therapy to Reduce SARS-CoV-2 Infection Progression and Respiratory Distress Caused by Cytokine Storm. PLoS ONE 2020, 15, e0240149. [Google Scholar] [CrossRef]
- Collet, P.; Rennard, J.-P. Stochastic Optimization Algorithms. In Intelligent Information Technologies: Concepts, Methodologies, Tools, and Applications; Idea Group Inc.: Hershey, PA, USA, 2008; pp. 1121–1137. [Google Scholar]
- Haritoglou, C.; Maier, M.; Augustin, A. Pathophysiology of Diabetic Macular Edema—A Background for Current Treatment Modalities. Expert Rev. Ophthalmol. 2018, 13, 273–281. [Google Scholar] [CrossRef]
- Rivals, I.; Personnaz, L.; Taing, L.; Potier, M.C. Enrichment or Depletion of a GO Category within a Class of Genes: Which Test? Bioinformatics 2007, 23, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Dimmer, E.C.; Huntley, R.P.; Alam-Faruque, Y.; Sawford, T.; O’Donovan, C.; Martin, M.J.; Bely, B.; Browne, P.; Mun Chan, W.; Eberhardt, R.; et al. The UniProt-GO Annotation Database in 2011. Nucleic Acids Res. 2012, 40, D565–D570. [Google Scholar] [CrossRef] [PubMed]
- Pujol, A.; Villanueva, J.J.; Alba, J.L. A Supervised Modification of the Hausdorff Distance for Visual Shape Classification. Int. J. Pattern Recognit. Artif. Intell. 2002, 16, 349–359. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanot, M.; Casaroli-Marano, R.P.; Mondéjar-Medrano, J.; Sallén, T.; Ramírez, E.; Segú-Vergés, C.; Artigas, L. Aflibercept Off-Target Effects in Diabetic Macular Edema: An In Silico Modeling Approach. Int. J. Mol. Sci. 2024, 25, 3621. https://doi.org/10.3390/ijms25073621
Blanot M, Casaroli-Marano RP, Mondéjar-Medrano J, Sallén T, Ramírez E, Segú-Vergés C, Artigas L. Aflibercept Off-Target Effects in Diabetic Macular Edema: An In Silico Modeling Approach. International Journal of Molecular Sciences. 2024; 25(7):3621. https://doi.org/10.3390/ijms25073621
Chicago/Turabian StyleBlanot, Morgane, Ricardo Pedro Casaroli-Marano, Jordi Mondéjar-Medrano, Thaïs Sallén, Esther Ramírez, Cristina Segú-Vergés, and Laura Artigas. 2024. "Aflibercept Off-Target Effects in Diabetic Macular Edema: An In Silico Modeling Approach" International Journal of Molecular Sciences 25, no. 7: 3621. https://doi.org/10.3390/ijms25073621
APA StyleBlanot, M., Casaroli-Marano, R. P., Mondéjar-Medrano, J., Sallén, T., Ramírez, E., Segú-Vergés, C., & Artigas, L. (2024). Aflibercept Off-Target Effects in Diabetic Macular Edema: An In Silico Modeling Approach. International Journal of Molecular Sciences, 25(7), 3621. https://doi.org/10.3390/ijms25073621