
Use of Enzymatically Converted Cell-Free DNA (cfDNA) Data for Copy Number Variation-Linked Fragmentation Analysis Allows for Early Colorectal Cancer Detection

 and
and
Abstract
1. Introduction
2. Results
2.1. Overall Approach
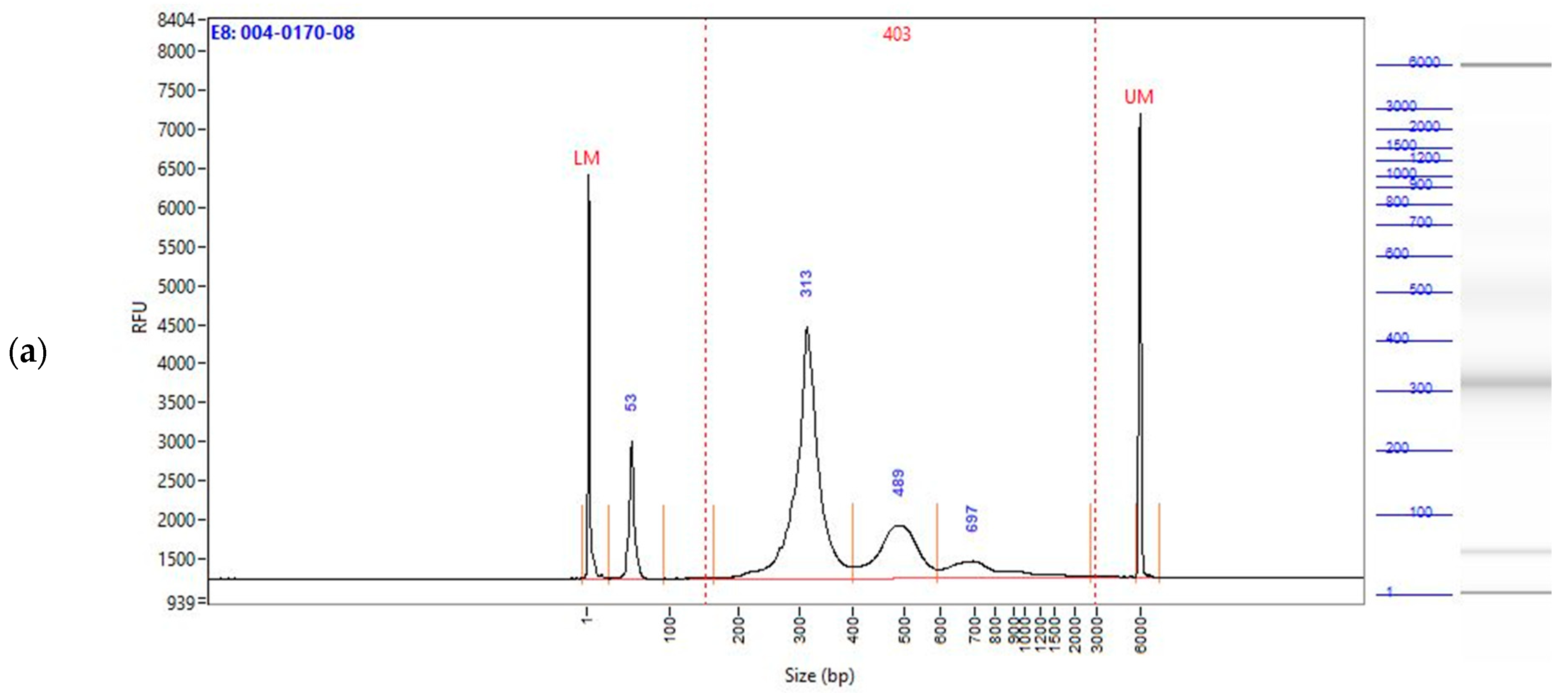
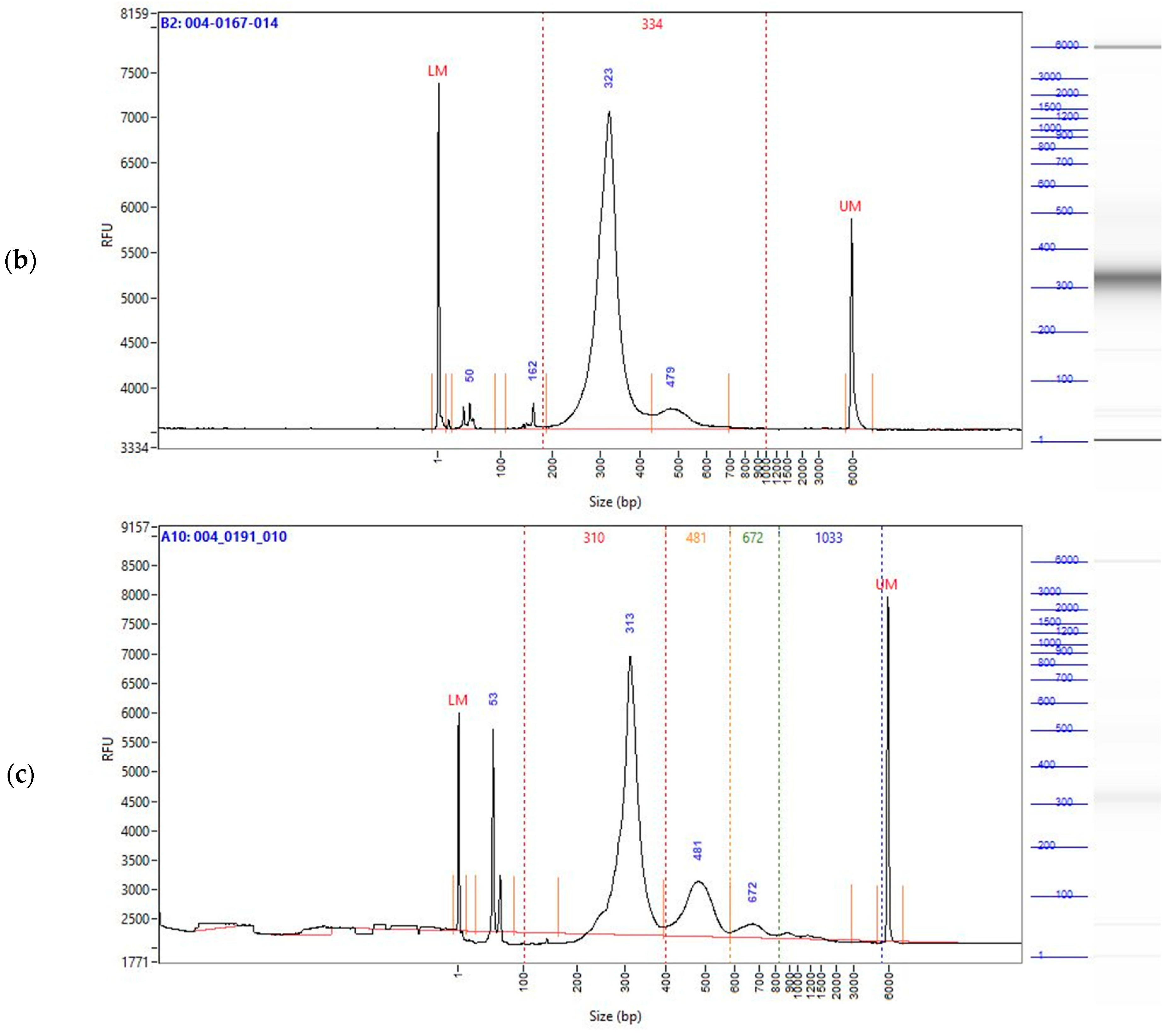
2.2. Enzymatically Converted cfDNA Conserves Biological Fragment Length Better than Bisulfite-Treated cfDNA
2.3. CNV Region Selection
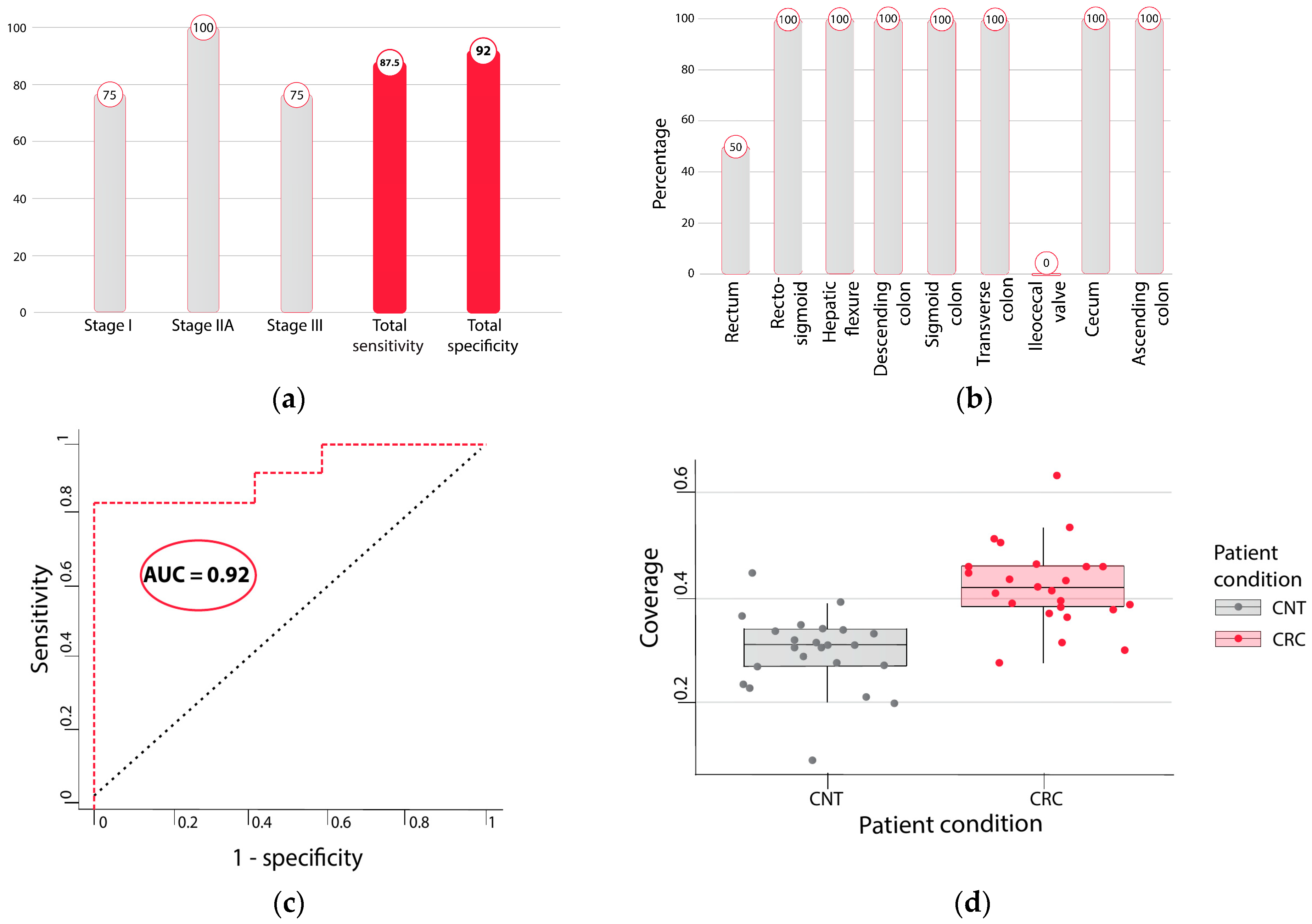
2.4. Detecting Cancer Samples with LDA Model Built over Enzymatically Converted Data
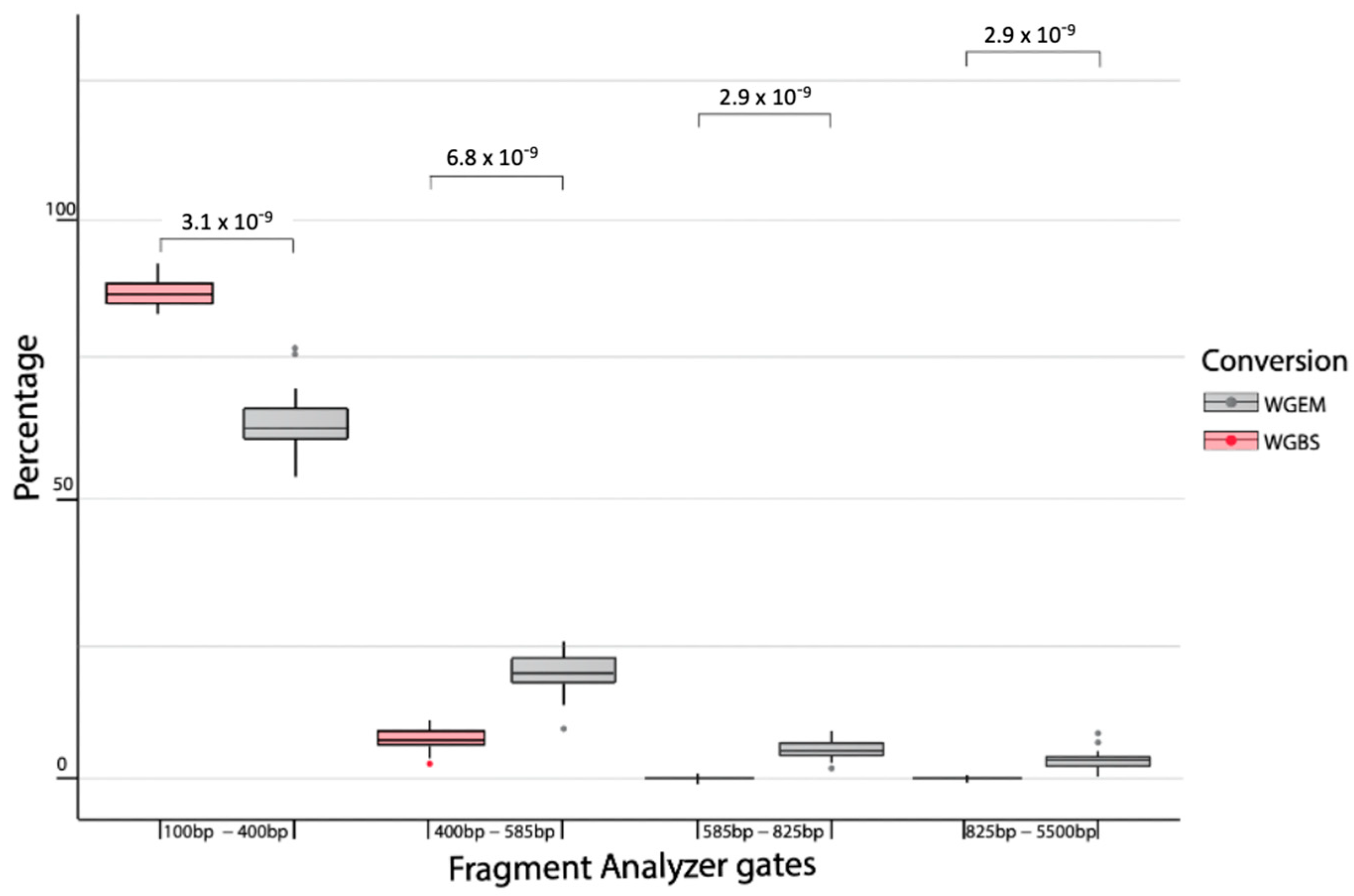
2.5. Whole Genome Enzymatic Sequencing Performs Better in Cancer Detection than Whole Genome Bisulfite Sequencing
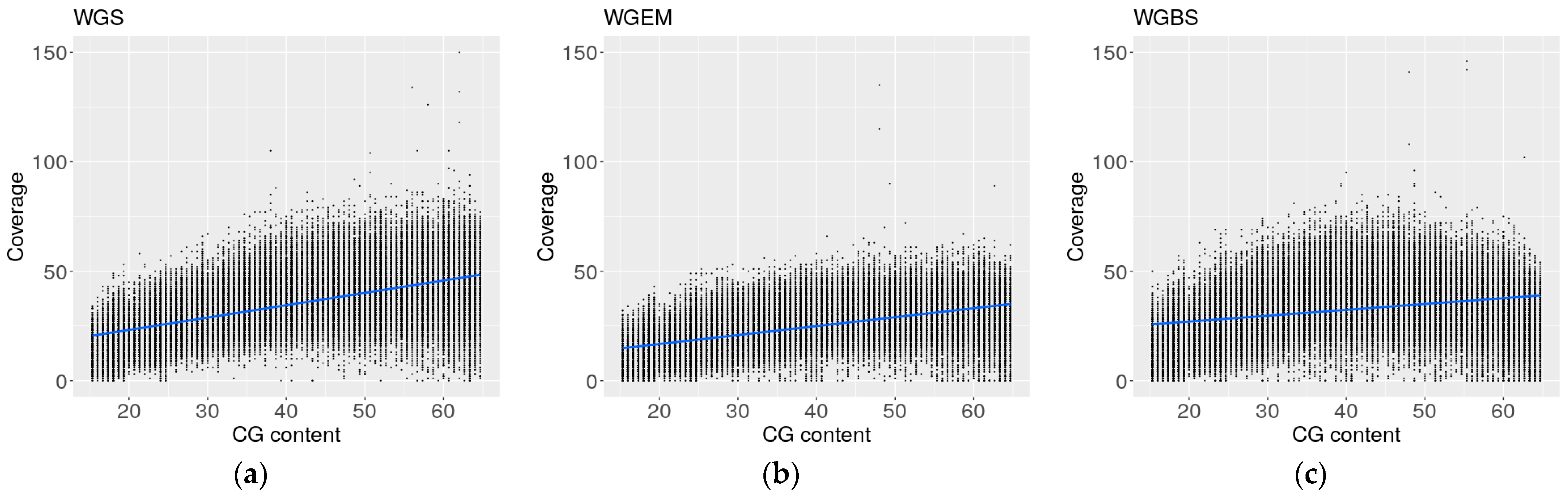
2.6. Coverage Profiles of Whole Genome Enzymatic Sequencing Are More Similar to Whole Genome Sequencing Profiles than Whole Genome Bisulfite Sequencing Profiles
3. Discussion
4. Materials and Methods
4.1. Sample Collection
Patient and Sample Characteristics
4.2. cfDNA Extraction and Library Preparation
4.3. Enzymatic Library Preparation
4.4. Bisulfite Library Preparation
4.5. Whole Genome (Non-Converted) Library Preparation
4.6. Sequencing
4.7. Alignment and Raw Data Processing
4.8. Fragment Analyzer Data Processing
4.9. Data Analysis
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- Baidoun, F.; Elshiwy, K.; Elkeraie, Y.; Merjaneh, Z.; Khoudari, G.; Sarmini, M.T.; Gad, M.; Al-Husseini, M.; Saad, A. Colorectal Cancer Epidemiology: Recent Trends and Impact on Outcomes. Curr. Drug Targets 2021, 22, 998–1009. [Google Scholar] [PubMed]
- Burnett-Hartman, A.N.; Lee, J.K.; Demb, J.; Gupta, S. An Update on the Epidemiology, Molecular Characterization, Diagnosis, and Screening Strategies for Early-Onset Colorectal Cancer. Gastroenterology 2021, 160, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Lech, G. Colorectal cancer tumour markers and biomarkers: Recent therapeutic advances. World J. Gastroenterol. 2016, 22, 1745. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2016, 66, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhu, L.; Song, J.; Wang, G.; Li, P.; Li, W.; Luo, P.; Sun, X.; Wu, J.; Liu, Y.; et al. Liquid biopsy at the frontier of detection, prognosis and progression monitoring in colorectal cancer. Mol. Cancer 2022, 21, 86. [Google Scholar] [CrossRef]
- Davidson, K.W.; Barry, M.J.; Mangione, C.M.; Cabana, M.; Caughey, A.B.; Davis, E.M.; Donahue, K.E.; Doubeni, C.A.; Krist, A.H.; Kubik, M.; et al. Screening for Colorectal Cancer. JAMA 2021, 325, 1965. [Google Scholar] [CrossRef]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9, eaan2415. [Google Scholar] [CrossRef]
- Arisi, M.F.; Dotan, E.; Fernandez, S.V. Circulating Tumor DNA in Precision Oncology and Its Applications in Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 4441. [Google Scholar] [CrossRef]
- Mandel, P.; Metais, P. Nuclear Acids in Human Blood Plasma. C. R. Seances Soc. Biol. Ses Fil. 1948, 142, 241–243. [Google Scholar]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Magbanua MJ, M.; Swigart, L.B.; Wu, H.-T.; Hirst, G.L.; Yau, C.; Wolf, D.M.; Tin, A.; Salari, R.; Shchegrova, S.; Pawar, H.; et al. Circulating tumor DNA in neoadjuvant-treated breast cancer reflects response and survival. Ann. Oncol. 2021, 32, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Husain, H.; Fanta, P.T.; Piccioni, D.E.; Kesari, S.; Schwab, R.B.; Patel, S.P.; Harismendy, O.; Ikeda, M.; Parker, B.A.; et al. Use of Liquid Biopsies in Clinical Oncology: Pilot Experience in 168 Patients. Clin. Cancer Res. 2016, 22, 5497–5505. [Google Scholar] [CrossRef] [PubMed]
- Vu, P.; Khagi, Y.; Riviere, P.; Goodman, A.; Kurzrock, R. Total Number of Alterations in Liquid Biopsies Is an Independent Predictor of Survival in Patients with Advanced Cancers. JCO Precis. Oncol. 2020, 4, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef]
- Liu, L.; Toung, J.M.; Jassowicz, A.F.; Vijayaraghavan, R.; Kang, H.; Zhang, R.; Kruglyak, K.M.; Huang, H.J.; Hinoue, T.; Shen, H.; et al. Targeted methylation sequencing of plasma cell-free DNA for cancer detection and classification. Ann. Oncol. 2018, 29, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Cummings, S.R.; Absalan, F.; Alexander, G.; Allen, B.; Amini, H.; et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, Y.; Hu, T.; He, X.; Zou, Y.; Deng, Q.; Ke, J.; Lian, L.; He, X.; Zhao, D.; et al. A novel cell-free DNA methylation-based model improves the early detection of colorectal cancer. Mol. Oncol. 2021, 15, 2702–2714. [Google Scholar] [CrossRef] [PubMed]
- Olova, N.; Krueger, F.; Andrews, S.; Oxley, D.; Berrens, R.V.; Branco, M.R.; Reik, W. Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data. Genome Biol. 2018, 19, 33. [Google Scholar] [CrossRef]
- Tanaka, K.; Okamoto, A. Degradation of DNA by bisulfite treatment. Bioorg. Med. Chem. Lett. 2007, 17, 1912–1915. [Google Scholar] [CrossRef]
- Morrison, J.; Koeman, J.M.; Johnson, B.K.; Foy, K.K.; Beddows, I.; Zhou, W.; Chesla, D.W.; Rossell, L.L.; Siegwald, E.J.; Adams, M.; et al. Evaluation of whole-genome DNA methylation sequencing library preparation protocols. Epigenet. Chromatin 2021, 14, 28. [Google Scholar] [CrossRef]
- Vaisvila, R.; Ponnaluri VK, C.; Sun, Z.; Langhorst, B.W.; Saleh, L.; Guan, S.; Dai, N.; Campbell, M.A.; Sexton, B.S.; Marks, K.; et al. Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res. 2021, 31, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Cristiano, S.; Leal, A.; Phallen, J.; Fiksel, J.; Adleff, V.; Bruhm, D.C.; Jensen, S.Ø.; Medina, J.E.; Hruban, C.; White, J.R.; et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019, 570, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Li, X.; Zhang, M.; Yang, Y.; Ge, G.; Wang, K.; Gong, Y.; Liang, Y.; Niu, H.; Ci, W. Enhanced Detection of Genitourinary Cancers Using Fragmentation and Copy Number Profiles Obtained from Urinary Cell-Free DNA. Clin. Chem. 2021, 67, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Sun, K.; Peng, W.; Cheng, S.H.; Ni, M.; Yeung, P.C.; Heung MM, S.; Xie, T.; Shang, H.; Zhou, Z.; et al. Plasma DNA End-Motif Profiling as a Fragmentomic Marker in Cancer, Pregnancy, and Transplantation. Cancer Discov. 2020, 10, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. At the dawn: Cell-free DNA fragmentomics and gene regulation. Br. J. Cancer 2021, 126, 379–390. [Google Scholar] [CrossRef]
- Martignano, F.; Munagala, U.; Crucitta, S.; Mingrino, A.; Semeraro, R.; Del Re, M.; Petrini, I.; Magi, A.; Conticello, S.G. Nanopore sequencing from liquid biopsy: Analysis of copy number variations from cell-free DNA of lung cancer patients. Mol. Cancer 2021, 20, 32. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.A.; Gibson, W.J.; Brown, M.S.; Kosmicki, J.A.; Busanovich, J.P.; Wei, H.; Urbanski, L.M.; Curimjee, N.; Berger, A.C.; Gao, G.F.; et al. Loss of heterozygosity of essential genes represents a widespread class of potential cancer vulnerabilities. Nat. Commun. 2020, 11, 2517. [Google Scholar] [CrossRef] [PubMed]
- Bui, V.M.H.; Mettling, C.; Jou, J.; Sun, H.S. Genomic amplification of chromosome 20q13.33 is the early biomarker for the development of sporadic colorectal carcinoma. BMC Med. Genom. 2020, 13 (Suppl. S10), 149. [Google Scholar] [CrossRef]
- Sarli, L.; Bottarelli, L.; Bader, G.; Iusco, D.; Pizzi, S.; Costi, R.; D’Adda, T.; Bertolani, M.; Roncoroni, L.; Bordi, C. Association Between Recurrence of Sporadic Colorectal Cancer, High Level of Microsatellite Instability, and Loss of Heterozygosity at Chromosome 18q. Dis. Colon Rectum 2004, 47, 1467–1482. [Google Scholar] [CrossRef]
- Popat, S.; Houlston, R.S. A systematic review and meta-analysis of the relationship between chromosome 18q genotype, DCC status and colorectal cancer prognosis. Eur. J. Cancer 2005, 41, 2060–2070. [Google Scholar] [CrossRef]
- Fearon, E.R.; Cho, K.R.; Nigro, J.M.; Kern, S.E.; Simons, J.W.; Ruppert, J.M.; Hamilton, S.R.; Preisinger, A.C.; Thomas, G.; Kinzler, K.W. Identification of a chromosome 18q gene that is altered in colorectal cancers. Science 1990, 247, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Ried, T.; Meijer, G.A.; Harrison, D.J.; Grech, G.; Sebastià Franch-Expósito Briffa, R.; Carvalho, B.; Camps, J. The landscape of genomic copy number alterations in colorectal cancer and their consequences on gene expression levels and disease outcome. Mol. Asp. Med. 2019, 69, 48–61. [Google Scholar] [CrossRef]
- Fleming, N.I.; Jorissen, R.N.; Mouradov, D.; Christie, M.; Sakthianandeswaren, A.; Palmieri, M.; Day, F.; Li, S.; Tsui, C.; Lipton, L.; et al. SMAD2, SMAD3 and SMAD4 Mutations in Colorectal Cancer. Cancer Res. 2012, 73, 725–735. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ding, N.; Luo, H.; Zhang, T.; Peng, T.; Yao, Y. Correlation between SMADs and Colorectal Cancer Expression, Prognosis, and Immune Infiltrates. Int. J. Anal. Chem. 2023, 2023, 8414040. [Google Scholar]
- Deletion and Down-Regulation of SMAD4 Gene in Colorectal Cancers in a Chinese Population—Chinese Journal of Cancer Research. Available online: https://www.cjcrcn.org/article/html_4877.html (accessed on 29 January 2024).
- Boulay, J.-L.; Mild, G.; Lowy, A.; Reuter, J.; Lagrange, M.; Terracciano, L.; Laffer, U.; Herrmann, R.; Rochlitz, C. SMAD7 is a prognostic marker in patients with colorectal cancer. Int. J. Cancer 2003, 104, 446–449. [Google Scholar] [CrossRef]
- Keino-Masu, K.; Masu, M.; Hinck, L.; Leonardo EDavid Chan, S.S.-Y.; Culotti, J.G.; Tessier-Lavigne, M. Deleted in Colorectal Cancer (DCC) Encodes a Netrin Receptor. Cell 1996, 87, 175–185. [Google Scholar] [CrossRef]
- Mehlen, P.; Fearon, E.R. Role of the Dependence Receptor DCC in Colorectal Cancer Pathogenesis. J. Clin. Oncol. 2004, 22, 3420–3428. [Google Scholar] [CrossRef]
- Reale, M.A.; Hu, G.; Zafar, A.I.; Getzenberg, R.H.; Levine, S.M.; Fearon, E.R. Expression and alternative splicing of the deleted in colorectal cancer (DCC) gene in normal and malignant tissues. Cancer Res. 1994, 54, 4493–4501. [Google Scholar]
- Gao, Q.; Zeng, Q.; Wang, Z.; Li, C.; Xu, Y.; Cui, P.; Zhu, X.; Lu, H.; Wang, G.; Cai, S.; et al. Circulating cell-free DNA for cancer early detection. Innovation 2022, 3, 100259. [Google Scholar] [CrossRef]
- Issa, I.A.; Noureddine, M. 2017. Colorectal cancer screening: An updated review of the available options. World J. Gastroenterol. 2017, 23, 5086. [Google Scholar] [CrossRef]
- Chen, H.; Li, N.; Ren, J.; Feng, X.; Lyu, Z.; Wei, L.; Li, X.; Guo, L.; Zheng, Z.; group of Cancer Screening Program in Urban China (CanSPUC); et al. Participation and yield of a population-based colorectal cancer screening programme in China. Gut 2019, 68, 1450–1457. [Google Scholar] [CrossRef] [PubMed]
- Hallermayr, A.; Wohlfrom, T.; Steinke-Lange, V.; Benet-Pagès, A.; Scharf, F.; Heitzer, E.; Mansmann, U.; Haberl, C.; de Wit, M.; Vogelsang, H.; et al. Somatic copy number alteration and fragmentation analysis in circulating tumor DNA for cancer screening and treatment monitoring in colorectal cancer patients. J. Hematol. Oncol. 2002, 15, 125. [Google Scholar] [CrossRef] [PubMed]
- Zhitnyuk, Y.V.; Koval, A.P.; Alferov, A.A.; Shtykova, Y.A.; Mamedov, I.Z.; Kushlinskii, N.E.; Chudakov, D.M.; Shcherbo, D.S. Deep cfDNA fragment end profiling enables cancer detection. Mol. Cancer 2022, 21, 26. [Google Scholar] [CrossRef] [PubMed]
- Nassar, R.; Thompson, L.; Fouquerel, E. Molecular mechanisms protecting centromeres from self-sabotage and implications for cancer therapy. NAR Cancer 2023, 5, zcad019. [Google Scholar] [CrossRef]
- Krueger, F. Trim Galore! Available online: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 19 November 2019).
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Range | % Total |
|---|---|---|
| WGEM | 100 bp to 400 bp | 60.6 |
| WGEM | 400 bp to 585 bp | 19.6 |
| WGEM | 585 bp to 825 bp | 5.9 |
| WGEM | 825 bp to 5500 bp | 3 |
| WGBS | 100 bp to 400 bp | 86.8 |
| WGBS | 400 bp to 585 bp | 9 |
| WGBS | 585 bp to 825 bp | 1 |
| WGBS | 825 bp to 5500 bp | 0.5 |
| WGS | 100 bp to 400 bp | 67.4 |
| WGS | 400 bp to 585 bp | 19.3 |
| WGS | 585 bp to 825 bp | 3.9 |
| WGS | 825 bp to 5500 bp | 1.3 |
| Characteristics | Controls | CRC |
|---|---|---|
| Age (years, mean (IQR)) | 68.9 (63–77) | 68.9 (63–77) |
| Female ((n (%)) | 12 (50%) | 13 (54.17%) |
| Male ((n (%)) | 12 (50%) | 11 (45.83%) |
| BMI (kg/m2, mean (IQR)) | 28.95 (25–29.3) | 29.6 (26.73–30.55) |
| CRC stage (n) | 24 | 24 |
| Stage 0 (n) | 3 | |
| Stage I (n) | 10 | |
| Stage IIA (n) | 5 | |
| Stage III (n) | 3 | |
| Stage IIIA (n) | 2 | |
| Stage IIIB (n) | 1 |
| Characteristics | Controls | CRC |
|---|---|---|
| Age (years, mean (IQR)) | 68.125 (60.75–75.50) | 68.125 (60.75–75.50) |
| Female ((n (%)) | 13 | 13 |
| Male ((n (%)) | 11 | 11 |
| BMI (kg/m2, mean (IQR)) | 29.01 (6 NA) (26.275–30.725) | 28.58 (7 NA) (25.6–28.7) |
| CRC stage (n) | 24 | 24 |
| Stage 0 (n) | 3 | |
| Stage I (n) | 6 | |
| Stage IIA (n) | 6 | |
| Stage III (n) | 4 | |
| Stage IIIA (n) | 1 | |
| Stage IIIB (n) | 1 | |
| Stage IV (n) | 3 |
| Characteristics | Controls | CRC |
|---|---|---|
| Age (years, mean (IQR)) | 65.17 (60.75–70.75) | 64.4 (61–69.5) |
| Female ((n (%)) | 6 (50%) | 5 (41.67%) |
| Male ((n (%)) | 6 (50%) | 7 (58.33%) |
| BMI (kg/m2, mean (IQR)) | 28.2 (24.95–30.85) | 28.92 (26.37–31.3) |
| CRC stage (n) | 12 | 12 |
| Stage I (n) | 4 | |
| Stage IIA (n) | 4 | |
| Stage III (n) | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Černoša, I.; Trincado-Alonso, F.; Canal-Noguer, P.; Kruusmaa, K.; Perera-Lluna, A. Use of Enzymatically Converted Cell-Free DNA (cfDNA) Data for Copy Number Variation-Linked Fragmentation Analysis Allows for Early Colorectal Cancer Detection. Int. J. Mol. Sci. 2024, 25, 3502. https://doi.org/10.3390/ijms25063502
Černoša I, Trincado-Alonso F, Canal-Noguer P, Kruusmaa K, Perera-Lluna A. Use of Enzymatically Converted Cell-Free DNA (cfDNA) Data for Copy Number Variation-Linked Fragmentation Analysis Allows for Early Colorectal Cancer Detection. International Journal of Molecular Sciences. 2024; 25(6):3502. https://doi.org/10.3390/ijms25063502
Chicago/Turabian StyleČernoša, Iva, Fernando Trincado-Alonso, Pol Canal-Noguer, Kristi Kruusmaa, and Alexandre Perera-Lluna. 2024. "Use of Enzymatically Converted Cell-Free DNA (cfDNA) Data for Copy Number Variation-Linked Fragmentation Analysis Allows for Early Colorectal Cancer Detection" International Journal of Molecular Sciences 25, no. 6: 3502. https://doi.org/10.3390/ijms25063502
APA StyleČernoša, I., Trincado-Alonso, F., Canal-Noguer, P., Kruusmaa, K., & Perera-Lluna, A. (2024). Use of Enzymatically Converted Cell-Free DNA (cfDNA) Data for Copy Number Variation-Linked Fragmentation Analysis Allows for Early Colorectal Cancer Detection. International Journal of Molecular Sciences, 25(6), 3502. https://doi.org/10.3390/ijms25063502